

Abstract
The CD6 membrane-proximal scavenger receptor cysteine-rich domain (SRCR3) includes the activated leucocyte cell adhesion molecule (ALCAM) binding site. CD6-ALCAM mediates a low-affinity interaction and their long-term engagement contributes to the immunological synapse. Their ligation may play a dual function, facilitating stable adhesion between the antigen-presenting cells and T cells during the early activation phase and later in the proliferative phase of the immune response. This study explored the strength of the CD6 co-stimulatory effect and whether CD6 co-stimulation with its natural ligand ALCAM also contributes to the lymphocyte effector differentiation. It was found that CD6–ALCAM interaction in vitro induced a synergistic co-stimulation of normal human peripheral blood mononuclear cells, defined by Bliss analysis. CD6 co-stimulation enhanced the CD3 proliferative efficacy by 23–34%. Moreover, a fivefold increment in the CD25 molecules number with a distinct gene transcription profile associated with cell activation, differentiation, survival and adhesion molecules was observed over CD3 single activation. Additionally, CD6 co-stimulation in excess interleukin (IL)-2 promotes a preferentially proinflammatory response. Besides, a CD6 membrane-distal domain (SRCR1)-specific non-depleting monoclonal antibody (mAb) inhibited the induced proliferation in the presence of ALCAM, reducing interferon-γ, IL-6 and tumour necrosis factor-α production. These results suggest that CD6 co-stimulation enhances the intrinsic activity of the CD3 activation pathway and contributes to the T helper type 1 subset commitment, enhancing the IL-2 sensitivity of recent activated human lymphocytes. It supports the role of CD6 as a susceptibility gene for pathological autoimmunity leading to tissue inflammation, and its relevance for targeted therapy.
Keywords: ALCAM, CD6, human, IL-2, SRCR
Introduction
Peripheral lymphocyte activation, division and effector differentiation integrates multiple signals from membrane-bound and soluble molecules. Productive T cell activation requires signalling through the antigen-specific receptor [T cell receptor (TCR)/CD3] complex and the co-ligation of other surface receptors [1,2]. CD3 recognition in vitro with specific monoclonal antibodies emulates antigen-specific stimulation, inducing monocyte-independent T cell activation and proliferation [3,4].
Identified as the prototypic co-stimulatory molecule, CD28 engagement promotes a quantitative change in activation leading to the proliferation and cytokine production by T cells [5]. Similarly, CD6 is a surface glycoprotein with co-stimulatory functions in mature T lymphocytes [6]. Dissected mainly through the cross-linking with monoclonal antibodies (mAb) [7,8], CD6 ligation enhances anti-CD3-induced human T cell proliferation comparable to the CD28 co-stimulation in vitro[9,10]. Distinctively, CD6- but not CD28-mediated proliferation is affected on interleukin (IL)-2 deficit [8,11–13]. Moreover, the anti-CD6 monoclonal antibody (mAb) promotes distinct functional responses depending upon the specific epitope recognized on the extracellular domains of the CD6 molecule [14,15], and potentially also on the mAb affinity.
The CD6 molecule contains three extracellular domains with the characteristic structure of the scavenger receptor cystein-rich (SRCR) superfamily [6]. Its membrane-proximal domain (SRCR3) includes the binding site for activated leucocyte cell adhesion molecule (ALCAM), a CD6 natural ligand member of the immunoglobulin supergene family [16,17]. CD6 mediates low-affinity binding to ALCAM, like most other leucocyte membrane protein interactions [18]. The physiological role of ALCAM in cell adhesion was identified early in vitro[17], and recently its role in leucocyte trafficking in the immunopathological process associated with an autoimmune disease model was explored in vivo[19].
The immunological synapse is a specialized cell structure site where the T cell activation process is not only triggered but also controlled. The outcome is determined by the molecular composition influenced by the local cytokine milieu [20,21]. Interestingly, it has been identified that the SRCR3 binding to ALCAM targets human CD6 at the immunological synapse [22]. Moreover, CD6 associates physically with the TCR/CD3 complex at the central supramolecular activation cluster, a topographic co-localization that suggests the potential relevance of CD6–ALCAM interaction in the modulation of T cell activation [9,23]. Indeed, interfering with this binding as explored by co-culturing human peripheral blood mononuclear cells (PBMCs) with allogeneic dendritic cells inhibits lymphocyte proliferation [9,23]. This finding supported a dual function of CD6, facilitating stable adhesion between the antigen-presenting cells and T cells during the early activation phase and later in the proliferative phase of the immune response [9,23]. Furthermore, the single CD6 ligation with an ALCAM-Fc chimera may promote intracellular pathway activation on T cells [10], although no further consequences for T cell immunobiology have been explored. Conversely, the bacterial binding properties of the CD6 ectodomain through the recognition of pathogen-associated molecular patterns have been identified recently. Displaying an interaction at the micromolar range, lipopolysaccharide-binding to membrane CD6 activates intracellular signalling cascades. Moreover, interfering with this interaction reduces the level of proinflammatory cytokines in plasma of an experimental animal model of septic shock [24]. This would suggest the implication of CD6 at the interface between innate and adaptive immunity.
Consequently, this study explored the strength of the CD6 co-stimulatory effect on recent activated human lymphocytes with a CD3-specific mAb and an immobilized ALCAM-Fc chimera, emulating the physiological conditions. Furthermore, we tested the hypothesis that CD6 co-stimulation with its natural ligand ALCAM also contributes to lymphocyte effector differentiation, modulated by IL-2. This is based on the consideration that cytokines function in a context-dependent manner [25], and their integration with heterologous receptor signalling pathways becomes critical in immune regulation [2,21]. IL-2 is secreted directionally into the immunological synapse, as defined recently, supporting its relevance to the priming and outcome of the immune response [26]. Finally, the potential CD6-mediated effect modulation with no interference in its adhesion to ALCAM was examined, using a well-defined CD6 membrane-distal domain (SRCR1)-specific mAb [27,28].
Here we show that CD6–ALCAM ligation induces a synergistic co-stimulation enhancing the intrinsic activity of the CD3 activation pathway in human lymphocytes. It contributes to the lymphocyte effector differentiation to the T helper type 1 (Th1) subset, enhancing the IL-2 sensitivity of recent activated cells. Targeting CD6 without interfering with ALCAM interaction modulates the immunoresponse, reducing cell activation, proliferation and proinflammatory response. These results suggest the involvement of the CD6 molecule in the fine tuning of the peripheral immune response, which may have therapeutic implications.
Materials and methods
Antibodies and reagents
The mouse immunoglobulin (Ig)G2a anti-human CD3 mAb (IOR-T3) was produced at the Center of Molecular Immunology, Havana, Cuba [29]. The T1h, a CD6 membrane-distal domain (SRCR1)-specific humanized IgG1 non-depleting mAb [27,28], and Nimotuzumab, an epidermal growth factor receptor-specific humanized IgG1 mAb [30],used as irrelevant isotype control, were produced at Biocon Ltd (Bangalore, India). Anti-human CD3-fluorescein isothiocyanate (FITC) (UCHT1 clone), CD4-Alexa (RPA-J4 clone), CD6-phycoerythrin (PE) (MEM98 clone), CD8-PE (LT8 clone), CD25-PE (MEM181 clone) and CD69-FITC (CH/4 clone) were obtained from AbD Serotec, Kidlington, Oxford, UK. Human recombinant IL-2 was obtained from Invitrogen Corporation, Carlsbad, CA, USA. The human ALCAM-Fc chimera was purchased from R&D Systems (Minneapolis, MN, USA).
Sampling and cell culture
The research programme including multi-centre studies in human subjects was approved by the relevant institutional ethic committees. Human PBMCs were obtained from peripheral venous samples from healthy volunteer blood donors after signing an informed consent form. Samples were subjected to 30 min standard density gradient centrifugation over Ficoll Paque (GE Healthcare Bio-sciences AB, Uppsala, Sweden). The purity of lymphocytes from this process was more than 70%, assessed by flow cytometry for CD3+ cells.
Cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 25 mM HEPES, 1% of 1 × 104 U/ml of penicillin and 1 × 104 µg/ml of streptomycin (Invitrogen Corporation). Cell culture was performed using 3 × 104 freshly isolated cells (100 µl) per well in triplicate or in quadruplicate in 96-well flat-bottomed Fluotrac 600 plates (Greiner Bio-One GmbH, Maybachstrasse, Frickenhausen, Germany) at 37°C and 5% CO2. Cells were incubated for 72 h for the proliferation assay and the flow cytometry analysis, and up to 96 h for cytokine analysis.
Cell proliferation
Anti-human CD3 mAb (0·0625–1 µg/ml) and human ALCAM-Fc, used at the highest assayed concentration (1 µg/ml) as there was no proliferation observed up to that amount, alone or in combination, were coated with 100 µl of coating buffer containing NaHCO3 8·4 g, Na2CO3 3·56 g (Sigma-Aldrich Co, St. Louis, MO, USA) in H2O 1000 ml, pH 9·5, and incubated at 4°C overnight. Plates were brought to room temperature and aspirated using a vaccusafe (Integra Biosciences AG, Landquart, Switzerland). Non-specific binding sites were blocked by adding 200 µl of 1% bovine serum albumin (BSA) (Sigma-Aldrich) in phosphate-buffered saline (PBS). Plates were incubated at 37°C for 1 h and washed twice with PBS/Tween20 (0·5%) (Sigma-Aldrich). The third wash was given with culture media. IL-2 was added at 2·5 ng/ml (based on titration in unstimulated cells, data not shown). CD6 membrane-distal domain-specific mAb (2·5–40 µg/ml) or the irrelevant isotype control were added in respective wells (100 µl) when required. Cells were cultured for 72 h and Alamar Blue solution (65 µl; Invitrogen, BioSource, Nivelles, Belgium) was added to the cultures for the last 18 h of incubation. Thereafter, cells were harvested and the fluorescence counted using a 96-well fluorimeter (Synergy HT with Gen 5 software; BioTek Instruments Inc, Highland Park, Winooski, VT, USA) with excitation at 530 nm and emission at 590 nm (sensitivity = 35).
