

Abstract
Drug resistance involves multiple mechanisms. Multidrug resistance (MDR) is the leading cause of treatment failure in cancer therapy. Elevated levels of MDR proteins [members of the ATP-binding cassette (ABC) transporter family] increase cellular efflux and decrease the effectiveness of chemotherapeutic agents. As a salvage approach to overcome drug resistance, inhibitors of MDR proteins have been developed, but have had limited success mainly due to undesired toxicities. Nuclear receptors (NRs), including pregnane X receptor (PXR), regulate the expression of proteins (including MDR proteins) involved in drug metabolism and drug clearance, suggesting that it is possible to overcome drug resistance by regulating NR. This review discusses the progress in the development of MDR inhibitors, with a focus on MDR1 inhibitors. Recent development of PXR antagonists to pharmacologically modulate PXR is also reviewed. The review proposes that selectively preventing the elevation of MDR levels by regulating NRs rather than non-selectively inhibiting the MDR activity by using MDR inhibitors can be a less toxic approach to overcome drug resistance during cancer therapy.
Keywords: Drug resistance, MDR1, PXR, CYP3A4, ABC transporters, Drug-metabolizing enzymes
1. Introduction
Drug resistance – the reduction in effectiveness of a drug in curing a disease or improving patient symptoms – can develop against antibiotics, antivirals, or chemotherapeutic agents for cancers. Drug resistance is a complex cellular response and target-specific and target-nonspecific mechanisms can be involved in the process.
In target-specific drug resistance, changes in a specific drug target that decrease the interaction between the target and drug might lead to drug resistance. For example, mutations in viral genes frequently lead to antiviral drug resistance [1], and loss of expression of the estrogen receptor (ER) can cause tamoxifen resistance in patients with breast cancer [2]. It can be difficult to predict, prevent, or overcome target-specific drug resistance without developing new therapeutic agents. On the other hand, in target-nonspecific drug resistance, changes in parameters not directly relevant to or dependent on the drug target contribute to drug resistance. For example, target cells or organisms might produce higher levels of drug-metabolizing enzymes (DMEs) to degrade the drug or increase their efflux capacity, resulting in decreased bioavailability and reduced effectiveness of drug [3].
Cases of target-nonspecific drug resistance have several features in common, which have been targeted by various approaches in order to overcome drug resistance, especially against chemotherapeutic agents. For example, a family of ATP-dependent drug pumps, known as ATP-binding cassette (ABC) transporter proteins, can increase the resistance to chemotherapeutic agents by increasing cellular efflux. Multidrug resistance (MDR) proteins belong to the ABC transporter protein family and play an important role in maintaining normal physiologic functions that protect human tissues from drugs and other xenobiotics. Elevated levels of MDR1, a key MDR protein [also known as P-glycoprotein (P-gp) or ABCB1], have been associated with drug-mediated drug resistance in cancer [4], making inhibition of MDR1 activity a logical approach to overcome MDR1-mediated drug resistance.
This review discusses the progress made in the development of MDR1 inhibitors in overcoming drug resistance in cancer. As the primary role of MDR1 is disposition of xenobiotics, the undesired toxicities resulting from the use of MDR1 inhibitors have posed a challenge in the development of MDR1 inhibitors for clinical applications. The problems encountered and the lessons learned in developing MDR1 inhibitors as salvage therapies to reverse drug resistance are reviewed.
The expression of MDR1 as well as other proteins involved in regulating the bioavailability of drugs is regulated by nuclear receptors (NRs), a family of ligand-activated transcription factors. The pregnane X receptor (PXR) is an NR that directly regulates the expression of MDR1 and other important proteins involved in drug metabolism and resistance. PXR can be activated by xenobiotics, including drugs involved in MDR, suggesting that drug resistance can be prevented instead of being reversed. The recent progress made in developing PXR antagonists to pharmacologically modulate PXR and thereby potentially prevent the elevation of MDR1 levels is also reviewed.
Recently, a new form of MDR – drug ratio–dependent MDR – has been reported in cancer therapy, which occurs at discrete drug:drug ratios of combined chemotherapeutic agents. Drug ratio–dependent MDR can be circumvented by systematically screening a wide range of drug ratios and concentrations and encapsulating the drug combination in a liposomal delivery vehicle at optimal synergistic ratios. This has been recently reviewed [5], and will not be discussed here.
2. Drug resistance in anticancer therapies
2.1 Cancer and drug resistance
Despite years of intensive research and development, cancer remains one of the leading causes of death worldwide. In 2009, there were an estimated 1.5 million new cases of and 560,000 deaths from cancer in the US [6]. Chemotherapy is the most commonly used treatment for cancer, as surgery and radiation are often not effective in treating cancer at every location where it spreads. MDR of cancer cells to chemotherapeutic agents – a complex cellular process – is the leading cause of failure of chemotherapy and the rise in cancer-related deaths [7].
A common feature among cases of resistance to anticancer drugs is the dynamic interactions among cancer cells, the human body (the “host”) that governs the systemic drug clearance, and the therapeutic agent (Fig. 1), which can be used to develop target-nonspecific approaches to address resistance to chemotherapeutic agents. To a healthy human body, xenobiotics or drugs are external stresses and these are disposed of via a highly regulated drug metabolism and drug clearance process. During this process, DMEs in the liver break down the drug, and NRs such as PXR and the therapeutic drugs play crucial roles in regulating the expression of DMEs [8,9]. Recently, cancer cells have also been shown to affect drug clearance by affecting the expression of DMEs [10].
Figure 1.
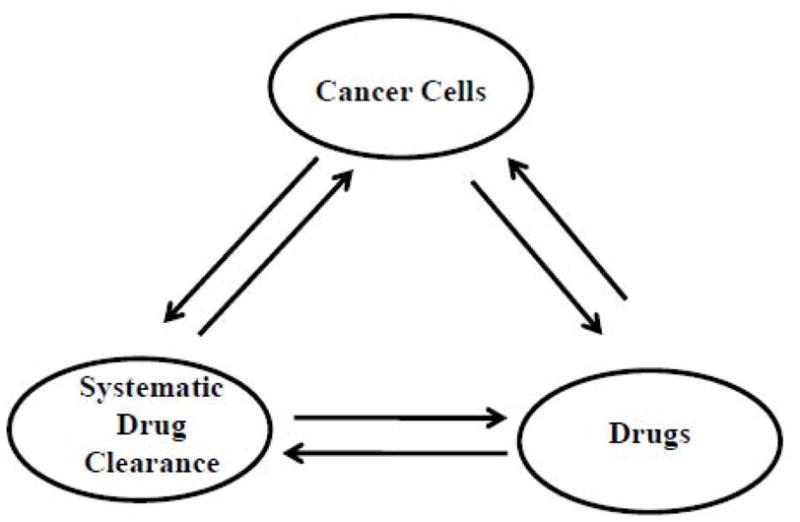
The ultimate efficacy of a drug is determined by the interactions among the drug, the target cancer cells, and the drug clearance system of the human body.
2.2 Proteins involved in resistance to cancer drugs
Changes in the expression levels of DMEs that break down drugs and ABC transporters that increase cellular efflux of chemotherapeutic agents have been associated with drug resistance in many cancers [7]. Among the 48 known human ABC transporters, MDR1, the multidrug resistance-associated protein 1 (MRP1; also known as ABCC1), and the breast cancer resistance protein (BCRP; also known as ABCG2) are major contributors to the MDR phenotype. There have been intensive investments in developing compounds that can reverse the MDR phenotype. Although laboratory research has led to promising results, efforts to translate them to clinical use have been somewhat disappointing (see sections 2.3 and 2.4 for details).
2.3 Approaches used to overcome cancer drug resistance
MDR1 is known to transport several cancer drugs [7], and its activity can be pharmacologically inhibited to prevent the efflux of cancer drugs and sensitize resistant cancer cells to cancer drugs both in vitro [11] and in the clinical setting [12]. These early data suggested that MDR1 can be a feasible target to reverse drug resistance, which was supported by the observation that loss of both Mdr1a and Mdr1b (there are 2 rodent Mdr1 genes but only 1 human MDR1 gene) does not result in an obvious phenotype. Significant efforts have since led to the development of 3 generations of MDR inhibitors.