Bliss analysis
Bliss analysis was performed to define whether the combination of anti-CD3 mAb with the ALCAM-Fc is synergistic or additive compared to each of them independently, as described previously [31,32]. A theoretical curve (dotted line) was calculated for combined proliferation by using the equation EBliss = EA + EB − EA × EB, where EA and EB are the fractional proliferation obtained by molecule A alone and molecule B alone. In this experiment, molecule A is the anti-CD3 mAb with a concentration range (0·0625–1 µg/ml) while molecule B is the ALCAM-Fc (1 µg/ml). All other proliferations at identical concentrations to anti-CD3 mAb were assumed to be equal to this. The theoretical curve thus generated is the EBliss, which is the fractional proliferation that would be expected if the effect of the two molecules were additive. If the experimentally measured curve was higher than this, then the combination was considered synergistic.
Flow cytometry analysis
Plates were coated with the anti-CD3 mAb (0·25 µg/ml) alone or in combination with the ALCAM-Fc (1 µg/ml). IL-2 was added at 2·5 ng/ml and the anti-CD6 mAb and the irrelevant isotype control mAb at 10 µg/ml. Cells from quadruplicate wells were pooled and spun down at 270 g for 5 min at 4°C; 1 × 105 cells in PBS (100 µl) were incubated with conjugated anti-human lymphocyte specific markers or irrelevant isotype control mAb for 30 min at 4°C. After washing in PBS, cells were resuspended in a final volume of 500 µl. Acquisition and analysis of 3000 gated lymphocytes were performed using a Cyan-ADP flow cytometer with the Summit version 4·3 software (Beckman Coulter, Fullerton, CA, USA). A culture plate under similar conditions was set up and cell proliferation evaluated by Alamar Blue, making the studies comparable.
Bead-based receptor quantification analysis
Bead-based receptor quantification was performed as reported previously [33]. Briefly, SPHEROTM RCP-30-5A (Spherotech Inc., Lake Forest, CA, USA) rainbow calibration particles were acquired with cells labelled with conjugated anti-human lymphocyte-specific marker mAbs, as described above, and at an identical voltage setting. According to the manufacturer's protocol, the relative number of fluorophores per particles were determined for every peak of RCP-30-5 in FL1 (FITC, MEFL), FL2 (RPE, MEPE), FL3 (RPE-Cy5, MEPCY) and FL4 (APC, MEAP) channels of the flow cytometer to plot the calibration graph. The calibration graph was used to check the linearity of the photomultiplier tube in each channel. In addition, the relative number of fluorophores could be cross-calibrated with cells or particles stained with a known number of spectral matching fluorophores such as FITC, PE or RPE-Cy5 to estimate the number of fluorophores on stained cells. The method of receptor quantification on the surface of cells is similar to the fluorescence mean channel number (i.e. relative brightness). The mean channel number was converted to the relative channel number using the following formula: (i) relative channel number (RCN) = (R/4) log (mean channel number × 10); and (ii) where R = resolution (i.e. 256). The RCN obtained from running the cells with the particular flurochrome was plotted on the Sphero calibration graph to quantify the CD3, CD4, CD8, CD25 and CD69 molecules. All antibodies were used at saturation concentration (data not shown).
Cytokine analysis
Plates were coated with the anti-CD3 mAb (0·25 µg/ml) alone or in combination with the ALCAM-Fc (1 µg/ml). IL-2 was added at 2·5 ng/ml and the anti-CD6 mAb and the irrelevant isotype control mAb at 10 µg/ml. Cultured cells from quadruplicate wells were pooled and spun down at 270 g for 5 min at 4°C, and the supernatant was collected. For cytokine analysis the Th1/Th2 CBA human kit (BD Biosciences, Pharmingen, San Jose, CA, USA) was used as described previously [34]. Briefly, one vial of lyophilized human Th1/Th2 cytokine standards was reconstituted with 0·2 ml of assay diluents to prepare a 10× bulk standard and allowed to equilibrate for at least 15 min before making dilutions. The capture beads were bottled individually and it was necessary to pool the bead reagents (A1–A6) immediately before mixing them together with the PE detection reagent, standards and samples. Each capture bead suspension was vortexed before mixing and 10 µl added for each assay tube to be analysed into a single tube labelled ‘mixed capture beads’ (e.g. 10 µl of IL-6 capture beads × 18 assay tubes = 180 µl of IL-6 capture beads required). Test samples were diluted by the desired dilution factor (i.e. 1:2, 1:10, or 1:100) using the appropriate volume of assay diluent. The sample dilutions were mixed thoroughly before transferring samples to the appropriate assay tubes containing mixed capture beads and PE detection reagent. Fifty µl of the mixed capture beads (prepared using the procedure described in the preparation of mixed human Th1/Th2 cytokine capture beads) were added to the appropriate assay tubes. The mixed capture beads were vortexed before adding to the assay tubes. Fifty µl of the human Th1/Th2-II PE detection reagent was added to the assay tubes. Fifty µl of the human Th1/Th2 cytokine standard dilutions was added to the control assay tubes. Fifty µl of each test sample was added to the test assay tubes. The assay tubes were incubated for 3 h at room temperature and protected from direct exposure to light. One ml of wash buffer was added to each assay tube and centrifuged at 200 g for 5 min. The supernatant was aspirated carefully and discarded from each assay tube; 300 µl of wash buffer was added to each assay tube to resuspend the bead pellet. Samples were analysed by using a Cyan-ADP flow cytometer with the Summit version 4·3 software (Beckman Coulter).
Signal transduction molecule analysis
Intracellular phospho-proteins were analysed by flow cytometry, as described previously [35]. Briefly, plates were coated with the anti-CD3 mAb (0·25 µg/ml) in 96-well plates. The anti-CD6 mAb and the irrelevant isotype control mAb were added at 10 µg/ml. After 24 h incubation, cells from quadruplicate wells were pooled and resuspended in 2% formaldehyde and fixed for 10 min at 37°C. Cells were permeabilized using ice-cold 100% methanol while gently vortexing, then washed in the incubation buffer (0·5 g BSA in 100 ml PBS) and resuspended in 100 µl incubation buffer. Specific antibodies to p44/42 mitogen-activated protein kinase (MAPK) (Erk1/2) (137F5 clone), Phospho-Stat3 (Tyr705) (D3A7 clone) and pAKT (Ser473) (193H12 clone) (Cell Signaling Technology, Danvers, MA, USA) were added and incubated overnight at 4°C. Cells were resuspended in incubation buffer and a secondary PE-conjugated antibody (Southern Biotech, Birmingham, AL, USA) was incubated for 30 min. Samples were rinsed in incubation buffer and finally resuspended in 0·5 ml PBS and acquired in the Beckman Coulter cyan cytometer. Mean fluorescence intensity was defined for 3000 gated lymphocytes compared to the irrelevant isotype control mAb.
Microarray analysis
The difference in gene expression between cells unstimulated and stimulated with the anti-CD3 mAb (0·25 µg/ml) alone or in combination with the ALCAM-Fc (1 µg/ml), IL-2 (2·5 ng/ml) or both was performed following a standard protocol [36]. After 72 h incubation, cells from quadruplicate wells were pooled and spun down at 270 g for 5 min at 4°C and collected in RNA later solution (Sigma, Ambion Inc., St Louis, MO, USA). A 1:1 ratio of PBS to RNA was added later and the cells spun down. Total RNA was isolated using Trizol reagent following standard protocol (Invitrogen, Carlsbad, CA, USA). One ml Trizol reagent was added to the cell pellet and mixed well. After 5 min incubation at room temperature, chloroform (200 µl) was added and the tubes were vortexed vigorously for 15 s and incubated at room temperature for 2 min. The upper aqueous (RNA) phase was collected following centrifugation (12 000 g for 10 min at 4°C) into a fresh tube and then precipitated into a pellet with isopropyl alcohol (∼350 µl) by incubating for 10 min at room temperature and centrifuging 12 000 g for 10 min at 4°C. After removal of the supernatant, the RNA pellet was washed once with 1 ml 75% ETOH by centrifugation at 7500 g for 10 min at 4°C. The RNA pellet was dried in a vacuum concentrator (Concentrator Plus, Eppendorf, Germany) for 3 min and RNA was resuspended in RNase free water (Ambion, AM9939). RNA concentration and purity was determined at an optical density ratio of 260/280 using the Nanodrop® ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE, USA). RNA was stored at − 80°C until use. Total RNA integrity was assessed using RNA 6000 Nano Lab Chip on the 2100 Bioanalyzer (Agilent, Palo Alto, CA, USA), following the manufacturer's protocol. Total RNA purity was assessed by the NanoDrop® ND-1000 UV-Vis Spectrophotometer (Nanodrop Technologies, Rockland, DE, USA). Total RNA with OD260/OD280>1·8 and OD260/OD270 ≥1·3 was used for microarray experiments. RNA was used when the rRNA 28S/18S ratios were greater than or equal to 1·5, with the rRNA contribution being 30% or more and an RNA integrity number (RIN) was ≥7·0. The samples for gene expression were labelled using Agilent Quick Amp Kit PLUS (part number: 5190-0442). Five hundred nanograms from each of the samples were incubated with reverse trancription mix at 42°C and converted to double-stranded cDNA primed by oligodeoxythymidylic acid (oligo-dT) with a T7 polymerase promoter. The cleaned-up double-stranded cDNA was used as a template for aRNA generation. aRNA was generated by in vitro transcription and the dyes Cy3 CTP (Agilent) were incorporated during this step. The cDNA synthesis and in vitro transcription steps were carried out at 40°C. Labelled aRNA was cleaned up and the quality assessed for yield and specific activity. The labelled aRNA samples were hybridized onto a human gene expression array 8 × 15K (AMADID: 16332); 600 ng of Cy3-labelled samples were fragmented and hybridized. Fragmentation of labelled aRNA and hybridization were performed using the Agilent gene expression hybridization kit (part no. 5188-5242). Hybridization was carried out in Agilent's Surehyb chambers at 65°C for 16 h. The hybridized slides were washed using Agilent gene expression wash buffers (part no. 5188-5327) and scanned using the Agilent microarray scanner G model G2565BA at 5 micron resolution. Data were analysed using the GeneSpringGX version 7·3·1 software (Agilent Technologies, Santa Clara, CA, USA). Data were deposited in the NCBI Gene Expression Omnibus database (GSE16331).
Statistical analysis
The GraphPad Prism version 4·00 for Windows (GraphPad Software, San Diego, CA, USA) program was used for statistical analysis. Experiments were repeated at least three times. One-way analysis of variance (anova) and the Tukey–Kramer multiple comparisons test were performed to assay significant differences between the experimental groups.
Results
ALCAM promotes synergistic co-stimulation of CD3-activated human peripheral blood mononuclear cells in vitro
In this study, the anti-CD3 mAb activity was defined in vitro at a range of five increasing concentrations using an Alamar Blue proliferation assay [37]. CD3 stimulation promotes human PBMC proliferation in a dose-dependent manner. Concentrations above 0·5 µg/ml of the plate-bound anti-CD3 mAb resulted in a stable increment in a more than fourfold proliferative response compared to cells where the mAb was not added. However, lower concentrations produced a substantially reduced effect, drastically shown at 0·125 µg/ml, where it only barely increased the proliferation to 25% compared to the control groups.
Suboptimal signalling through the T cell receptor complex influences lymphocyte fate. Therefore, other molecules contributing to the immunological synapse provide additional co-stimulatory signals tuning the lymphocyte response [3,5]. The CD6 antigen co-stimulatory effect on T cells has been established based upon its interaction with specific monoclonal antibodies [7,8]. Based upon the proliferation assay system described above, we observed that a single CD6 interaction with plate-bound human recombinant ALCAM-Fc molecule at up to 1 µg/ml showed no detectable activity as a single agent (Fig. 1a). Having defined the proliferative activity of each single agent, the pairwise combination was tested maintaining the fixed maximal concentration of the immobilized ALCAM-Fc molecule. Consequently, the combinatorial effect was assessed quantitatively for additivity, synergy or eventually antagonism based on the Bliss algorithm [31]. This model allows evaluation of the nature of interactions of single agents and may reflect variability within data directly, indicating whether the extent of synergy is significant [31,32,38].
Fig. 1.
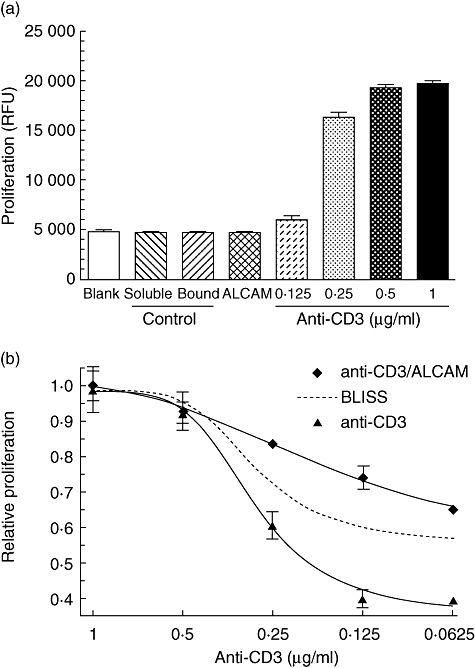
CD6 co-stimulation with activated leucocyte-cell adhesion molecule (ALCAM) induces a synergistic proliferative response of CD3-activated human peripheral blood mononuclear cells (PBMC) in vitro. (a) The immobilized anti-CD3 monoclonal antibody (mAb) induced proliferation in a dose-dependent manner but not the ALCAM-Fc molecule alone. (b) The immobilized anti-CD3 mAb with ALCAM-Fc induced a synergistic proliferative response of human PBMCs. The Bliss theoretical curve is depicted as a dotted line, which is the functional proliferation that would be expected if both the molecules are additive. Because ALCAM-Fc at the highest concentration did not induce proliferation over the cell background, all other proliferations at identical concentrations to anti-CD3 mAb are assumed to be equal to this. The combination of the anti-CD3 mAb and ALCAM was synergistic, as the actual curve lies above the theoretical Bliss curve. Immobilized anti-CD3 mAb, ALCAM-Fc (1 µg/ml) and a soluble or immobilized irrelevant isotype control mAb (1 µg/ml) were incubated with 3 × 104 freshly isolated human PBMCs. Cells were cultured for 72 h and proliferation evaluated using Alamar Blue, added for the last 18 h of incubation. Fluorescence was read at 530 nm excitation and emission at 590 nm.
Interestingly, the combination of CD3 and CD6 ligation acts synergistically stimulating human PBMC proliferation (Fig. 1b). That effect was evidenced for the dose–response expressed as percentage increase in cell growth above the expected theoretical curve wherein the combination is strictly additive in nature, which was calculated for varying doses of anti-CD3 mAb when combined with a constant dose of ALCAM. Thus, the proliferative efficacy across suboptimal concentrations of CD3 as a single activation pathway increases approximately by 23–34%, even at doses where the anti-CD3 mAb alone induces a negligible effect (i.e. 0·0625 µg/ml). In addition, the agonistic potency for the anti-CD3 mAb evaluated as the half-maximal effective concentration (EC50) shifted from 290 ng/ml to 180 ng/ml in the combination, as estimated by a four-parameter logistic curve. In particular, it would suggest that CD6 ligation by its natural ligand enhances the intrinsic activity of the CD3 activation pathway.
ALCAM co-stimulation enhances CD25 expression in CD3-activated human peripheral blood mononuclear cells in vitro
Lymphocyte activation is accompanied by the increment of different surface molecule expression [4]. Considering that CD6 co-stimulation induced a synergistic proliferative response of CD3-activated cells, we evaluated whether that effect may also be associated with the increment of defined activation markers. After incubation of human PBMCs with plate-bound anti-CD3 mAb (0·25 µg/ml) and ALCAM-Fc (1 µg/ml), the percentage of cells expressing a particular surface molecule as well as its mean fluorescence intensity (MFI) was evaluated by flow cytometry.
A significant increment in the proportion of the activation markers CD25 and CD69 was evidenced after activation, which was not observed with other lymphocyte molecules such as CD3, CD4, CD8 and CD6 (Table 1). Non-activated PBMCs in culture contained about 20·3% of CD25+ lymphocytes; however, in the anti-CD3 mAb-activated group that subpopulation increased to 98·7%. In addition, the CD69+ subpopulation increased from 7·2% to 28·3% after CD3 activation. Under the same conditions, ALCAM co-stimulation in the presence of anti-CD3 mAb does not increase further the proportion of lymphocytes expressing those markers, being 99·2% and 35·7% for CD25 and CD69, respectively.
Table 1.
CD6 co-stimulation with activated leucocyte-cell adhesion molecule (ALCAM) enhances preferentially the CD25 expression in CD3-activated human human peripheral blood mononuclear cells (PBMC) in vitro.
| Protein expression† |
||||
|---|---|---|---|---|
| CD marker | Condition* | Positive cells‡ | Molecules§ | Gene expression¶ |
| CD3 | CD3 | 1·12 | 0·93 | 1·59 |
| CD3/ALCAM | 1·12 | 0·96 | 1·48 | |
| CD4 | CD3 | 1·56 | 1·98 | 0·75 |
| CD3/ALCAM | 1·62 | 1·83 | 0·64 | |
| CD8 | CD3 | 1·63 | 1·21 | 2·19 |
| CD3/ALCAM | 1·72 | 1·15 | 2·13 | |
| CD6 | CD3 | 1·06 | 0·73 | 1·00 |
| CD3/ALCAM | 1·05 | 0·68 | 0·68 | |
| CD25 | CD3 | 4·86 | 7·75 | 9·31 |
| CD3/ALCAM | 4·89 | 12·73 | 14·46 | |
| CD69 | CD3 | 3·94 | 0·92 | 0·79 |
| CD3/ALCAM | 4·96 | 1·01 | 1·39 | |
Freshly isolated human PBMCs (3 × 104) were stimulated with the immobilized anti-CD3 monoclonal antibody (mAb) (0·25 µg/ml) alone or with ALCAM-Fc (1 µg/ml). Lymphocyte specific markers fold-expression compared to unstimulated cells is shown.
Difference at the protein level defined by flow cytometry.
Analysis of the percentage of labelled cells using specific conjugated mAb.
Definition of the surface molecules number defined by a bead-based receptor quantification analysis, as described in Materials and methods.
Difference at the gene transcription level defined by microarray analysis.
Furthermore, it was shown that CD25 MFI increased from 65 in non-activated PBMCs to 491 after CD3 activation. However, that sevenfold increment was amplified further to 12-fold following CD6 co-stimulation, with an MFI value of 800. Hence, from an average of 2·5 × 103 CD25 molecules per cell expressed in non-activated PBMCs, it rose to approximately 2 × 104 molecules subsequent to CD3 activation, which was boosted to more than 3·2 × 104 molecules per cells promoted by CD6 co-stimulation (Table 1).
However, despite an enhancement in the proportion of CD69+ cells, the MFI for this molecule did not change substantially after activation. The divergence between the CD25 and CD69 molecules expression described at the protein level was also confirmed defining the number of transcripts for the corresponding genes. CD3 activation promoted a ninefold enhancement of the CD25, which reached a 14-fold increase following ALCAM co-stimulation. In summary, this quantitative assessment extends the contribution of CD6 as a co-stimulatory molecule on T cells, as shown by up-regulation of the early activation markers CD25 and CD69 upon ALCAM co-ligation in CD3 activated PBMCs.
CD6 co-stimulation with ALCAM induces a distinct gene transcription profile which is amplified further by IL-2
Cell-to-cell interaction is a central event for the immune system, with consequences for lymphocyte biology and ultimately the individual immune response. The molecular basis of cell interaction is a fundamental issue for immunobiology due to the diversity of outcomes that may be driven during this process, which is currently being explored actively [3]. Lymphocyte activation essentially requires the spatial and temporal localization of molecules involved in the antigen-specific recognition and co-stimulatory receptors, depicting the immunological synapse. Each component may contribute to qualitatively different signalling [39].
The CD6 molecule contributes to this elaborate structure by long-term engagement with ALCAM to promote lymphocyte proliferation [9]. The particular enhancement of CD25 expression upon ALCAM co-stimulation in CD3 activated PBMCs, together with the recent finding on the directional IL-2 secretion into the immunological synapse [26], prompted us to evaluate the impact of exogenous addition of that cytokine on co-stimulated ALCAM cells. For this purpose human PBMCs were stimulated in vitro with the plate-bound CD3-specific mAb and either co-stimulated with ALCAM-Fc (1 µg/ml), human recombinant IL-2 (2·5 ng/ml) or both. The number of transcripts for the multiple genes was evaluated.