First-generation MDR1 inhibitors are compounds that have already been approved by the Food and Drug Administration (FDA) for other clinical applications. These non-specific MDR1 inhibitors, such as verapamil, quinine, and cyclosporine A, generally fail to show clinical efficacy, mainly because they have toxic side effects at doses required to inhibit MDR1 activity [13]. However, a few positive outcomes [14] encouraged the development of second-generation MDR1 inhibitors, and efforts were centered on increasing the potency for MDR1 while decreasing toxicities, using pharmacophores of the first-generation MDR1 inhibitors. PSC-833, a cyclosporine D analog with high-affinity for MDR1 and no immunosuppressive side effects, is representative of second-generation MDR1 inhibitors. However, the inhibition of MDR1 decreased the systemic clearance of drugs and increased the exposure of both normal and cancerous tissues to the toxic effect of drugs. In addition, PSC-833 and other MDR1 inhibitors inhibited cytochrome p450 3A (CYP3A) function and decreased CYP3A-mediated drug metabolism. These undesired pharmacokinetic interactions led to drug-associated adverse effects. Therefore, although PSC-833 enhanced the therapeutic effect of certain chemotherapeutic drugs (e.g., etoposide, cytarabine, and daunorubicin) in patients with acute myeloid leukemia (AML) [15], its use was associated with high rates of mortality in other phase III trials [16], and its development was therefore discontinued. The development of another second-generation MDR1 inhibitor, biricodar, was discontinued because of similar adverse effects [17]. Efforts to develop third-generation MDR1 inhibitors have focused on increasing the affinity for MDR1 and lowering pharmacokinetic interactions (i.e., not inhibiting CYP3A function and normal CYP3A-mediated drug metabolism). Therefore, unlike first- and second-generation MDR1 inhibitors, which were developed from compounds known to target other biologic functions, third-generation MDR1 inhibitors are derived from new compounds generated by combinatorial chemistry. Laniquidar, OC144-093, zosuquidar, elacridar, tariquidar and CBT-1 are examples of third-generation MDR1 inhibitors that have a high affinity for MDR1 without having a CYP3A inhibitory effect [7]. Tariquidar was being tested in phase III clinical trials as adjunctive therapy in combination with first-line chemotherapy in patients with non-small-cell lung cancer (NSCLC), but was discontinued because of treatment-associated toxicities. It is important to note that the rationale for choosing patients with NSCLC in the studies was not clear, since there was no convincing data suggesting that the target of tariquidar, MDR1, is significantly expressed in NSCLC. In addition, the dose used for the combination therapy was higher than the maximum tolerated dose previously determined [18]. Newly exploratory trials with tariquidar are currently ongoing; zosuquidar is also being tested in phase II trials in women with metastatic and locally recurrent breast cancer [19].
Some third-generation MDR1 inhibitors are less toxic, do not affect the pharmacokinetics of anti-cancer drugs, and have better outcomes in clinical trials than first- and second-generation MDR1 inhibitors. In addition to chemical inhibitors, other MDR-reversing agents aimed at inhibiting the activity of MDR, including antibodies, have been developed [7]; however, whether the activity of MDR1 can be inhibited without causing undesired toxicity remains unclear.
2.4 Lessons learned
First- and second-generation MDR1 inhibitors have been developed based on compounds previously discovered to act on targets other than MDR1. These non-specific MDR1 inhibitors also inhibited the activity of CYP3A, affected drug metabolism and clearance, and failed in clinical trials due to undesired toxicity. Third-generation MDR1 inhibitors that are specific and potent for MDR1 and devoid of CYP3A inhibitory effect have been developed. Again, early trials in clinics failed due to undesired toxicities. The inappropriate study design of earlier trials on third-generation MDR1 inhibitors might have contributed to the failure of these trials; therefore, with appropriate study design, the approach to develop reversing agents for ABC drug transporters might have an optimistic future, suggesting that overcoming drug resistance by down-regulating MDR1 remains a feasible strategy [7]. Overcoming drug resistance by countering the elevated levels of MDR1 (due to drug-mediated over-expression) is a salvage approach. MDR1 is constitutively expressed in many normal tissues (e.g., adrenal gland, liver, kidney, intestinal mucosa, muscle, and endothelial cells of the blood brain barrier [20]) and plays an essential role in protecting normal tissues from drugs and other xenobiotics. MDR1 is over-expressed in cancer cells and causes drug resistance. MDR1 inhibitors inhibit the activity of MDR1, regardless whether it is the drug-mediated over-expressed MDR1 (which causes drug resistance) or the constitutively-expressed MDR1 (which is required for normal protecting function). To date, it has not been possible to avoid the toxicities associated with inhibition of MDR1 activity, so it remains to be studied whether drug-mediated over-expression of MDR1 can be selectively prevented. Studies on the regulation of MDR1 expression can help address the question of whether drug-mediated over-expression of MDR1 can be prevented.
The expression of MDR1 is regulated at the transcriptional level by multiple signaling mechanisms, including those mediated by hypoxia-inducible factor-1α (HIF-1α) [21], p53 [22], and even chromosomal rearrangement [23]. MDR1 expression is also regulated by epigenetic mechanisms such as methylation [24,25] and acetylation [26]. Post-transcriptional regulation of MDR1 expression by microRNA has been reported recently [27,28].
Recently, the expression of MDR1 has been shown to be regulated by xenobiotic receptor PXR [29–31], suggesting a role of NRs in regulating inducible drug resistance and a possible new strategy to overcome drug resistance by preventing the induction of MDR1 over-expression during drug therapy instead of inhibiting the activity of total MDR1.
3. Nuclear receptors and drug resistance
3.1 Regulation of drug resistance by nuclear receptors
MDR1, MRP1, and BCRP – the ABC transporters that mediate the ATP-dependent cellular export of drugs – have high expression levels in liver, intestine, kidney, and blood-brain barrier. Their normal physiologic function is to protect the body from cytotoxicity caused by drugs or other xenobiotics. This protecting function is coordinated with the DMEs, which first break down the drugs in most cases. MDR1, MRP1, and BCRP, which partially overlap in their substrate specificity, are the major ABC transporters involved in cancer drug resistance. MDR1 was the first ABC transporter identified in Chinese hamster ovary cells selected for resistance to the cytotoxic agent colchicine [32]. MRP1 was discovered in a multi-drug-resistant human lung cancer cell line [33] and BRCP in a multi-drug-resistant human breast cancer cell line [34].
There is only 1 gene for MDR1 in humans, but 2 genes (Mdr1a and Mdr1b) in rodents [35]. MDR1, which was first discovered as a protein associated with cancer cell resistance to cytotoxic compounds [32], was subsequently found to be expressed in normal cells from various tissues [36–39] and playing key roles such as elimination of drugs from the system by exporting drugs into the lumen of the gut [39], biliary excretion in the liver [39,40], renal elimination [41], and limiting drug uptakes into the central nervous system (CNS) [42–45]. MDR1 transports a broad range of hydrophobic compounds, including anticancer drugs, anti-HIV drugs, antibiotics, cardiac drugs, calcium channel blockers, and immunosuppressants [7,46–48].
There are 13 MRPs in humans. MRP1 was found to be amplified in multiple drug-resistant cancer cells [33]. MRP1 transports anticancer cytotoxic drugs [47,49]. BCRP also confers resistance to many anticancer drugs [49,50].
NRs have been shown to regulate the expressions of MDR1 and BCRP at the transcription level. PXR [29,31] and constitutive androstane receptor (CAR) [51] bind to and activate the promoter of MDR1. The promoter of BCRP contains response elements for both ER [52] and proliferator-activated receptor γ (PPARγ) [53], suggesting the role of NR in regulating BCRP expression. Whether NRs regulate the expression of MRP1 is unknown. The regulation of the MDR1 expression by PXR has been well-studied, and is the focus of this review.
3.2 PXR and drug resistance
PXR and CAR are master xenobiotic receptors that regulate the expression of genes involved in drug metabolism and clearance, including DMEs and transporters. Although no physiologic ligand has been definitively identified for PXR, PXR can bind to many structurally diverse chemicals (a characteristic referred to as “ligand promiscuity”), including anticancer drugs such as paclitaxel [54–57]. PXR is expressed not only in normal tissues such as liver, intestine, colon, kidney, brain [58–61], breast [62], prostate [63], peripheral mononuclear blood cells [64,65], heart, bone marrow, spinal cord [66], stomach, ovary, placenta [58,67] and the immune cells [68], but also in many human cancers, including breast [62, 69], prostate [63], colon [70], osteosarcoma [71], ovarian [72], and endometrial [73,74] cancers. Activation of PXR induces expression of DMEs and transporters, including MDR1, suggesting a significant role of PXR in cancer drug resistance.
NRs are ligand-activated transcription factors that regulate target gene activation [75,76]. PXR, a member of the NR superfamily, was discovered in 1998 by multiple groups [59,60,77,78]. Similar to other NRs, PXR has a highly variable N-terminal domain, a conserved DNA-binding domain (DBD), and a C-terminal ligand-binding domain (LBD) (Fig. 2).
Figure 2.

A schematic comparison of the domain structures of a typical nuclear receptor and PXR. AF-1, activation function 1; DBD, DNA binding domain; H, hinge region; LBD, ligand binding domain; AF-2, transactivation function 2.