A characteristic gene profile was induced by CD3 activation (Fig. 2a). Moreover, activation in the presence of ALCAM co-stimulation or exogenous IL-2 addition separately induced distinctive increment of transcripts, with eventual further modification in cells activated with both together. Detailed analysis showed modifications of particular genes associated with apoptosis, chemokine, cytokine and receptor activities, as well as related to DNA and protein binding activities, signalling pathways and members of the tumour necrosis factor (TNF) superfamily, among others (Fig. 2b).
Fig. 2.
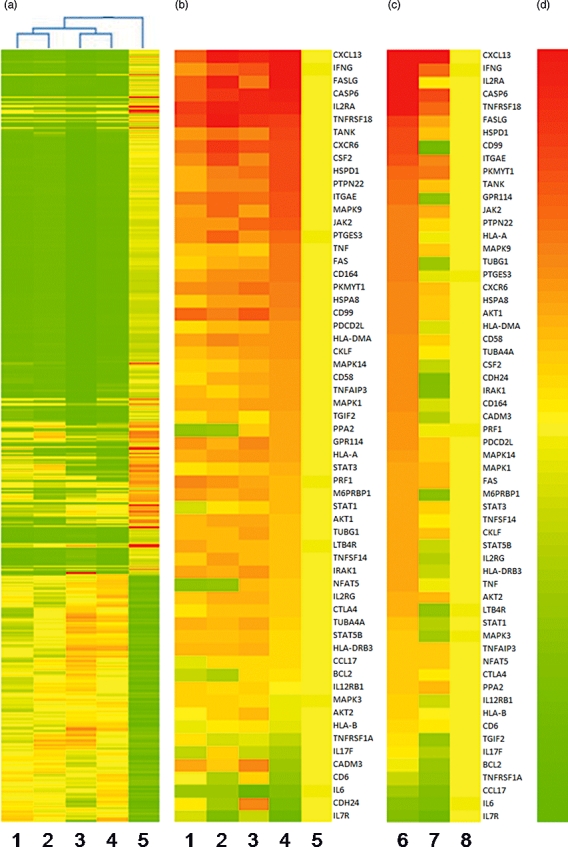
CD6 co-stimulation with activated leucocyte-cell adhesion molecule (ALCAM) induces a distinct gene transcription profile which is amplified further by interleukin (IL)-2. Difference in gene expression profile between 3 × 104 freshly isolated human peripheral blood mononuclear cells (PBMC) stimulated for 72 h with the immobilized anti-CD3 monoclonal antibody (mAb) (0·25 µg/ml) alone (lane 1), with ALCAM-Fc (1 µg/ml) (lane 2), interleukin (IL)-2 (2·5 ng/ml) (lane 4) or both (lane 3) or compared to unstimulated cells (lane 5). (a) Clustering of 15 000 genes or (b) 60 selected genes regulated differentially relative to the unstimulated cells are depicted. (c) Selected genes expression in CD3-activated cells incubated with a soluble CD6 membrane-distal domain specific humanized immunoglobulin (Ig)G1 non-depleting mAb (lane 7) or an irrelevant isotype control mAb (lane 6) at 10 µg/ml, compared to unstimulated cells (lane 8). (d) Colour bar ranging from 0 to 15-fold change relative to control. RNA was extracted by using Trizol reagent protocol. The labelled RNA was hybridized onto the human gene expression array 8 × 15 K. Hybridization was carried out in Agilent's Surehyb Chambers at 65°C for 16 h. The hybridized slides were washed and scanned using Agilent microarray scanner G Model G2565BA at 5 micron resolution.
CD6 co-stimulation enhanced the transcription of multiple genes over CD3 activation. Several cytokine receptors, including IL-21 (1·5-fold), IL-5 (1·6-fold) and the gamma chain of the IL-2 receptor complex (1·3-fold), were enhanced in addition to the described increment in the CD25 gene (1·6-fold), which may reinforce the relevance of IL-2 in the physiological process mediated by ALCAM binding. Moreover, CD6 co-stimulation also enhanced gene transcription of a variety of cytokines, in particular the interferon (IFN)-γ gene transcription, which increased 2·0- and 2·2-fold after ALCAM ligation or IL-2 excess, respectively, although in the presence of both together IFN-γ increased 5·6-fold compared to single CD3 activation, suggesting their concurrent effect promoting a proinflammatory profile on PBMCs. Furthermore, we explored whether a CD6-specific monoclonal antibody may modulate the gene profile observed upon lymphocyte activation. Interestingly, incubation with soluble anti-CD6 mAb reduced the transcription of multiple genes unregulated in activated cells (Fig. 2c).
In summary, these results reinforce the involvement of the CD6 co-stimulatory pathway in lymphocyte activation denoted by the increment of heat-shock proteins and signalling pathway gene transcription. Similarly, further molecules relevant for cell migration and interaction are also increased. Distinctively, the detected increment in the transcription for cytokine genes, in particular for IFN-γ and members of the TNF superfamily, may suggest that CD6 co-stimulation promotes preferentially a proinflammatory response which is amplified further by excess IL-2.
ALCAM in the presence of IL-2 promotes proinflammatory cytokine production by human PBMC in vitro
Co-stimulatory signals are essential for the initiation of the immune response, up-regulating the expression of cytokines and promoting the expansion and differentiation of T cells [40]. For instance, the CD28 molecule has been implicated in the early development and differentiation of the Th2 subset in humans, associated basically with enhanced production of IL-4 and IL-5 cytokines [41]. Thus, to define further the potential effect of CD6 co-stimulation with ALCAM skewing the lymphocyte differentiation to a proinflammatory response, human PBMCs were co-incubated with plate-bound anti-CD3 mAb (0·25 µg/ml) and ALCAM-Fc (1 µg/ml), human recombinant IL-2 (2·5 ng/ml) or both. After 5 days in culture, the simultaneous quantitation of characteristic Th1/Th2 cytokines was performed in the supernatant from different experimental conditions assayed, using a flow cytometry assay (Fig. 3).
Fig. 3.
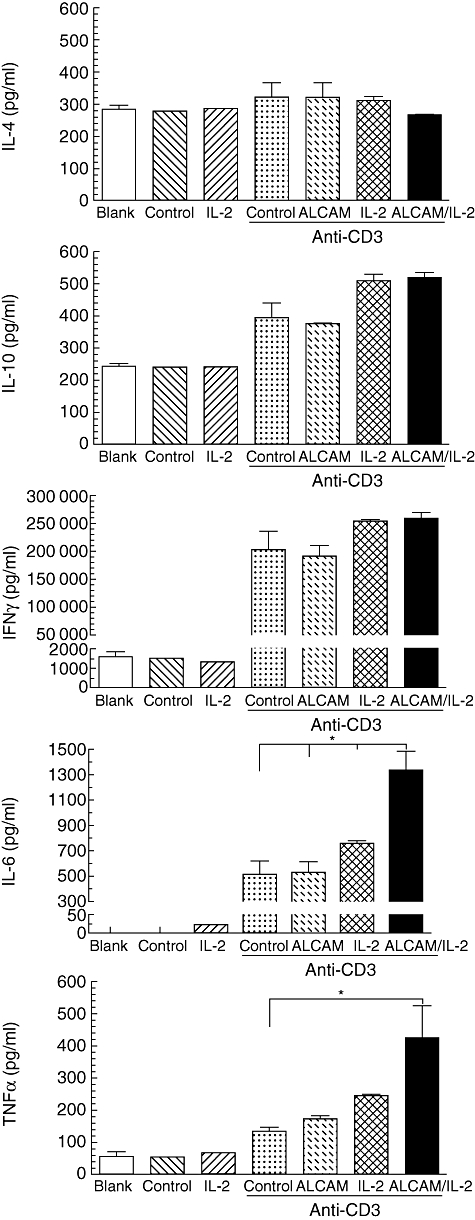
CD6 co-stimulation with activated leucocyte-cell adhesion molecule (ALCAM) in the presence of interleukin (IL)-2 promotes proinflammatory cytokines production by human peripheral blood mononuclear cells (PBMCs) in vitro. Immobilized anti-CD3 monoclonal antibody (mAb) (0·25 µg/ml) with the ALCAM-Fc or an irrelevant isotype control (1 µg/ml), IL-2 (2·5 ng/ml) and both were incubated with 3 × 104 freshly isolated human PBMCs for 96 h. Culture supernatant was collected after centrifugation at 270 g for 5 min at 4°C. Samples run in duplicate were evaluated using the T helper type 1 (Th1)/Th2 CBA human kit BD and analysed by flow cytometry. The actual cytokine concentration (pg/ml) was calculated after measuring the mean value and comparing to the standard curve generated by the standards. Mean (±standard error of the mean) and results for one-way analysis of variance test and the Tukey–Kramer multiple comparisons test are shown. *P < 0·05.
Cytokine production at the protein level was defined upon CD3 activation for TNF-α (134·0 ± 12·5 pg/ml), IL-4 (321·7 ± 45·6 pg/ml), IL-10 (396·4 ± 62·6 pg/ml), IL-6 (510·3 ± 106·1 pg/ml) and IFN-γ (202·8 ± 33·1 ng/ml). The IFN-γ production increased to the same extent after the addition of exogenous IL-2 (253·6 ± 3·4 ng/ml) or co-stimulation with ALCAM in the presence of excess IL-2 (259·3 ± 10·7 ng/ml). Production of IL-6, despite an increase of about 50% in the presence of exogenous IL-2, reached 1·3 ± 0·1 ng/ml in cells also co-incubated with ALCAM, which represent a 2·6-fold increment compared with cells activated with an anti-CD3 mAb alone. Of particular interest was the secretion of TNF-α which, in the presence of ALCAM ligation, increased by 30% compared to cells activated with the anti-CD3 mAb alone (173·1 ± 8·9 pg/ml) and increased further when IL-2 was added (244·7 ± 4·7 pg/ml). However, it escalated to 425·4 ± 99·7 pg/ml when co-stimulation with ALCAM occurred in the presence of IL-2 excess, resulting in a 3·2-fold increase over the CD3 single activation.
The production of IL-10 increased by 30% upon CD3 activation in the presence of exogenous IL-2 alone (509·9 ± 19·8 pg/ml), or together with ALCAM (519·8 ± 16·2 pg/ml). The secretion of IL-4 did not differ under the conditions evaluated for co-stimulation. In summary, these results align with the distinct amplification in the cytokine genes transcription described above, and together may suggest that CD6 co-stimulation enhances the intrinsic activity of the CD3 activation pathway and contributes to the Th1 commitment, enhancing IL-2 sensitivity of recently activated lymphocytes.
Activation and proliferation of human PBMC induced by CD6 co-stimulation can be inhibited without interfering with ALCAM interaction even in the presence of excess IL-2
The contribution of CD6 co-stimulation in T cell activation and proliferation has been demonstrated experimentally, while co-culturing total human PBMCs with allogenic mature dendritic cells. Moreover, it has been shown that soluble recombinant ALCAM-Fc may inhibit the proliferation in vitro[9]. Furthermore, it has been shown in vivo that blocking ALCAM expressed on blood–brain barrier cells restricts the transmigration of leucocytes, ameliorating the autoimmune response [19], supporting the immunopathological relevance of this interaction. Despite the clear contribution of ALCAM as an adhesion molecule with a role in leucocyte trafficking, it has been found that anti-ALCAM antibodies are less efficient in inhibiting T cell proliferation induced by mature dendritic cells compared to an anti-CD6 mAb [9].
Considering that the binding site for ALCAM is located in the membrane-proximal domain (SRCR3) of the CD6 molecule [16,17], together with the above-described evidence, we decided to dissect the effect of a well-defined CD6 membrane-distal domain (SRCR1)-specific mAb on lymphocyte activation and proliferation induced in the presence of ALCAM. T1h is a CD6-specific humanized IgG1 non-depleting mAb [27]. It does not block the CD6–ALCAM interaction and recognizes a conformational epitope independent of the N-glycosylation of the CD6 molecule, with an affinity constant of about 6 × 10−8 M. The epitope contains a RXE/Q consensus motif located in the membrane-distal SRCR1 [28].
Freshly isolated total human PBMCs used as the T cell source were incubated with an immobilized anti-CD3 mAb (0·25 µg/ml) and the soluble anti-CD6 mAb was added to define its influence on lymphocyte activation. It was found that after 24 h in culture the intracellular phospho-proteins pMAPK, pSTAT3 and pAKT were reduced by 37·5, 43·3 and 24·3%, respectively, compared to an irrelevant isotype control mAb (data not shown). Moreover, the anti-CD6 mAb inhibited T cell proliferation from 2·5 µg/ml to 40 µg/ml with a maximal inhibition of 70% at the highest evaluated concentration. To the same extent, T1h mAb reduced the proliferation of cells co-stimulated in the presence of ALCAM-Fc (1 µg/ml), showing a sigmoidal dose–response effect (Fig. 4a). To explore this result further we evaluated whether PBMC activation induced with plate-bound anti-CD3 mAb and ALCAM-Fc in the presence of human recombinant IL-2 (2·5 ng/ml) can be inhibited with the anti-CD6 mAb (10 µg/ml), compared to an irrelevant isotype control mAb (Fig. 4b). Consistently, the anti-CD6 mAb reduces significantly the lymphocyte proliferation even in the presence of additional exogenous IL-2 (P < 0·05). In summary, these results corroborate the unexpected capacity of CD6 specific mAb to inhibit T cell proliferation [9], defined in this experiment with a CD6 membrane-distal domain (SRCR1)-specific mAb. Moreover, the persistent anti-proliferative result in the presence of excess IL-2 demonstrates the strength of this effect and documents the potential contribution of the CD6 molecule in cell physiology, beyond leucocyte trafficking through its interaction with ALCAM.
Fig. 4.
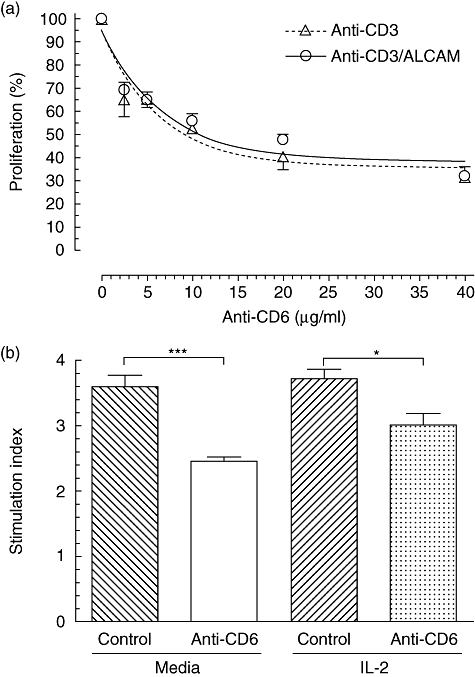
Human peripheral blood mononuclear cells (PBMCs) proliferation induced by CD6 co-stimulation in the presence of interleukin (IL)-2 can be inhibited without blocking activated leucocyte-cell adhesion molecule (ALCAM) interaction. Immobilized anti-CD3 monoclonal antibody (mAb) (0·25 µg/ml) alone or with ALCAM-Fc (1 µg/ml) were incubated with 3 × 104 freshly isolated human PBMCs. Cells were cultured for 72 h and proliferation evaluated using Alamar Blue, added for the last 18 h of incubation. Fluorescence was read at 530 nm excitation and emission at 590 nm. (a) Relative proliferation inhibition induced by a soluble CD6 membrane-distal domain-specific humanized IgG1 non-depleting mAb at different concentrations, in CD3-activated cells (anti-CD3) or co-stimulated with ALCAM (anti-CD3/ALCAM) cells. (b) Inhibition of proliferation induced by the anti-CD6 mAb compared to an irrelevant isotype control mAb (10 µg/ml) in CD3-activated cells which were co-stimulated with ALCAM in the presence or not of IL-2 (2·5 ng/ml). Mean (± standard error of the mean) and results for one-way analysis of variance test and the Tukey–Kramer multiple comparisons test are shown. *P < 0·05; ***P < 0·001.
CD6 domain 1-specific monoclonal antibody inhibits ALCAM co-stimulation in the presence of IL-2 and reduces proinflammatory cytokine production
The distinct lymphocyte proliferation inhibition mediated by the anti-CD6 mAb on human PBMCs co-stimulated with ALCAM raised the issue of whether that effect was associated with a reduction in cell numbers and/or further modification in the immunobiology of the target cells. T1h mAb does not induce lymphodepletion of naive cells [28], which was corroborated in our experimental conditions on recently activated lymphocytes with the anti-CD3 mAb and ALCAM in vitro (data not shown). Consequently, whether the anti-CD6 mAb at 10 µg/ml may modify the expression of the alpha chain of the IL-2 receptor complex was evaluated, an early event observed in freshly isolated total human PBMCs after activation with immobilized anti-CD3 mAb (0·25 µg/ml) and ALCAM-Fc (1 µg/ml). The anti-CD6 mAb decreased the CD25 MFI more than 50% either in cells activated with the anti-CD3 mAb alone or in combination with the ALCAM-Fc molecule. Moreover, that effect was also observed in cells activated in the presence of exogenous human recombinant IL-2 (2·5 ng/ml), although to a lesser extent (approximately 20%) compared with the above experimental groups (Fig. 5a).
Fig. 5.
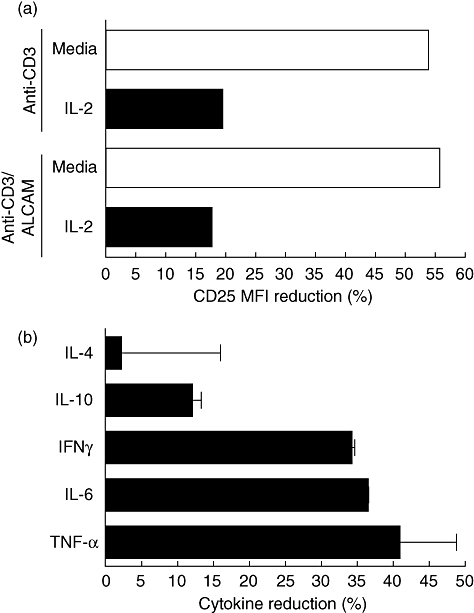
CD6 domain 1-specific monoclonal antibody inhibits activated leucocyte-cell adhesion molecule (ALCAM) co-stimulation in the presence of interleukin (IL)2 and reduces proinflammatory cytokine production by human peripheral blood mononuclear cells (PBMCs) in vitro. Freshly isolated human PBMCs (3 × 104) were incubated with the immobilized anti-CD3 monoclonal antibody (mAb) (0·25 µg/ml) alone or with ALCAM-Fc (1 µg/ml), in the presence or not of IL-2 (2·5 ng/ml). (a) After 72 h in culture, cells were stained with anti-CD25 phycoerythrin (PE)-conjugated mAb (MEM181 clone) at saturating concentration for 30 min at 4°C and acquired in the Cyan-ADP flow cytometer. Mean fluorescence intensity reduction was defined in gated lymphocytes. (b) Cytokine concentration (pg/ml) was calculated in CD3-activated cells co-stimulated with ALCAM in the presence or not of IL-2 after 96 h in culture. Supernatant was collected after centrifugation at 270 g for 5 min at 4°C. Samples run in duplicate were evaluated using the T helper type 1 (Th1)/Th2 CBA human kit BD and analysed in the Cyan-ADP flow cytometer. Mean (±standard error of the mean) reduction of the lymphocyte surface molecule expression and cytokine secretion induced by the soluble CD6 membrane-distal domain-specific mAb (10 µg/ml) estimated by comparison with an irrelevant isotype control mAb are shown.
Therefore, this result motivated us to assess whether the apparent decline on lymphocyte CD25 expression lessens IL-2 sensitivity and in turn may impact upon the proinflammatory profile commitment promoted by CD6 co-stimulation with ALCAM on PBMC. The percentage reduction of cytokine production was defined in recently activated lymphocytes for TNF-α (60·6 ± 0·5%), IFN-γ (47·8 ± 1·0%) and IL-6 (39·9 ± 8·9%) induced by the anti-CD6 mAb, compared to an irrelevant isotype control mAb (Fig. 5b). Additionally, in those cells also activated in the presence of exogenous IL-2, a decline was corroborated by the secretion of TNF-α (40·9 ± 7·9%), IL-6 (36·5 ± 0·1%) and IFN-γ (34·2 ± 0·4%) promoted by the anti-CD6 mAb. The production of IL-10 was comparatively less affected, in particular after incubation in the presence of exogenous IL-2 (12·0 ± 1·3%) compared to a 24·3 ± 9·1% reduction observed in PBMCs activated with the anti-CD3 mAb and ALCAM-Fc alone. IL-4 concentrations were not modified in these experimental conditions. In summary, these results may support that with the inhibition of the lymphocyte proliferation, a reduction in the expression of the CD25 molecule and the secretion of proinflammatory cytokines associated with the Th1 differentiation profile can be induced by a CD6 membrane-distal domain (SRCR1)-specific mAb in recently activated cells co-stimulated with ALCAM.