Although the sub-cellular localization of un-liganded PXR remains controversial [79–82], it is clear that PXR binds to the promoter of its target gene as a heterodimer with retinoid X receptor α (RXRα) [75,83]. The consensus sequence, 5′ AG(G/T)TCA 35′, that the PXR DBD interacts with [77,78] can be arranged as direct repeats separated by 3–5 nucleotides (DR3, DR4, or DR5), everted repeats separated by 6 or 8 nucleotides (ER6 or ER8), or inverted repeats separated by 6 or no nucleotides (IR6 or IR0). Two most important PXR target genes, CYP3A4 and MDR1, contain DR3/ER6 [78,84] and DR4/ER6 [31] in their promoter regions, respectively.
Depending on the ligand-regulated conformation of the LBD, the activation function 2 (AF-2) region interacts with either corepressors or coactivators, resulting in transcriptional repression or activation [75,76]. Example of coactivators are steroid receptor coactivator-1 (SRC-1), glucocorticoid receptor interacting protein 1 (GRIP1), activator for thyroid hormone and retinoid receptors (ACTR), and PPARγ coactivator 1-α (PGC-1α) [60,77,78,85]. Nuclear receptor corepressor (NCoR) and silencing mediator of retinoid and thyroid hormone receptors (SMRT) are corepressors that regulate PXR [81,86]. In the absence of PXR agonist, PXR associates with corepressors, resulting in transcriptional repression. The binding of an agonist to PXR changes its conformation, allowing coactivators to interact with the AF-2 and resulting in transcriptional activation of the target genes of PXR [75]. The ultimate outcome of transcriptional activation of a target gene for PXR depends on the PXR agonist, the promoter of the target gene for PXR, and the specific tissue- and cellular context (availability of corepressors and coactivators, cell cycle status, etc.) [71,87].
Because of its unique structure of the LBD [88,89], PXR is a “promiscuous” xenobiotics receptor that can bind to a wide variety of structurally and chemically diverse compounds [90]. Endobiotics such as endogenous steroids and bile acids [60,77,78], cholesterol, and metabolites [91] have been shown to activate PXR. In addition, xenobiotics such as antibiotics rifampicin [59], cholesterol-lowering agent SR12813 [59], anticancer drug paclitaxel [55], anti-HIV drugs, and calcium channel modulators [92], are among an expanding list of drugs that can bind to and activate PXR. The activation of PXR is likely to affect the effectiveness of many drugs.
It has been clearly demonstrated that PXR directly regulates the transcriptional activation of MDR1. Geick et al. [31] first identified a distal enhancer region −7.8 kb from the transcriptional start site of the MDR1 promoter that mediates the induction of MDR1 expression by rifampicin. By using LS174T, a colon cancer cell line that expresses PXR, and a reporter gene under the control of the MDR1 promoter, Geick et al. showed that the promoter region between −8.0 and −7.7 kb mediates the induction by rifampicin. An electrophoretic mobility shift assay (EMSA) confirmed the binding of PXR/RXRα to 3 DR4 (I, II, and III) and an ER6/DR4(III). Mutational analysis demonstrated that DR4(I) is essential for the rifampicin-mediated induction of MDR1 in LS174T cells. These studies elucidated the molecular mechanism responsible for PXR-mediated induction of MDR1 expression by rifampicin. Geick et al. subsequently demonstrated that MDR1 is also regulated by CAR, through the DR4(I), and, to a lesser extent, the ER6/DR(III) [51]. Interestingly, CAR also binds to the DR(II) as a monomer. Both DR4(I) and DR4(II) are required for the maximal induction of MDR1 by CAR.
The expression of MDR1 and CYP3A4 is induced by PXR agonists both in vivo [93,94] and in vitro [95]. Although the level of in vitro induction of MDR1 and other transporters is lower than that of CYP3A4 and other CYPs [95], the in vivo inductions of CYP3A4 and MDR1 are similar [93,94,96]. The difference in in vitro induction might result from the significantly reduced basal expression level of CYP3A4 and other CYPs, but not that of MDR1 and other transporters, in in vitro systems [95,97]. These observations indicate that PXR-mediated induction of MDR1 plays important roles in modulating drug clearance, and also suggest that in vitro systems (e.g., cancer cell lines or isolated primary hepatocytes) might be suitable to study the regulation of MDR1 before conducting in vivo experiments.
The significant role of induction of MDR1 in increasing drug clearance has also been illustrated in human volunteers treated with various PXR agonists and MDR1 substrates. PXR agonists such as rifampicin, St. John’s Wort, and carbamazepine can induce the expression of intestinal MDR1 and decrease the bioavailability and plasma levels of compounds transported by MDR1 (e.g., digoxin, talinolol, and fexofenadine) [93,94,98–101]. Rifampicin can also increase digoxin clearance into bile [39], and carbamazepine can increase the renal clearance of talinolol [99]. To emphasize the role of transporters in drug clearance, digoxin, talinolol, and fexofenadine were chosen in these studies because they are transported by MDR1 but minimally metabolized by DMEs.
These studies demonstrate that induction of drug transporters such as MDR1 affects drug disposition and ultimately reduces the plasma concentration of drugs. Although induction of MDR1 and other transporters is a mechanism to protect the body against potentially toxic chemicals, it also reduces the effectiveness of therapeutic drugs in curing a disease or improving patients’ symptoms, thereby contributing to drug resistance.
The undesired toxicity associated with inhibition of the physiologic function of MDR1 has limited the success of MDR1 inhibitors in clinical applications. Because MDR1 can be induced by drugs through activation of PXR, a feasible option is to pharmacologically antagonize the drug-mediated activation of PXR and PXR-induced expression of MDR1 to increase the bioavailability of drugs and minimize toxicity.
3.3 Preventing drug resistance by regulating PXR
The concept that down-regulating PXR in PXR-expressing cancers can sensitize cancer cells to chemotherapeutic agents has been proposed and investigated in several recent studies. Chen et al. detected the expression of PXR in both normal and cancerous prostate tissues and in prostate cancer cell lines [63]. In the prostate cancer cell line PC-3, treatment with the PXR agonist SR12813 activated PXR and increased both the expression of MDR1 and the resistance of PC-3 cells to the anticancer drugs paclitaxel and vinblastine. The targeted knock-down of PXR by using short hairpin RNA (shRNA) enhanced the sensitivity of PC-3 to paclitaxel and vinblastine, suggesting that the effectiveness of anticancer drugs can be enhanced in PXR-positive cancers by blocking the activity of PXR.
Masuyama et al. showed that PXR is expressed in endometrial cancer. Down-regulation of PXR by small interfering RNA (siRNA) in the endometrial cancer cell line HEC-1 decreased the expression of MDR1 and sensitized cells to anticancer agent and PXR agonist paclitaxel and cisplatin [73]. In contrast, increased expression level of PXR led to increased resistance of HEC-1 cells to paclitaxel and cisplatin.
The correlation between the activity of PXR and drug resistance observed in the studies discussed previously has also been reported in osteosarcoma [71], in which the effectiveness of etoposide was reduced due to activation of PXR. Furthermore, co-administration of PXR agonists enhanced the clearance of all-trans-retinoic acid (ATRA), which could potentially contribute to ATRA resistance in the treatment of acute promyelocytic leukemia (APL) and several solid tumors [102].
Because of its ligand promiscuity, PXR can be activated by many anticancer drugs, such as tamoxifen, Taxol [55,30,103], and vincristine [55]. Most patients with cancer are usually administered many other drugs in addition to anticancer drugs while undergoing chemotherapy, which further increases the possibility of drug-mediated PXR activation. As PXR regulates the expression of proteins involved in drug metabolism and drug transport, activation of PXR can lead to undesired drug interactions. In PXR-expressing cancers, the anticancer drug that activates PXR might compromise the effectiveness of the drug itself as well as that of other drugs in combination therapy. The ability to activate PXR is therefore considered an undesirable property for a lead compound for development as a drug [104]. One approach to overcome the PXR activation of a lead compound is to chemically modify the compound to remove the PXR activating function without compromising the target activity. This has been shown to be possible in principle in a few studies. For example, paclitaxel and docetaxel, both inhibitors of microtubule disassembly, have minor structural difference and are equally potent in inhibiting microtubule depolymerization and cancer cell proliferation. However, paclitaxel, but not docetaxel, significantly activates PXR and induces MDR1 expression [30]. Recently, Zimmermann et al. reported the chemical modifications of their first generation IGF-1R inhibitors to reduce PXR transactivation while maintaining potency against IGF-1R [104]. However, given the agonist promiscuity of PXR, tremendous efforts are needed in drug development programs to remove the PXR activity while maintaining the target activity for many lead compounds. In addition, it is highly likely that other properties of compounds might have also changed because of the chemical modifications to remove the PXR activating function. Furthermore, many anticancer drugs with PXR agonistic activity continue to be used in the clinical setting. In light of these considerations, efforts need to focus on developing compounds that can antagonize PXR-mediated MDR1 expression and enhance the effectiveness of anticancer drugs.
A few compounds previously known to target various biological pathways can inhibit PXR function (Table 1). Here, PXR inhibitors refer to compounds that inhibit the agonist-mediated activation of PXR, but whether they bind to PXR is unknown. PXR antagonists refer to PXR inhibitors that have been shown to competitively bind to PXR in in vitro binding assays. Ecteinascidin-743 (ET-743), an antineoplastic agent, has been shown to inhibit PXR transactivation [30]. Ketoconazole, an inhibitor of CYP3A4 enzyme activity, can inhibit multiple NRs, including PXR, by disrupting the NR–coactivator interaction [105]. A-792611, an HIV protease inhibitor, inhibits PXR-mediated CYP3A4 expression [106]. Sulforaphane (SFN), an inhibitor of histone deacetylases and an inducer of phase II DMEs such as glutathione S-transferases (GSTs), appears to be a PXR antagonist [107]. SFN down-regulates CYP3A4 expression by directly binding to PXR and inhibiting coactivator recruitment. Coumestrol, a potent agonist of ERα and ERβ (EC50 21 – 67 nM), antagonizes PXR at high concentrations (EC50 12 μM) [108]. Camptothecin, an inhibitor of topoisomerase I, inhibits PXR-mediated transcriptional activation of CYP3A4 by disrupting the interaction of PXR with SRC-1 without competing with agonist for binding to PXR [109]. The effect of camptothecin is not specific for PXR, because camptothecin also inhibits CAR-mediated, but activates vitamin D receptor (VDR)-mediated transactivation [109]. Although all known PXR inhibitors or antagonists have an activity other than inhibiting PXR, these studies suggest that it is feasible to antagonize the inducible activity of PXR and to enhance the effectiveness of drugs. In a recent study, Raynal et al. showed that activation of PXR reduced the chemosensitivity of colorectal cancer cells to irinotecan. Interestingly, the reduction in chemosensitivity was reversed by the PXR antagonist SFN [110].
Table 1.
Chemical structures and known activities of PXR inhibitors/antagonists
| Compound | Structure | Other known activity | References |
|---|---|---|---|
| ET-743 | 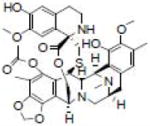 |
Antineoplastic | Synold et al. [30] |
| Ketoconazole | 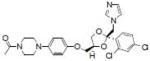 |
Inhibiting CYP3A4 enzyme activity | Huang et al. [105] |
| Sulforaphane |  |
Inhibiting histone deacetylases: inducing Phase II enzymes | Zhou et al. [107] |
| A-792611 | 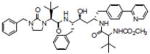 |
Inhibiting HIV protease | Healan-Greenberg et al. [106] |
| Coumestrol | 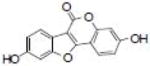 |
Agonist for estrogen receptors | Wang et al. [108] |
| Camptothecin | 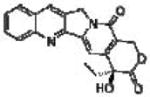 |
Inhibiting topoisomerase I | Chen et al. [109] |
In addition to test compounds with known bioactivity for their PXR antagonistic activity, other groups used a computational approach to study PXR antagonism. Ekins et al. investigated pharmacophores for both PXR agonists and antagonists, and suggested that agonists and antagonists might bind to distinct regions of PXR [111]. Ekins et al. used computational pharmacophore and docking tools to discover PXR antagonists in the low micromolar range [112]. In a study of the crystal structure of PXR with the agonist T-1317, Xue et al. suggested that because of the ligand promiscuity of PXR it may be difficult to design an effective antagonist that targets the ligand-binding pocket of PXR [113].
As several studies support the existence of PXR antagonists, the development of specific and non-toxic PXR antagonists as codrugs hold promise in order to prevent the activation of PXR and induction of MDR1 during drug therapies and thereby prevent drug resistance. Such specific PXR antagonists might have broad applications in overcoming drug resistance. For example, a PXR-like pathway regulating multidrug resistance in fungi has been reported by Thakur et al. [114]. The authors showed that drug resistance during treatment of fungal infections is often due to upregulation of drug efflux pumps by a fungal transcription factor that directly binds to xenobiotics, including PXR agonists, and suggest that a PXR antagonist can be used to treat multidrug-resistant fungal infections.
4. Conclusions
Drug resistance involves multiple mechanisms and targets; it is therefore impossible to overcome drug resistance by targeting a single protein. MDR1 is an important protein involved in target-nonspecific drug resistance. Inhibition of MDR1 to overcome drug resistance has had limited success due to toxicity. MDR1 expression can be regulated by several mechanisms. The recent discovery that the expression of MDR1 is induced by PXR, a xenobiotic receptor activated by many compounds, including anticancer drugs, suggests that it is possible to antagonize the drug-induced activation of PXR to prevent the drug-mediated expression of MDR1. The identification of PXR antagonists further suggests that pharmaceutical agents can be developed to enhance the efficacy of anticancer drugs. All known PXR inhibitors or antagonists have activities other than inhibiting PXR. Future studies need to focus on identifying specific PXR antagonists that target the agonist-induced activation of PXR. Such specific PXR antagonists will not interfere with the basal activity of PXR and might have minimal toxicity. Owing to the ligand promiscuity of PXR, it might be difficult to design such PXR antagonists. Large-scale high-throughput screening, using a large collection of structurally diverse compounds, might provide the most effective approach to identify and develop PXR antagonists.
Non-toxic, specific, and potent PXR antagonists can be used to improve the efficacy of anticancer drugs in PXR-positive cancers. Such specific PXR antagonists might have broad applications in overcoming drug resistance, including treating multidrug-resistant fungal infections.
Acknowledgments
I thank Drs. Wenwei Lin, Satya Pondugula, Su Sien Ong, Yueming Wang and other members of the Chen research laboratory for their valuable discussions, and Dr. Vani Shanker for editing the manuscript. This work was supported in part by the National Institutes of Health National Institute of General Medical Sciences [Grant GM086415] (to T.C.); the National Institutes of Health National Cancer Institute [Grant P30-CA027165]; the American Lebanese Syrian Associated Charities; and St. Jude Children’s Research Hospital.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nguyen MH, Garcia RT, Trinh HN, Nguyen HA, Nguyen KK, Nguyen LH, Levitt B. Prevalence of hepatitis B virus DNA polymerase mutations in treatment-naïve patients with chronic hepatitis B. Aliment Pharmacol Ther. 2009;30:1150–1158. doi: 10.1111/j.1365-2036.2009.04151.x. [DOI] [PubMed] [Google Scholar]
- 2.Clarke R, Liu MC, Bouker KB, Gu Z, Lee RY, Zhu Y, Skaar TC, Gomez B, O’Brien K, Wang Y, Hilakivi-Clarke LA. Antiestrogen resistance in breast cancer and the role of estrogen receptor signaling. Oncogene. 2003;22:7316–7339. doi: 10.1038/sj.onc.1206937. [DOI] [PubMed] [Google Scholar]
- 3.Mellor HR, Callaghan R. Resistance to chemotherapy in cancer: a complex and integrated cellular response. Pharmacology. 2008;81:275–300. doi: 10.1159/000115967. [DOI] [PubMed] [Google Scholar]
- 4.Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov. 2006;5:219–234. doi: 10.1038/nrd1984. [DOI] [PubMed] [Google Scholar]
- 5.Harasym TO, Liboiron BD, Mayer LD. Drug ratio-dependent antagonism: a new category of multidrug resistance and strategies for its circumvention. Methods Mol Biol. 2010;596:291–323. doi: 10.1007/978-1-60761-416-6_13. [DOI] [PubMed] [Google Scholar]