Discussion
Leucocyte membrane molecules participating in diverse cellular interactions may have a role as co-stimulatory molecules in the immunological synapse, modulating the immune response with different outcomes [42]. CD6 glycoprotein contributes to lymphocyte adhesion, activation, differentiation and survival processes [18,43,44]. Those effects have been associated essentially with the extracellular engagement between CD6 and its ALCAM ligand, although they mediate a low-affinity binding interaction [18].
The contribution of CD6 co-stimulation to the CD3 activation pathway has been described using specific monoclonal antibodies [7,8]. However, a detailed analysis of the potency of this interaction and the consequences for lymphocyte immunobiology after binding with a natural ligand was undefined. Assessed comparatively, it was found that in contrast to the anti-CD3 mAb, which induced detectable lymphoproliferative activity on its own [4], the ALCAM-Fc molecule does not promote cell growth in human peripheral blood mononuclear cells in vitro (Fig. 1a), despite its reported effect activating intracellular activation pathways [10]. Therefore, this result prompted us to identify whether CD6 co-stimulation induces a CD3 potency shift or increases its intrinsic activity. Using a standard reference algorithm for additivism, the potential synergy of CD6 ligation upon CD3 activation was explored. The Bliss additivism model is an effect-based synergy model which makes no assumptions about the functional form of the dose–response curves, and does not require dose–response information that lies outside the range sampled by each screening matrix [31,32].
A fine threshold below was distinctively identifiable in which CD3 recognition becomes a poor stimulatory pathway as a single agent in this assay (Fig. 1a). ALCAM ligation boosted the proliferative efficacy of CD3 from approximately 23% to 34% (Fig. 1b). The combination shifted the anti-CD3 mAb potency, reducing the threshold for its half-maximal effective concentration from 290 to 180 ng/ml. Altogether, using this immunopharmacological approach it was found that ALCAM synergizes with anti-CD3 mAb stimulating human PBMC proliferation, denoting that CD6 co-stimulation may increase the intrinsic activity of the CD3 signalling, due probably to the connectivity between their intracellular physiological pathways [45]. This result, together with the CD6-mediated proliferation in resting T cells induced by simultaneous cross-linking with the CD28 receptor, supports the strength of this co-stimulatory molecule [11].
CD25 molecule up-regulation characterizes CD3-induced activation, assuming the consequent proliferation to be mediated via the IL-2-dependent pathway [4]. We found that activation of human PBMCs in the presence of ALCAM substantially enhances the number of CD25 molecules on the cell surface, over the up-regulation induced by the CD3 stimulation alone, but not other markers – including the CD69 molecule (Table 1). It endorses the potential contribution of IL-2 in the lymphocyte response involving the CD6 molecule, and aligns with early evidence obtained in different experimental conditions. T cell activation induced by the simultaneous cross-linking of CD6 and CD28, without CD3 activation, is also accompanied by the increment of the CD25 molecule [11]. Moreover, the CD6-mediated T cell proliferation is inhibited in the presence of cyclosporin A, an inhibitor of the IL-2 gene transcription [46], distinctly different to the CD28-mediated effect which is completely resistant to this compound [12,13]. That effect has been corroborated using a neutralizing anti-IL-2 mAb [11] and a CD25 blocking mAb [8]. The addition of exogenous IL-2 had a limited impact on the proliferative response induced by the immobilized anti-CD3, as reported previously [4]. In addition, we observed that co-stimulation with ALCAM in the presence of exogenous IL-2 did not enhance the proliferation further (Fig. 4), as documented in another conditions promoting CD6 activation [8]. Interestingly, in that latter report the secreted IL-2 was detected only when the anti-CD25 mAb was also present in the culture [8], which may indicate the elevated consumption of IL-2 after CD6 activation.
However, in this study the microarray analysis revealed that an IL-2 excess amplified a distinctive gene transcription profile promoted after CD6 co-stimulation with ALCAM, encoding chemokines and particularly inflammatory cytokines (Fig. 2). Although the immobilized anti-CD3 mAb initiated the production of IFN-γ, as reported [4], the additional increment of IL-6 and TNF-α (Fig. 3) supports a particular contribution of the CD6 molecule to the Th1 commitment of human T cells. This potential role modulating T cell activation and differentiation signals may also be supported by the recent finding on the bacterial binding capability of the CD6 ectodomains triggering intracellular signalling pathways, associated with a burst of proinflammatory cytokine release [24]. In addition to the recently verified contribution of the CD6 molecule in leucocyte trafficking thorough the interaction with its ligand ALCAM [19], our unexpected finding may distinguish CD6 from membrane receptors with a predominant adhesion function [47]. Although most membrane molecules contribute to cell adhesion, those with a defined co-stimulatory T cell function are limited [1]. Furthermore, while the CD28 molecule prototypically provides a potent synergistic signal in naive cells, it favours mainly the Th2-cell differentiation [5,40,41,48]. Hence, altogether the present data may support a novel role for the CD6 co-stimulatory pathway controlling the immune response through a Th1 effector differentiation of recently activated cells. It is particularly relevant considering that CD6/ALCAM interaction is effective at a micromolar range [18]. Therefore, it may exemplify a highly efficient low-affinity interaction between a scavenger receptor cysteine-rich domain in the CD6 molecule with an ALCAM immunoglobulin-like domain. Moreover, CD6 as an evolutionary conserved molecule may contribute to the host protection against infectious pathogens [24], promoting a rapid and optimal activation and effector function differentiation of the immune response.
Conversely, these results also converge into recent knowledge integrating the cytokine and heterologous receptor signalling pathways [2]. IL-2 induces expression of its high-affinity receptor, turning into an autocrine and paracrine loop which guarantees T cell proliferation and survival [49,50]. Interestingly, IL-2 is secreted directionally into the immunological synapse [26], the functional structure where CD6 and ALCAM are necessarily recruited, and co-localize with the TCR/CD3 complex to promote an efficient T cell response [9,23]. Thus, we suggest that the integration between the TCR/CD3 complex with the CD6 and IL-2 receptors may regulate the immunological synapse, a process that other cytokines, including IL-6 [51], may collaborate functionally, influencing the consolidation of the Th1 cell phenotype differentiation and eventually the duration of the response, which has to be corroborated. These results, supporting the contribution of CD6 co-stimulation to the human T cell linage commitment, reinforces the relevance of the molecular composition of the immunological synapse in the T cell fate decision, fine-tuning the peripheral immune response on the spatial and temporal co-ordinate influence of multiple membrane receptors [20,21]. In general, it may exemplify a meaningful cross-talk between heterologous receptors for human peripheral lymphocyte immunobiology, where the intracellular signalling interpathway cross-communication has to be defined.
The functional role of CD6 in cell adhesion has been documented associated with the identification of different ligands with varied expression, including lymphoid and non-lymphoid tissues [52–55]. In particular, the wide distribution of ALCAM, together with the blocking effect of monoclonal antibodies interfering with the CD6–ALCAM interaction with physiological implications in vitro, supported that assumption initially [56]. Moreover, the recent demonstration that ALCAM blockade may restrict the migration of CD6 cells into the central nervous system additionally sustains a relevant role for its adhesion function in the physiopathology of autoimmune diseases [19].
Furthermore, other emergent data suggested not ruling out other CD6 implications in T cell immunobiology during lymphocyte selection and the immune response in the periphery [43]. The reduced activation of memory T cells mediated by the recombinant CD6 molecule or a CD6–ALCAM blocking mAb supports the impact of such engagement in the immune response [18,57], assumed as a direct consequence of their interference on the CD6 localization at the immunological synapse [22] or inhibiting the maturation of this structure [23]. Similarly, the in vitro inhibition of human T cell clone autoreactivity [14] and T cell proliferation [9] with anti-CD6 mAb provided evidence of a direct role of CD6 modulating an ongoing activation process in recently activated T cells, although it was not defined whether or not those mAbs functioned blocking the CD6–ALCAM binding. However, the sustained repression of the proliferative response mediated by those mAbs indicated that CD6-ALCAM-mediated adhesion is important throughout the T cell proliferation process [9]. Nevertheless, it would also suggest that such interaction may induce the functional differentiation of T cells that would eventually be modified in the presence of a non-blocking CD6–ALCAM interaction molecule. In addition, it suggests that beyond the ALCAM binding site located in the membrane-proximal domain (SRCR3) [17] interaction with other epitopes, including those related to the bacterial binding properties of the CD6 ectodomains, may display different effects on T cells [14].
Indeed, here we found that a CD6 membrane-distal domain (SRCR1)-specific humanized IgG1 non-depleting mAb [28] reduced the intracellular phospho-proteins pMAPK, pSTAT3 and pAKT, which have been implicated downstream in the CD6-mediated activation pathways [9,10]. Furthermore, it is able to inhibit T cell proliferation even in the presence of ALCAM and excess IL-2 (Fig. 4). The dose-dependent effect is associated with a reduction in CD25 molecule expression and the production of IFN-γ, IL-6 and TNF-α (Fig. 5). These results, obtained with an anti-CD6 mAb which does not interfere with the ALCAM interaction, support a dual role of the CD6 molecule on lymphocyte biology. These data may also converge with the finding that administration of a recombinant soluble CD6 protein interfering with the lipopolysaccharide-binding to the lymphocyte-expressed CD6 molecule induced a beneficial effect in an experimental model of septic shock, linked to the serum level reduction of those proinflammatory cytokines [24].
The unique feature of lymphocyte-expressed SCRC modulating human T cell immunobiology suggests a convergent pathway between the innate and adaptive immunity mediated by evolutionarily conserved receptors [24,58,59]. Sequentially, adhesion of CD6-expressing lymphocytes to ALCAM may make them migrate to the inflammation sites, but that interaction also promotes a co-stimulatory signal synergizing the antigen-specific receptor (TCR/CD3) complex activation pathways. Then, the consequent increment of the CD25 molecule enhances the IL-2 sensitivity of recently activated human lymphocytes which, in turn, triggers or amplifies the production of cytokines, chemokines and other adhesion molecules increasing the local inflammatory environment during the course of an immune response (Fig. 2). Physiologically at this point, CD6 molecule expression may decrease to prevent further activation, as evidenced by a reduction in the number of its gene transcripts and surface molecules (Table 1). Although a preferential reduction in the CD6 expression on CD8+ cells has been shown upon mitogen-mediated activation [60], it remains to be defined whether ALCAM may promote a distinct effect on lymphocyte subpopulations. However, under particular conditions the activation process may progress to an uncontrolled tissue inflammation, represented characteristically by the autoimmune immunopathology.