- 6.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 7.Lee CH. Reversing agents for ATP-binding cassette drug transporters. Methods Mol Biol. 2010;596:325–340. doi: 10.1007/978-1-60761-416-6_14. [DOI] [PubMed] [Google Scholar]
- 8.Zhang B, Xie W, Krasowski MD. PXR: a xenobiotic receptor of diverse function implicated in pharmacogenetics. Pharmacogenomics. 2008;9:1695–1709. doi: 10.2217/14622416.9.11.1695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pondugula SR, Dong H, Chen T. Phosphorylation and protein-protein interactions in PXR-mediated CYP3A repression. Expert Opin Drug Metab Toxicol. 2009;5:861–873. doi: 10.1517/17425250903012360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robertson GR, Liddle C, Clarke SJ. Inflammation and altered drug clearance in cancer: transcriptional repression of a human CYP3A4 transgene in tumor-bearing mice. Clin Pharmacol Ther. 2008;83:894–897. doi: 10.1038/clpt.2008.55. [DOI] [PubMed] [Google Scholar]
- 11.Dano K. Active outward transport of daunomycin in resistant Ehrlich ascites tumor cells. Biochem Biophys Acta. 1973;323:466–483. doi: 10.1016/0005-2736(73)90191-0. [DOI] [PubMed] [Google Scholar]
- 12.Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y. Overcoming of vincrintine resistance in PP388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res. 1981;41:1967–1972. [PubMed] [Google Scholar]
- 13.Daenen S, van der Holt B, Verhoef GE, Löwenberg B, Wijermans PW, Huijgens PC, van Marwijk Kooy R, Schouten HC, Kramer MH, Ferrant A, van den Berg E, Steijaert MM, Verdonck LF, Sonneveld P. Addition of cyclosporin A to the combination of mitoxantrone and etoposide to overcome resistance to chemotherapy in refractory or relapsing acute myeloid leukaemia: a randomised phase II trial from HOVON, the Dutch-Belgian Haemato-Oncology Working Group for adults. Leuk Res. 2004;28:1057–1067. doi: 10.1016/j.leukres.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Wattel E, Solary E, Hecquet B, Caillot D, Ifrah N, Brion A, Milpied N, Janvier M, Guerci A, Rochant H, Cordonnier C, Dreyfus F, Veil A, Hoang-Ngoc L, Stoppa AM, Gratecos N, Sadoun A, Tilly H, Brice P, Lioure B, Desablens B, Pignon B, Abgrall JP, Leporrier M, Fenaux P, et al. Quinine improves results of intensive chemotherapy (IC) in myelodysplastic syndromes (MDS) expressing P-glycoprotein (PGP). Updated results of a randomized study. Groupe Français des Myélodysplasies (GFM) and Groupe GOELAMS. Adv Exp Med Biol. 1999;457:35–46. doi: 10.1007/978-1-4615-4811-9_5. [DOI] [PubMed] [Google Scholar]
- 15.Kolitz JE, George SL, Dodge RK, Hurd DD, Powell BL, Allen SL, Velez-Garcia E, Moore JO, Shea TC, Hoke E, Caligiuri MA, Vardiman JW, Bloomfield CD, Larson RA. Dose escalation studies of cytarabine, daunorubicin, and etoposide with and without multidrug resistance modulation with PSC-833 in untreated adults with acute myeloid leukemia younger than 60 years: final induction results of Cancer and Leukemia Group B Study 9621. J Clin Oncol. 2004;22:4290–4301. doi: 10.1200/JCO.2004.11.106. [DOI] [PubMed] [Google Scholar]
- 16.Baer MR, George SL, Dodge RK, O’Loughlin KL, Minderman H, Caligiuri MA, Anastasi J, Powell BL, Kolitz JE, Schiffer CA, Bloomfield CD, Larson RA. Phase 3 study of the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of age and older with acute myeloid leukemia: Cancer and Leukemia Group B Study 9720. Blood. 2002;100:1224–1232. [PubMed] [Google Scholar]
- 17.Goldman B. Multidrug resistance: can new drugs help chemotherapy score against cancer? J Natl Cancer Inst. 2003;95:255–257. doi: 10.1093/jnci/95.4.255. [DOI] [PubMed] [Google Scholar]
- 18.Fox E, Bates SE. Tariquidar (XR9576): a P-glycoprotein drug efflux pump inhibitor. Expert Rev Anticancer Ther. 2007;7:447–459. doi: 10.1586/14737140.7.4.447. [DOI] [PubMed] [Google Scholar]
- 19.Ruff P, Vorobiof DA, Jordaan JP, Demetriou GS, Moodley SD, Nosworthy AL, Werner ID, Raats J, Burgess LJ. A randomized, placebo-controlled, double-blind phase 2 study of docetaxel compared to docetaxel plus zosuquidar ( LY335979) in women with metastatic or locally recurrent breast cancer who have received one prior chemotherapy regimen. Cancer Chemother Pharmacol. 2009;64:763–768. doi: 10.1007/s00280-009-0925-9. [DOI] [PubMed] [Google Scholar]
- 20.Cordon-Cardo C, O’Brien JP, Boccia J, Casals D, Bertino JR, Melamed MR. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J Histochem Cytochem. 1990;38:1277–1287. doi: 10.1177/38.9.1974900. [DOI] [PubMed] [Google Scholar]
- 21.Comerford KM, Wallace TJ, Karhausen J, Louis NA, Montalto MC, Colgan SP. Hypoxia-inducible factor-1-dependent regulation of the multidrug resistance (MDR1) gene. Cancer Res. 2002;62:3387–3394. [PubMed] [Google Scholar]
- 22.Sampath J, Sun D, Kidd VJ, Grenet J, Gandhi A, Shapiro LH, Wang Q, Zambetti GP, Schuetz JD. Mutant p53 cooperates with ETS and selectively up-regulates human MDR1 not MRP1. J Biol Chem. 2001;276:39359–39367. doi: 10.1074/jbc.M103429200. [DOI] [PubMed] [Google Scholar]
- 23.Huff LM, Lee JS, Robey RW, Fojo T. Characterization of gene rearrangements leading to activation of MDR-1. J Biol Chem. 2006;281:36501–36509. doi: 10.1074/jbc.M602998200. [DOI] [PubMed] [Google Scholar]
- 24.El-Osta A, Kantharidis P, Zalcberg JR, Wolffe AP. Precipitous release of methyl-CpG binding protein 2 and histone deacetylase 1 from the methylated human multidrug resistance gene (MDR1) on activation. Mol Cell Biol. 2002;22:1844–1857. doi: 10.1128/MCB.22.6.1844-1857.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tada Y, Wada M, Kuroiwa K, Kinugawa N, Harada T, Nagayama J, Nakagawa M, Naito S, Kuwano M. MDR1 gene overexpression and altered degree of methylation at the promoter region in bladder cancer during chemotherapeutic treatment. Clin Cancer Res. 2000;6:4618–4627. [PubMed] [Google Scholar]
- 26.Baker EK, Johnstone RW, Zalcberg JR, El-Osta A. Epigenetic changes to the MDR1 locus in response to chemotherapeutic drugs. Oncogene. 2005;24:8061–8675. doi: 10.1038/sj.onc.1208955. [DOI] [PubMed] [Google Scholar]
- 27.Gómez-Martínez A, García-Morales P, Carrato A, Castro-Galache MD, Soto JL, Carrasco-García E, García-Bautista M, Guaraz P, Ferragut JA, Saceda M. Post-transcriptional regulation of P-glycoprotein expression in cancer cell lines. Mol Cancer Res. 2007;5:641–653. doi: 10.1158/1541-7786.MCR-06-0177. [DOI] [PubMed] [Google Scholar]
- 28.Zhu H, Wu H, Liu X, Evans BR, Medina DJ, Liu CG, Yang JM. Role of MicroRNA miR-27a and miR-451 in the regulation of MDR1/P-glycoprotein expression in human cancer cells. Biochem Pharmacol. 2008;76:582–588. doi: 10.1016/j.bcp.2008.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cerveny L, Svecova L, Anzenbacherova E, Vrzal R, Staud F, Dvorak Z, Ulrichova J, Anzenbacher P, Pavek P. Valproic acid induces CYP3A4 and MDR1 gene expression by activation of constitutive androstane receptor and pregnane X receptor pathways. Drug Metab Dispos. 2007;35:1032–1041. doi: 10.1124/dmd.106.014456. [DOI] [PubMed] [Google Scholar]
- 30.Synold TW, Dussault I, Forman BM. The orphan nuclear receptor SXR coordinately regulates drug metabolism and efflux. Nat Med. 2001;7:584–590. doi: 10.1038/87912. [DOI] [PubMed] [Google Scholar]
- 31.Geick A, Eichelbaum M, Burk O. Nuclear receptor response elements mediate induction of intestinal MDR1 by rifampin. J Biol Chem. 2001;276:14581–14587. doi: 10.1074/jbc.M010173200. [DOI] [PubMed] [Google Scholar]
- 32.Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta. 1976;455:152–162. doi: 10.1016/0005-2736(76)90160-7. [DOI] [PubMed] [Google Scholar]