The quantitative and qualitative impact of CD6 co-stimulation in the immunoresponse described in this work converge with the recent finding of CD6 as a susceptibility locus in multiple sclerosis, a prototypic autoimmune disease [61]. This, with the previous description of the IL-2RA between other susceptibility genes [62], as well as the dysregulated proinflammatory response pathways [63], may support a central role for the CD6 molecule in the emergence of pathological autoimmunity. Thereby, further than blocking the ALCAM binding, modulation of the lymphocyte effector differentiation induced by anti-CD6 may have therapeutic implications in those conditions. Initial attempts with a CD6-depleting monoclonal antibody in multiple sclerosis patients [64], followed by its systematic evaluation as an ex vivo depletion therapeutic strategy in patients undergoing bone marrow transplantation for prophylaxis against graft-versus-host disease [65], showed clinical benefit. That the use of non-depleting CD6 mAbs with immunomodulatory effects may result in an alternative option, although preliminarily encouraging [66,67], remains to be elucidated.
Acknowledgments
We thank Drs H. Iyer and A. Anand for insightful discussions and Drs S. Rao and M. Vasudevan from Genotypic Technology Pvt Limited, Bangalore, India for their support on the gene microarray data analysis. We also acknowledge Clinigene International Limited for guaranteeing the quality of the blood donation process following the ethical guidelines.
Disclosure
The authors P. Nair, R. Melarkode and D. Rajkumar are employees at Biocon Ltd; E. Montero does not have potential conflicts of interest.
References
- 1.Zhu Y, Chen L. Turning the tide of lymphocyte costimulation. J Immunol. 2009;182:2557–8. doi: 10.4049/jimmunol.0990006. [DOI] [PubMed] [Google Scholar]
- 2.Bezbradica JS, Medzhitov R. Integration of cytokine and heterologous receptor signaling pathways. Nat Immunol. 2009;10:333–9. doi: 10.1038/ni.1713. [DOI] [PubMed] [Google Scholar]
- 3.Smith-Garvin JE, Koretzky GA, Jordan MS. T cell activation. Annu Rev Immunol. 2009;27:591–619. doi: 10.1146/annurev.immunol.021908.132706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van Lier RA, Brouwer M, Rebel VI, van Noesel CJ, Aarden LA. Immobilized anti-CD3 monoclonal antibodies induce accessory cell-independent lymphokine production, proliferation and helper activity in human T lymphocytes. Immunology. 1989;68:45–50. [PMC free article] [PubMed] [Google Scholar]
- 5.Acuto O, Michel F. CD28-mediated co-stimulation: a quantitative support for TCR signalling. Nat Rev Immunol. 2003;3:939–51. doi: 10.1038/nri1248. [DOI] [PubMed] [Google Scholar]
- 6.Aruffo A, Melnick MB, Linsley PS, Seed B. The lymphocyte glycoprotein CD6 contains a repeated domain structure characteristic of a new family of cell surface and secreted proteins. J Exp Med. 1991;174:949–52. doi: 10.1084/jem.174.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Osorio LM, Garcia CA, Jondal M, Chow SC. The anti-CD6 mAb, IOR-T1, defined a new epitope on the human CD6 molecule that induces greater responsiveness in T cell receptor/CD3-mediated T cell proliferation. Cell Immunol. 1994;154:123–33. doi: 10.1006/cimm.1994.1062. [DOI] [PubMed] [Google Scholar]
- 8.Gangemi RM, Swack JA, Gaviria DM, Romain PL. Anti-T12, an anti-CD6 monoclonal antibody, can activate human T lymphocytes. J Immunol. 1989;143:2439–47. [PubMed] [Google Scholar]
- 9.Zimmerman AW, Joosten B, Torensma R, Parnes JR, van Leeuwen FN, Figdor CG. Long-term engagement of CD6 and ALCAM is essential for T-cell proliferation induced by dendritic cells. Blood. 2006;107:3212–20. doi: 10.1182/blood-2005-09-3881. [DOI] [PubMed] [Google Scholar]
- 10.Ibanez A, Sarrias MR, Farnos M, et al. Mitogen-activated protein kinase pathway activation by the CD6 lymphocyte surface receptor. J Immunol. 2006;177:1152–9. doi: 10.4049/jimmunol.177.2.1152. [DOI] [PubMed] [Google Scholar]
- 11.Osorio LM, Rottenberg M, Jondal M, Chow SC. Simultaneous cross-linking of CD6 and CD28 induces cell proliferation in resting T cells. Immunology. 1998;93:358–65. doi: 10.1046/j.1365-2567.1998.00442.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ledbetter JA, June CH, Grosmaire LS, Rabinovitch PS. Crosslinking of surface antigens causes mobilization of intracellular ionized calcium in T lymphocytes. Proc Natl Acad Sci USA. 1987;84:1384–8. doi: 10.1073/pnas.84.5.1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bjorndahl JM, Sung SS, Hansen JA, Fu SM. Human T cell activation: differential response to anti-CD28 as compared to anti-CD3 monoclonal antibodies. Eur J Immunol. 1989;19:881–7. doi: 10.1002/eji.1830190515. [DOI] [PubMed] [Google Scholar]
- 14.Singer NG, Richardson BC, Powers D, et al. Role of the CD6 glycoprotein in antigen-specific and autoreactive responses of cloned human T lymphocytes. Immunology. 1996;88:537–43. [PMC free article] [PubMed] [Google Scholar]
- 15.Bott CM, Doshi JB, Morimoto C, Romain PL, Fox DA. Activation of human T cells through CD6: functional effects of a novel anti-CD6 monoclonal antibody and definition of four epitopes of the CD6 glycoprotein. Int Immunol. 1993;5:783–92. doi: 10.1093/intimm/5.7.783. [DOI] [PubMed] [Google Scholar]
- 16.Bodian DL, Skonier JE, Bowen MA, et al. Identification of residues in CD6 which are critical for ligand binding. Biochemistry. 1997;36:2637–41. doi: 10.1021/bi962560+. [DOI] [PubMed] [Google Scholar]
- 17.Whitney GS, Starling GC, Bowen MA, Modrell B, Siadak AW, Aruffo A. The membrane-proximal scavenger receptor cysteine-rich domain of CD6 contains the activated leukocyte cell adhesion molecule binding site. J Biol Chem. 1995;270:18187–90. doi: 10.1074/jbc.270.31.18187. [DOI] [PubMed] [Google Scholar]
- 18.Hassan NJ, Barclay AN, Brown MH. Frontline: optimal T cell activation requires the engagement of CD6 and CD166. Eur J Immunol. 2004;34:930–40. doi: 10.1002/eji.200424856. [DOI] [PubMed] [Google Scholar]
- 19.Cayrol R, Wosik K, Berard JL, et al. Activated leukocyte cell adhesion molecule promotes leukocyte trafficking into the central nervous system. Nat Immunol. 2008;9:137–45. doi: 10.1038/ni1551. [DOI] [PubMed] [Google Scholar]
- 20.Friedl P, den Boer AT, Gunzer M. Tuning immune responses: diversity and adaptation of the immunological synapse. Nat Rev Immunol. 2005;5:532–45. doi: 10.1038/nri1647. [DOI] [PubMed] [Google Scholar]
- 21.Maldonado RA, Irvine DJ, Schreiber R, Glimcher LH. A role for the immunological synapse in lineage commitment of CD4 lymphocytes. Nature. 2004;431:527–32. doi: 10.1038/nature02916. [DOI] [PubMed] [Google Scholar]
- 22.Castro MA, Oliveira MI, Nunes RJ, et al. Extracellular isoforms of CD6 generated by alternative splicing regulate targeting of CD6 to the immunological synapse. J Immunol. 2007;178:4351–61. doi: 10.4049/jimmunol.178.7.4351. [DOI] [PubMed] [Google Scholar]
- 23.Gimferrer I, Calvo M, Mittelbrunn M, et al. Relevance of CD6-mediated interactions in T cell activation and proliferation. J Immunol. 2004;173:2262–70. doi: 10.4049/jimmunol.173.4.2262. [DOI] [PubMed] [Google Scholar]
- 24.Sarrias MR, Farnos M, Mota R, et al. CD6 binds to pathogen-associated molecular patterns and protects from LPS-induced septic shock. Proc Natl Acad Sci USA. 2007;104:11724–9. doi: 10.1073/pnas.0702815104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nathan C, Sporn M. Cytokines in context. J Cell Biol. 1991;113:981–6. doi: 10.1083/jcb.113.5.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huse M, Lillemeier BF, Kuhns MS, Chen DS, Davis MM. T cells use two directionally distinct pathways for cytokine secretion. Nat Immunol. 2006;7:247–55. doi: 10.1038/ni1304. [DOI] [PubMed] [Google Scholar]
- 27.Roque-Navarro L, Mateo C, Lombardero J, et al. Humanization of predicted T-cell epitopes reduces the immunogenicity of chimeric antibodies: new evidence supporting a simple method. Hybrid Hybridomics. 2003;22:245–57. doi: 10.1089/153685903322328974. [DOI] [PubMed] [Google Scholar]
- 28.Alonso R, Huerta V, de Leon J, et al. Towards the definition of a chimpanzee and human conserved CD6 domain 1 epitope recognized by T1 monoclonal antibody. Hybridoma. 2008;27:291–301. doi: 10.1089/hyb.2008.0007. [DOI] [PubMed] [Google Scholar]
- 29.IOR-T3 (CD3 human leukocyte differentiation antigen) Hybridoma. 1997;16:588. [Google Scholar]
- 30.Mateo C, Moreno E, Amour K, Lombardero J, Harris W, Perez R. Humanization of a mouse monoclonal antibody that blocks the epidermal growth factor receptor: recovery of antagonistic activity. Immunotechnology. 1997;3:71–81. doi: 10.1016/s1380-2933(97)00065-1. [DOI] [PubMed] [Google Scholar]