- 33.Duncan AM, Deeley RG, Cole SP, Bhardwaj G, Gerlach JH, Mackie JE, Grant CE, Almquist KC, Stewart AJ, Kurz EU. Overexpression of a transporter gene in a multidrug-resistant human lung cancer cell line. Science. 1992;258:1650–1654. doi: 10.1126/science.1360704. [DOI] [PubMed] [Google Scholar]
- 34.Doyle LA, Yang W, Abruzzo LV, Krogmann T, Gao Y, Rishi AK, Ross DD. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc Natl Acad Sci U S A. 1998;95:15665–15670. doi: 10.1073/pnas.95.26.15665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hsu SI, Lothstein L, Horwitz SB. Differential overexpression of three mdr gene family members in multidrug-resistant J774.2 mouse cells. Evidence that distinct P-glycoprotein precursors are encoded by unique mdr genes. J Biol Chem. 1989;264:12053–12062. [PubMed] [Google Scholar]
- 36.Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC. Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci U S A. 1987;84:7735–7738. doi: 10.1073/pnas.84.21.7735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Terao T, Hisanaga E, Sai Y, Tamai I, Tsuji A. Active secretion of drugs from the small intestinal epithelium in rats by P-glycoprotein functioning as an absorption barrier. J Pharm Pharmacol. 1996;48:1083–1089. doi: 10.1111/j.2042-7158.1996.tb05904.x. [DOI] [PubMed] [Google Scholar]
- 38.Stephens RH, O’Neill CA, Bennett J, Humphrey M, Henry B, Rowland M, Warhurst G. Resolution of P-glycoprotein and non-P-glycoprotein effects on drug permeability using intestinal tissues from mdr1a (−/−) mice. Br J Pharmacol. 2002;135:2038–2046. doi: 10.1038/sj.bjp.0704668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Drescher S, Glaeser H, Mürdter T, Hitzl M, Eichelbaum M, Fromm MF. P-glycoprotein-mediated intestinal and biliary digoxin transport in humans. Clin Pharmacol Ther. 2003;73:223–231. doi: 10.1067/mcp.2003.27. [DOI] [PubMed] [Google Scholar]
- 40.Annaert PP, Turncliff RZ, Booth CL, Thakker DR, Brouwer KL. P-glycoprotein-mediated in vitro biliary excretion in sandwich-cultured rat hepatocytes. Drug Metab Dispos. 2001;29:1277–1283. [PubMed] [Google Scholar]
- 41.de Lannoy IA, Mandin RS, Silverman M. Renal secretion of vinblastine, vincristine and colchicine in vivo. Pharmacol Exp Ther. 1994;268:388–395. [PubMed] [Google Scholar]
- 42.Cordon-Cardo C, O’Brien JP, Casals D, Rittman-Grauer L, Biedler JL, Melamed MR, Bertino JR. Multidrug-resistance gene (P-glycoprotein) is expressed by endothelial cells at blood-brain barrier sites. Proc Natl Acad Sci U S A. 1989;86:695–698. doi: 10.1073/pnas.86.2.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tatsuta T, Naito M, Oh-hara T, Sugawara I, Tsuruo T. Functional involvement of P-glycoprotein in blood-brain barrier. J Biol Chem. 1992;267:20383–20391. [PubMed] [Google Scholar]
- 44.Schinkel AH, Smit JJ, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CA, van der Valk MA, Robanus-Maandag EC, te Riele HP, et al. Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood-brain barrier and to increased sensitivity to drugs. Cell. 1994;77:491–502. doi: 10.1016/0092-8674(94)90212-7. [DOI] [PubMed] [Google Scholar]
- 45.Tahara H, Kusuhara H, Fuse E, Sugiyama Y. P-glycoprotein plays a major role in the efflux of fexofenadine in the small intestine and blood-brain barrier, but only a limited role in its biliary excretion. Drug Metab Dispos. 2005;33:963–968. doi: 10.1124/dmd.105.004192. [DOI] [PubMed] [Google Scholar]
- 46.Zhang Y Y, Benet LZ. The gut as a barrier to drug absorption: combined role of cytochrome P450 3A and P-glycoprotein. Clin Pharmacokinet. 2001;40:159–168. doi: 10.2165/00003088-200140030-00002. [DOI] [PubMed] [Google Scholar]
- 47.Chan LM, Lowes S, Hirst BH. The ABCs of drug transport in intestine and liver: efflux proteins limiting drug absorption and bioavailability. Eur J Pharm Sci. 2004;21:25–51. doi: 10.1016/j.ejps.2003.07.003. [DOI] [PubMed] [Google Scholar]
- 48.Dietrich CG, Geier A, Oude Elferink RP. ABC of oral bioavailability: transporters as gatekeepers in the gut. Gut. 2003;52:1788–1795. doi: 10.1136/gut.52.12.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haimeur A, Conseil G, Deeley RG, Cole SP. The MRP-related and BCRP/ABCG2 multidrug resistance proteins: biology, substrate specificity and regulation. Curr Drug Metab. 2004;5:21–53. doi: 10.2174/1389200043489199. [DOI] [PubMed] [Google Scholar]
- 50.Leslie EM, Deeley RG, Cole SP. Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense. Toxicol Appl Pharmacol. 2005;204:216–237. doi: 10.1016/j.taap.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 51.Burk O, Arnold KA, Geick A, Tegude H, Eichelbaum M. A role for constitutive androstane receptor in the regulation of human intestinal MDR1 expression. Biol Chem. 2005;386:503–513. doi: 10.1515/BC.2005.060. [DOI] [PubMed] [Google Scholar]
- 52.Ee PL, Kamalakaran S, Tonetti D, He X, Ross DD, Beck WT. Identification of a novel estrogen response element in the breast cancer resistance protein (ABCG2) gene. Cancer Res. 2004;64:1247–1251. doi: 10.1158/0008-5472.can-03-3583. [DOI] [PubMed] [Google Scholar]
- 53.Szatmari I, Vámosi G, Brazda P, Balint BL, Benko S, Széles L, Jeney V, Ozvegy-Laczka C, Szántó A, Barta E, Balla J, Sarkadi B, Nagy L. Peroxisome proliferator-activated receptor gamma-regulated ABCG2 expression confers cytoprotection to human dendritic cells. J Biol Chem. 2006;281:23812–23823. doi: 10.1074/jbc.M604890200. [DOI] [PubMed] [Google Scholar]
- 54.Huang R, Murry DJ, Kolwankar D, Hall SD, Foster DR. Vincristine transcriptional regulation of efflux drug transporters in carcinoma cell lines. Biochem Pharmacol. 2006;71:1695–1704. doi: 10.1016/j.bcp.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 55.Mani S, Huang H, Sundarababu S, Liu W, Kalpana G, Smith AB, Horwitz SB. Activation of the steroid and xenobiotic receptor (human pregnane X receptor) by nontaxane microtubule-stabilizing agents. Clin Cancer Res. 2005;11:6359–6369. doi: 10.1158/1078-0432.CCR-05-0252. [DOI] [PubMed] [Google Scholar]
- 56.Nallani SC, Goodwin B, Maglich JM, Buckley DJ, Buckley AR, Desai PB. Induction of cytochrome P450 3A by paclitaxel in mice: pivotal role of the nuclear xenobiotic receptor, pregnane X receptor. Drug Metab Dispos. 2003;31:681–684. doi: 10.1124/dmd.31.5.681. [DOI] [PubMed] [Google Scholar]
- 57.Gustafson DL, Long ME, Bradshaw EL, Merz AL, Kerzic PJ. P450 induction alters paclitaxel pharmacokinetics and tissue distribution with multiple dosing. Cancer Chemother Pharmacol. 2005;56:248–254. doi: 10.1007/s00280-004-0988-6. [DOI] [PubMed] [Google Scholar]
- 58.Staudinger JL, Goodwin B, Jones SA, Hawkins-Brown D, MacKenzie KI, LaTour A, Liu Y, Klaassen CD, Brown KK, Reinhard J, Willson TM, Koller BH, Kliewer SA. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc Natl Acad Sci U S A. 2001;98:3369–3374. doi: 10.1073/pnas.051551698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lehmann JM, McKee DD, Watson MA, Willson TM, Moore JT, Kliewer SA. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J Clin Invest. 1998;102:1016–1023. doi: 10.1172/JCI3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bertilsson G, Heidrich J, Svensson K, Asman M, Jendeberg L, Sydow-Bäckman M, Ohlsson R, Postlind H, Blomquist P, Berkenstam A. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc Natl Acad Sci U S A. 1998;95:12208–12213. doi: 10.1073/pnas.95.21.12208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bauer B, Hartz AM, Fricker G, Miller DS. Pregnane X receptor up-regulation of P-glycoprotein expression and transport function at the blood-brain barrier. Mol Pharmacol. 2004;66:413–419. doi: 10.1124/mol.66.3.. [DOI] [PubMed] [Google Scholar]