- 31.Berenbaum MC. Criteria for analyzing interactions between biologically active agents. Adv Cancer Res. 1981;35:269–335. doi: 10.1016/s0065-230x(08)60912-4. [DOI] [PubMed] [Google Scholar]
- 32.Borisy AA, Elliott PJ, Hurst NW, et al. Systematic discovery of multicomponent therapeutics. Proc Natl Acad Sci USA. 2003;100:7977–82. doi: 10.1073/pnas.1337088100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hultin LE, Matud JL, Giorgi JV. Quantitation of CD38 activation antigen expression on CD8+ T cells in HIV-1 infection using CD4 expression on CD4+ T lymphocytes as a biological calibrator. Cytometry. 1998;33:123–32. doi: 10.1002/(sici)1097-0320(19981001)33:2<123::aid-cyto6>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 34.Hodge G, Hodge S, Haslam R, et al. Rapid simultaneous measurement of multiple cytokines using 100 microl sample volumes – association with neonatal sepsis. Clin Exp Immunol. 2004;137:402–7. doi: 10.1111/j.1365-2249.2004.02529.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Krutzik PO, Nolan GP. Intracellular phospho-protein staining techniques for flow cytometry: monitoring single cell signaling events. Cytometry A. 2003;55:61–70. doi: 10.1002/cyto.a.10072. [DOI] [PubMed] [Google Scholar]
- 36.Verma-Gaur J, Rao SN, Taya T, Sadhale P. Genomewide recruitment analysis of Rpb4, a subunit of polymerase II in Saccharomyces cerevisiae, reveals its involvement in transcription elongation. Eukaryot Cell. 2008;7:1009–18. doi: 10.1128/EC.00057-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ahmed SA, Gogal RM, Jr, Walsh JE. A new rapid and simple non-radioactive assay to monitor and determine the proliferation of lymphocytes: an alternative to [3H]thymidine incorporation assay. J Immunol Methods. 1994;170:211–24. doi: 10.1016/0022-1759(94)90396-4. [DOI] [PubMed] [Google Scholar]
- 38.Buck E, Eyzaguirre A, Brown E, et al. Rapamycin synergizes with the epidermal growth factor receptor inhibitor erlotinib in non-small-cell lung, pancreatic, colon, and breast tumors. Mol Cancer Ther. 2006;5:2676–84. doi: 10.1158/1535-7163.MCT-06-0166. [DOI] [PubMed] [Google Scholar]
- 39.Bromley SK, Burack WR, Johnson KG, et al. The immunological synapse. Annu Rev Immunol. 2001;19:375–96. doi: 10.1146/annurev.immunol.19.1.375. [DOI] [PubMed] [Google Scholar]
- 40.Lenschow DJ, Walunas TL, Bluestone JA. CD28/B7 system of T cell costimulation. Annu Rev Immunol. 1996;14:233–58. doi: 10.1146/annurev.immunol.14.1.233. [DOI] [PubMed] [Google Scholar]
- 41.King CL, Stupi RJ, Craighead N, June CH, Thyphronitis G. CD28 activation promotes Th2 subset differentiation by human CD4+ cells. Eur J Immunol. 1995;25:587–95. doi: 10.1002/eji.1830250242. [DOI] [PubMed] [Google Scholar]
- 42.Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–47. doi: 10.1146/annurev.immunol.25.022106.141618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singer NG, Fox DA, Haqqi TM, et al. CD6: expression during development, apoptosis and selection of human and mouse thymocytes. Int Immunol. 2002;14:585–97. doi: 10.1093/intimm/dxf025. [DOI] [PubMed] [Google Scholar]
- 44.Lenschow DJ, Herold KC, Rhee L, et al. CD28/B7 regulation of Th1 and Th2 subsets in the development of autoimmune diabetes. Immunity. 1996;5:285–93. doi: 10.1016/s1074-7613(00)80323-4. [DOI] [PubMed] [Google Scholar]
- 45.Osorio LM, Ordonez C, Garcia CA, Jondal M, Chow SC. Evidence for protein tyrosine kinase involvement in CD6-induced T cell proliferation. Cell Immunol. 1995;166:44–52. doi: 10.1006/cimm.1995.0006. [DOI] [PubMed] [Google Scholar]
- 46.Kunz J, Hall MN. Cyclosporin A, FK506 and rapamycin: more than just immunosuppression. Trends Biochem Sci. 1993;18:334–8. doi: 10.1016/0968-0004(93)90069-y. [DOI] [PubMed] [Google Scholar]
- 47.Girard JP, Springer TA. Cloning from purified high endothelial venule cells of hevin, a close relative of the antiadhesive extracellular matrix protein SPARC. Immunity. 1995;2:113–23. doi: 10.1016/1074-7613(95)90083-7. [DOI] [PubMed] [Google Scholar]
- 48.Rulifson IC, Sperling AI, Fields PE, Fitch FW, Bluestone JA. CD28 costimulation promotes the production of Th2 cytokines. J Immunol. 1997;158:658–65. [PubMed] [Google Scholar]
- 49.Malek TR. The biology of interleukin-2. Annu Rev Immunol. 2008;26:453–79. doi: 10.1146/annurev.immunol.26.021607.090357. [DOI] [PubMed] [Google Scholar]
- 50.Montero E, Alonso L, Perez R, Lage A. Interleukin-2 mastering regulation in cancer and autoimmunity. Ann NY Acad Sci. 2007;1107:239–50. doi: 10.1196/annals.1381.026. [DOI] [PubMed] [Google Scholar]
- 51.Pasare C, Medzhitov R. Toll pathway-dependent blockade of CD4+CD25+ T cell-mediated suppression by dendritic cells. Science. 2003;299:1033–6. doi: 10.1126/science.1078231. [DOI] [PubMed] [Google Scholar]
- 52.Wee S, Wang WC, Farr AG, et al. Characterization of a CD6 ligand(s) expressed on human- and murine-derived cell lines and murine lymphoid tissues. Cell Immunol. 1994;158:353–64. doi: 10.1006/cimm.1994.1282. [DOI] [PubMed] [Google Scholar]
- 53.Singer NG, Mitra R, Lialios F, et al. CD6 dependent interactions of T cells and keratinocytes: functional evidence for a second CD6 ligand on gamma-interferon activated keratinocytes. Immunol Lett. 1997;58:9–14. doi: 10.1016/s0165-2478(97)02707-7. [DOI] [PubMed] [Google Scholar]
- 54.Joo YS, Singer NG, Endres JL, et al. Evidence for the expression of a second CD6 ligand by synovial fibroblasts. Arthritis Rheum. 2000;43:329–35. doi: 10.1002/1529-0131(200002)43:2<329::AID-ANR12>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 55.Saifullah MK, Fox DA, Sarkar S, et al. Expression and characterization of a novel CD6 ligand in cells derived from joint and epithelial tissues. J Immunol. 2004;173:6125–33. doi: 10.4049/jimmunol.173.10.6125. [DOI] [PubMed] [Google Scholar]
- 56.Bowen MA, Patel DD, Li X, et al. Cloning, mapping, and characterization of activated leukocyte-cell adhesion molecule (ALCAM), a CD6 ligand. J Exp Med. 1995;181:2213–20. doi: 10.1084/jem.181.6.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hassan NJ, Simmonds SJ, Clarkson NG, et al. CD6 regulates T-cell responses through activation-dependent recruitment of the positive regulator SLP-76. Mol Cell Biol. 2006;26:6727–38. doi: 10.1128/MCB.00688-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vera J, Fenutria R, Canadas O, et al. The CD5 ectodomain interacts with conserved fungal cell wall components and protects from zymosan-induced septic shock-like syndrome. Proc Natl Acad Sci USA. 2009;106:1506–11. doi: 10.1073/pnas.0805846106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lenz LL. CD5 sweetens lymphocyte responses. Proc Natl Acad Sci USA. 2009;106:1303–4. doi: 10.1073/pnas.0812579106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bank I, Dardik R, Levy V, Goldstein I, Shoham J. Differential expression and regulation of CD6 on T-cell subsets revealed by monoclonal antibody (MAb) CH11. Hybridoma. 2001;20:75–84. doi: 10.1089/02724570152057562. [DOI] [PubMed] [Google Scholar]
- 61.De Jager PL, Jia X, Wang J, et al. Meta-analysis of genome scans and replication identify CD6, IRF8 and TNFRSF1A as new multiple sclerosis susceptibility loci. Nat Genet. 2009;41:776–82. doi: 10.1038/ng.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rubio JP, Stankovich J, Field J, et al. Replication of KIAA0350, IL2RA, RPL5 and CD58 as multiple sclerosis susceptibility genes in Australians. Genes Immun. 2008;9:624–30. doi: 10.1038/gene.2008.59. [DOI] [PubMed] [Google Scholar]
- 63.Degre M, Dahl H, Vandvik B. Interferon in the serum and cerebrospinal fluid in patients with multiple sclerosis and other neurological disorders. Acta Neurol Scand. 1976;53:152–60. doi: 10.1111/j.1600-0404.1976.tb04333.x. [DOI] [PubMed] [Google Scholar]
- 64.Hafler DA, Fallis RJ, Dawson DM, Schlossman SF, Reinherz EL, Weiner HL. Immunologic responses of progressive multiple sclerosis patients treated with an anti-T-cell monoclonal antibody, anti-T12. Neurology. 1986;36:777–84. doi: 10.1212/wnl.36.6.777. [DOI] [PubMed] [Google Scholar]
- 65.Lee SJ, Weller E, Alyea EP, Ritz J, Soiffer RJ. Efficacy and costs of granulocyte colony-stimulating factor in allogeneic T-cell depleted bone marrow transplantation. Blood. 1998;92:2725–9. [PubMed] [Google Scholar]
- 66.Montero E, Falcon L, Morera Y, Delgado J, Amador JF, Perez R. CD6 molecule may be important in the pathological mechanisms of lymphocytes adhesion to human skin in psoriasis and ior t1 MAb a possible new approach to treat this disease. Autoimmunity. 1999;29:155–6. doi: 10.3109/08916939908995386. [DOI] [PubMed] [Google Scholar]
- 67.Montero E, Reyes G, Guibert M, et al. Immunodiagnosis and therapeutic immunosuppression in rheumatoid arthritis with ior t1 (anti-CD6) monoclonal antibody. Arthritis Res. 2002;4:114. [Google Scholar]