- 62.Dotzlaw H, Leygue E, Watson P, Murphy LC. The human orphan receptor PXR messenger RNA is expressed in both normal and neoplastic breast tissue. Clin Cancer Res. 1999;5:2103–2107. [PubMed] [Google Scholar]
- 63.Chen Y, Tang Y, Wang MT, Zeng S, Nie D. Human pregnane X receptor and resistance to chemotherapy in prostate cancer. Cancer Res. 2007;67:10361–10367. doi: 10.1158/0008-5472.CAN-06-4758. [DOI] [PubMed] [Google Scholar]
- 64.Owen A, Chandler B, Back DJ, Khoo SH. Expression of pregnane-X-receptor transcript in peripheral blood mononuclear cells and correlation with MDR1 mRNA. Antivir Ther. 2004;9:819–821. [PubMed] [Google Scholar]
- 65.Albermann N, Schmitz-Winnenthal FH, Z’graggen K, Volk C, Hoffmann MM, Haefeli WE, Weiss J. Expression of the drug transporters MDR1/ABCB1, MRP1/ABCC1, MRP2/ABCC2, BCRP/ABCG2, and PXR in peripheral blood mononuclear cells and their relationship with the expression in intestine and liver. Biochem Pharmacol. 2005;70:949–958. doi: 10.1016/j.bcp.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 66.Lamba V, Yasuda K, Lamba JK, Assem M, Davila J, Strom S, Schuetz EG. PXR (NR1I2): splice variants in human tissues, including brain, and identification of neurosteroids and nicotine as PXR activators. Toxicol Appl Pharmacol. 2004;199:251–265. doi: 10.1016/j.taap.2003.12.027. [DOI] [PubMed] [Google Scholar]
- 67.Masuyama H, Hiramatsu Y, Mizutani Y, Inoshita H, Kudo T. The expression of pregnane X receptor and its target gene, cytochrome P450 3A1, in perinatal mouse. Mol Cell Endocrinol. 2001;172:47–56. doi: 10.1016/s0303-7207(00)00395-6. [DOI] [PubMed] [Google Scholar]
- 68.Dubrac S, Elentner A, Ebner S, Horejs-Hoeck J, Schmuth M. Modulation of T lymphocyte function by the pregnane X receptor. J Immunol. 2010;184:2949–2957. doi: 10.4049/jimmunol.0902151. [DOI] [PubMed] [Google Scholar]
- 69.Miki Y, Suzuki T, Kitada K, Yabuki N, Shibuya R, Moriya T, Ishida T, Ohuchi N, Blumberg B, Sasano H. Expression of the steroid and xenobiotic receptor and its possible target gene, organic anion transporting polypeptide-A, in human breast carcinoma. Cancer Res. 2006;66:535–542. doi: 10.1158/0008-5472.CAN-05-1070. [DOI] [PubMed] [Google Scholar]
- 70.Zhou J, Liu M, Zhai Y, Xie W. The antiapoptotic role of pregnane X receptor in human colon cancer cells. Mol Endocrinol. 2008;22:868–880. doi: 10.1210/me.2007-0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mensah-Osman EJ, Thomas DG, Tabb MM, Larios JM, Hughes DP, Giordano TJ, Lizyness ML, Rae JM, Blumberg B, Hollenberg PF, Baker LH. Expression levels and activation of a PXR variant are directly related to drug resistance in osteosarcoma cell lines. Cancer. 2007;109:957–965. doi: 10.1002/cncr.22479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gupta D, Venkatesh M, Wang H, Kim S, Sinz M, Goldberg GL, Whitney K, Longley C, Mani S. Expanding the roles for pregnane X receptor in cancer: proliferation and drug resistance in ovarian cancer. Clin Cancer Res. 2008;14:5332–5340. doi: 10.1158/1078-0432.CCR-08-1033. [DOI] [PubMed] [Google Scholar]
- 73.Masuyama H, Hiramatsu Y, Kodama J, Kudo T T. Expression and potential roles of pregnane X receptor in endometrial cancer. J Clin Endocrinol Metab. 2003;88:4446–4454. doi: 10.1210/jc.2003-030203. [DOI] [PubMed] [Google Scholar]
- 74.Masuyama H, Nakatsukasa H, Takamoto N, Hiramatsu Y. Down-regulation of pregnane X receptor contributes to cell growth inhibition and apoptosis by anticancer agents in endometrial cancer cells. Mol Pharmacol. 2007;72:1045–1053. doi: 10.1124/mol.107.037937. [DOI] [PubMed] [Google Scholar]
- 75.Sonoda J, Pei L, Evans RM. Nuclear receptors: decoding metabolic disease. FEBS Lett. 2008;582:2–9. doi: 10.1016/j.febslet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen T. Nuclear receptor drug discovery. Curr Opin Chem Biol. 2008;12:418–426. doi: 10.1016/j.cbpa.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 77.Kliewer SA, Moore JT, Wade L, Staudinger JL, Watson MA, Jones SA, McKee DD, Oliver BB, Willson TM, Zetterström RH, Perlmann T, Lehmann JM. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998;92:73–82. doi: 10.1016/s0092-8674(00)80900-9. [DOI] [PubMed] [Google Scholar]
- 78.Blumberg B, Sabbagh W, Jr, Juguilon H, Bolado J, Jr, van Meter CM, Ong ES, Evans RM. SXR, a novel steroid and xenobiotic-sensing nuclear receptor. Genes Dev. 1998;12:3195–3205. doi: 10.1101/gad.12.20.3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Squires EJ, Sueyoshi T, Negishi M. Cytoplasmic localization of pregnane X receptor and ligand-dependent nuclear translocation in mouse liver. J Biol Chem. 2004;279:49307–49314. doi: 10.1074/jbc.M407281200. [DOI] [PubMed] [Google Scholar]
- 80.Saradhi M, Sengupta A, Mukhopadhyay G, Tyagi RK. Pregnane and Xenobiotic Receptor (PXR/SXR) resides predominantly in the nuclear compartment of the interphase cell and associates with the condensed chromosomes during mitosis. Biochim Biophys Acta. 2005;1746:85–94. doi: 10.1016/j.bbamcr.2005.10.004. [DOI] [PubMed] [Google Scholar]
- 81.Johnson DR, Li CW, Chen LY, Ghosh JC, Chen JD. Regulation and Binding of Pregnane X Receptor by Nuclear Receptor Corepressor Silencing Mediator of Retinoid and Thyroid Hormone Receptors (SMRT) Mol Pharmacol. 2006;69:99–108. doi: 10.1124/mol.105.013375. [DOI] [PubMed] [Google Scholar]
- 82.Pondugula SR, Brimer-Cline C, Wu J, Schuetz EG, Tyagi RK, Chen T. A phosphomimetic mutation at threonine-57 abolishes transactivation activity and alters nuclear localization pattern of human pregnane x receptor. Drug Metab Dispos. 2009;37:719–730. doi: 10.1124/dmd.108.024695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gong H, Sinz MW, Feng Y, Chen T, Venkataramanan R, Xie W. Animal models of xenobiotic receptors in drug metabolism and diseases. Methods Enzymol. 2005;400:598–618. doi: 10.1016/S0076-6879(05)00034-0. [DOI] [PubMed] [Google Scholar]
- 84.Goodwin B, Hodgson E, Liddle C. The orphan human pregnane X receptor mediates the transcriptional activation of CYP3A4 by rifampicin through a distal enhancer module. Mol Pharmacol. 1999;56:1329–1339. doi: 10.1124/mol.56.6.1329. [DOI] [PubMed] [Google Scholar]
- 85.Timsit YE, Negishi M. CAR and PXR: the xenobiotic-sensing receptors. Steroids. 2007;72:231–246. doi: 10.1016/j.steroids.2006.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hu X X, Lazar MA. The CoRNR motif controls the recruitment of corepressors by nuclear hormone receptors. Nature. 1999;402:93–96. doi: 10.1038/47069. [DOI] [PubMed] [Google Scholar]
- 87.Lin W, Wu J, Dong H, Bouck D, Zeng FY, Chen T. Cyclin-Dependent Kinase 2 Negatively Regulates Human Pregnane X Receptor-Mediated CYP3A4 Gene Expression in HepG2 Liver Carcinoma Cells. J Biol Chem. 2008;283:30650–30657. doi: 10.1074/jbc.M806132200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Watkins RE, Wisely GB, Moore LB, Collins JL, Lambert MH, Williams SP, Willson TM, Kliewer SA, Redinbo MR. The human nuclear xenobiotic receptor PXR: structural determinants of directed promiscuity. Science. 2001;292:2329–2333. doi: 10.1126/science.1060762. [DOI] [PubMed] [Google Scholar]
- 89.Watkins RE, Noble SM, Redinbo MR. Structural insights into the promiscuity and function of the human pregnane X receptor. Curr Opin Drug Discov Devel. 2002;5:150–158. [PubMed] [Google Scholar]
- 90.Sinz M, Kim S, Zhu Z, Chen T, Anthony M, Dickinson K, Rodrigues AD. Evaluation of 170 xenobiotics as transactivators of human pregnane X receptor (hPXR) and correlation to known CYP3A4 drug interactions. Curr Drug Metab. 2006;7:375–388. doi: 10.2174/138920006776873535. [DOI] [PubMed] [Google Scholar]
- 91.Sonoda J, Chong LW, Downes M, Barish GD, Coulter S, Liddle C, Lee CH, Evans RM. Pregnane X receptor prevents hepatorenal toxicity from cholesterol metabolites. Proc Natl Acad Sci U S A. 2005;102:2198–2203. doi: 10.1073/pnas.0409481102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Dussault I, Lin M, Hollister K, Wang EH, Synold TW, Forman BM. Peptide mimetic HIV protease inhibitors are ligands for the orphan receptor SXR. J Biol Chem. 2001;276:33309–33312. doi: 10.1074/jbc.C100375200. [DOI] [PubMed] [Google Scholar]
- 93.Dürr D, Stieger B, Kullak-Ublick GA, Rentsch KM, Steinert HC, Meier PJ, Fattinger K. St John’s Wort induces intestinal P-glycoprotein/MDR1 and intestinal and hepatic CYP3A4. Clin Pharmacol Ther. 2000;68:598–604. doi: 10.1067/mcp.2000.112240. [DOI] [PubMed] [Google Scholar]
- 94.Greiner B, Eichelbaum M, Fritz P, Kreichgauer HP, von Richter O, Zundler J, Kroemer HK. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest. 1999;104:147–153. doi: 10.1172/JCI6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jigorel E, Le Vee M, Boursier-Neyret C, Parmentier Y, Fardel O. Differential regulation of sinusoidal and canalicular hepatic drug transporter expression by xenobiotics activating drug-sensing receptors in primary human hepatocytes. Drug Metab Dispos. 2006;34:1756–1763. doi: 10.1124/dmd.106.010033. [DOI] [PubMed] [Google Scholar]
- 96.Oscarson M, Burk O, Winter S, Schwab M, Wolbold R, Dippon J, Eichelbaum M, Meyer UA. Effects of rifampicin on global gene expression in human small intestine. Pharmacogenet Genomics. 2007;17:907–918. doi: 10.1097/FPC.0b013e3280143dfc. [DOI] [PubMed] [Google Scholar]
- 97.Richert L, Liguori MJ, Abadie C, Heyd B, Mantion G, Halkic N, Waring JF. Gene expression in human hepatocytes in suspension after isolation is similar to the liver of origin, is not affected by hepatocyte cold storage and cryopreservation, but is strongly changed after hepatocyte plating. Drug Metab Dispos. 2006;34:870–879. doi: 10.1124/dmd.105.007708. [DOI] [PubMed] [Google Scholar]
- 98.Westphal K, Weinbrenner A, Zschiesche M, Franke G, Knoke M, Oertel R, Fritz P, von Richter O, Warzok R, Hachenberg T, Kauffmann HM, Schrenk D, Terhaag B, Kroemer HK, Siegmund W. Induction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: a new type of drug/drug interaction. Clin Pharmacol Ther. 2000;68:345–355. doi: 10.1067/mcp.2000.109797. [DOI] [PubMed] [Google Scholar]
- 99.Giessmann T, May K, Modess C, Wegner D, Hecker U, Zschiesche M, Dazert P, Grube M, Schroeder E, Warzok R, Cascorbi I, Kroemer HK, Siegmund W. Carbamazepine regulates intestinal P-glycoprotein and multidrug resistance protein MRP2 and influences disposition of talinolol in humans. Clin Pharmacol Ther. 2004;76:192–200. doi: 10.1016/j.clpt.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 100.Johne A, Brockmöller J, Bauer S, Maurer A, Langheinrich M, Roots I. Pharmacokinetic interaction of digoxin with an herbal extract from St John’s wort (Hypericum perforatum) Clin Pharmacol Ther. 1999;66:338–345. doi: 10.1053/cp.1999.v66.a101944. [DOI] [PubMed] [Google Scholar]
- 101.Hamman MA, Bruce MA, Haehner-Daniels BD, Hall SD. The effect of rifampin administration on the disposition of fexofenadine. Clin Pharmacol Ther. 2001;69:114–121. doi: 10.1067/mcp.2001.113697. [DOI] [PubMed] [Google Scholar]
- 102.Wang T, Ma X, Krausz KW, Idle JR, Gonzalez FJ. Role of pregnane X receptor in control of all-trans retinoic acid (ATRA) metabolism and its potential contribution to ATRA resistance. J Pharmacol Exp Ther. 2008;324:674–684. doi: 10.1124/jpet.107.131045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Desai PB, Nallani SC, Sane RS, Moore LB, Goodwin BJ, Buckley DJ, Buckley AR. Induction of cytochrome P450 3A4 in primary human hepatocytes and activation of the human pregnane X receptor by tamoxifen and 4-hydroxytamoxifen. Drug Metab Dispos. 2002;30:608–612. doi: 10.1124/dmd.30.5.608. [DOI] [PubMed] [Google Scholar]
- 104.Zimmermann K, Wittman MD, Saulnier MG, Velaparthi U, Sang X, Frennesson DB, Struzynski C, Seitz SP, He L, Carboni JM, Li A, Greer AF, Gottardis M, Attar RM, Yang Z, Balimane P, Discenza LN, Lee FY, Sinz M, Kim S, Vyas D. SAR of PXR transactivation in benzimidazole-based IGF-1R kinase inhibitors. Bioorg Med Chem Lett. 2010;20:1744–1748. doi: 10.1016/j.bmcl.2010.01.087. [DOI] [PubMed] [Google Scholar]
- 105.Huang H, Wang H, Sinz M, Zoeckler M, Staudinger J, Redinbo MR, Teotico DG, Locker J, Kalpana GV, Mani S. Inhibition of drug metabolism by blocking the activation of nuclear receptors by ketoconazole. Oncogene. 2007;26:258–268. doi: 10.1038/sj.onc.1209788. [DOI] [PubMed] [Google Scholar]
- 106.Healan-Greenberg C, Waring JF, Kempf DJ, Blomme EA, Tirona RG, Kim RB. A human immunodeficiency virus protease inhibitor is a novel functional inhibitor of human pregnane X receptor. Drug Metab Dispos. 2008;36:500–507. doi: 10.1124/dmd.107.019547. [DOI] [PubMed] [Google Scholar]
- 107.Zhou C, Poulton EJ, Grün F, Bammler TK, Blumberg B, Thummel KE, Eaton DL. The dietary isothiocyanate sulforaphane is an antagonist of the human steroid and xenobiotic nuclear receptor. Mol Pharmacol. 2007;71:220–229. doi: 10.1124/mol.106.029264. [DOI] [PubMed] [Google Scholar]
- 108.Wang H, Li H, Moore LB, Johnson MD, Maglich JM, Goodwin B, Ittoop OR, Wisely B, Creech K, Parks DJ, Collins JL, Willson TM, Kalpana GV, Venkatesh M, Xie W, Cho SY, Roboz J, Redinbo M, Moore JT, Mani S. The phytoestrogen coumestrol is a naturally occurring antagonist of the human pregnane X receptor. Mol Endocrinol. 2008;22:838–857. doi: 10.1210/me.2007-0218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen Y, Tang Y, Robbins GT, Nie D. Camptothecin attenuates cytochrome P450 3A4 induction by blocking the activation of human pregnane X receptor. J Pharmacol Exp Ther. 2010 May 26; doi: 10.1124/jpet.110.168294. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Raynal C, Pascussi JM, Leguelinel G, Breuker C, Kantar J, Lallemant B, Poujol S, Bonnans C, Joubert D, Hollande F, Lumbroso S, Brouillet JP, Evrard A. Pregnane X Receptor (PXR) expression in colorectal cancer cells restricts irinotecan chemosensitivity through enhanced SN-38 glucuronidation. Mol Cancer. 2010;9:46. doi: 10.1186/1476-4598-9-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ekins S, Chang C, Mani S, Krasowski MD, Reschly EJ, Iyer M, Kholodovych V, Ai N, Welsh WJ, Sinz M, Swaan PW, Patel R, Bachmann K K. Human pregnane X receptor antagonists and agonists define molecular requirements for different binding sites. Mol Pharmacol. 2007;72:592–603. doi: 10.1124/mol.107.038398. [DOI] [PubMed] [Google Scholar]
- 112.Ekins S, Kholodovych V, Ai N, Sinz M, Gal J, Gera L, Welsh WJ, Bachmann K, Mani S. Computational discovery of novel low micromolar human pregnane X receptor antagonists. Mol Pharmacol. 2008;74:662–672. doi: 10.1124/mol.108.049437. [DOI] [PubMed] [Google Scholar]
- 113.Xue Y, Moore LB, Orans J, Peng L, Bencharit S, Kliewer SA, Redinbo MR. Crystal structure of the pregnane X receptor-estradiol complex provides insights into endobiotic recognition. Mol Endocrinol. 2007;21:1028–1038. doi: 10.1210/me.2006-0323. [DOI] [PubMed] [Google Scholar]
- 114.Thakur JK, Arthanari H, Yang F, Pan SJ, Fan X, Breger J, Frueh DP, Gulshan K, Li DK, Mylonakis E, Struhl K, Moye-Rowley WS, Cormack BP, Wagner G, Näär AM. A nuclear receptor-like pathway regulating multidrug resistance in fungi. Nature. 2008;452:604–609. doi: 10.1038/nature06836. [DOI] [PubMed] [Google Scholar]