

Abstract
The non-innocent behaviors of NHC ligands have attracted wide attention due to their important implications for catalyst designs and reaction mechanisms. Herein, we report facile Ccarbene–halogen reductive eliminations from NHC copper halide complexes at RT under oxidative conditions. Density functional calculations on a simplified model system suggest that the reactions occur through oxidation of Cu(I) to Cu(III) species followed by Ccarbene–halogen reductive eliminations from NHC Cu(III) halide complexes. Remarkably short Ccarbene–chloride contacts and rare interactions between the chloride lone pair electrons and the Ccarbene pπ orbital were found for the calculated NHC Cu(III) chlorides. The facile Ccarbene–X reductive elimination reported here warrants consideration as a potential decomposition pathway in reactions involving NHC-supported high-valent metal complexes, especially with late transition metals.
N-heterocyclic carbene (NHC) metal complexes are used widely as catalysts in organic reactions.1 Compared to other neutral type ligands, NHCs usually form stronger bonds with metals due to excellent σ donating ability. The conjugation between the carbene carbon (Ccarbene) pπ orbital and nitrogen lone pair electrons in the heterocycle further stabilizes the metal–Ccarbene bonds. As a result, NHCs are often better at suppressing catalyst decompositions. The metal–Ccarbene bonds are remarkably inert compared to other metal–carbon bonds at catalytic metal centers, which are prone to undergo various reactions such as migratory insertion and olefin metathesis.1 In contrast, only a small number of elementary organomellic reactions, mostly limited to Ccarbene–C reductive elimination and ligand dissociation/displacement, are documented with the metal–Ccarbene bonds.2–4
Carbon-halogen (C–X) reductive elimination of metal–Calkyl or metal–Caryl bonds is an important elementary organometallic reaction,5 but no well-defined example is reported for Ccarbene–X reductive elimination from NHC metal halide complexes,6 although such complexes are ubiquitous in NHC chemistry.1 Herein, we report facile formation of 2-halo-imidazoliums from NHC Cu(I) halide complexes at room temperature (RT) under oxidative conditions as well as computational evidence to support a mechanism involving Ccarbene–X reductive elimination from NHC Cu(III) halide complexes.
As a part of our continuing efforts in the characterization of reactivity of Cu(III) complexes,7 we postulated that NHC Cu(III) complexes might be isolable because of the relatively inert Cu–Ccarbene bond. In that regard, oxidations of IPrCuICl (1Cl, IPr: 1, 3-bis(2, 6-diisopropylphenyl)imidazol-2-ylidene) by various oxidants were investigated. No significant reaction was observed between 1Cl and [Ph2I]+PF6− or [Cp2Fe]+PF6− (0.64 V vs NHE)8 in acetonitrile (MeCN). By contrast, mixing 1Cl with Selectfluor® (≥ 1.5 eq.) or CuII(CF3SO3)2 (≥ 2.0 eq., 1.30 V vs NHE)9 in MeCN at RT rapidly and quantitatively forms 2-chloro-imidazolium 2Cl (Scheme 1).10 Quantitative formation of 2-halo-imidazolium 2Br or 2I was realized for IPrCuIBr (1Br) or IPrCuII (1I) under similar conditions.11
Scheme 1.

These reactions appear to occur through either an inner- or outer-sphere oxidation followed by Ccarbene-X reductive elimination. An outer-sphere oxidation is supported by the ca. 80 % formation of 2Cl by reacting 1Cl with [(1, 10-phenanthroline)3FeIII]3+ (≥ 2 eq., 1.22 V vs NHE)12 in MeCN at RT.13 However, all these reactions are fast even at low temperatures (t1/2 ~ seconds at ca. −40 °C), and no intermediate species could be detected by UV-Vis spectroscopy on a timescale of seconds.
DFT calculations provide mechanistic insights into the oxidation by Selectfluor®. The calculated free-energy profiles corresponding to Ccarbene-X reductive elimination from either a Cu(II) or Cu(III) species using a simplified model system (3) are shown in Scheme 2.14 The oxidation of 3 by Selectfluor® to form 5, a three-coordinate Cu(III) species, is thermodynamically favorable.15 The Cu(III) species is further stabilized by coordination of an MeCN ligand to form 7.16 The activation barrier for Ccarbene–Cl reductive elimination from 7 via TS1 to form 2-chloro-imidazolium 10 is remarkably low at 3.5 kcal mol−1.17 The overall reaction is favorable by −54.2 kcal mol−1. Other possible Cu(III) species18 are less stable than 7 and lead to higher activation barriers. Alternatively, 7 could react with another equivalent of 3 to form Cu(II) intermediates 8 and 9, the two most stable Cu(II) species.19 Although no transition state from 9 to 10 could be located (the energy monotonically increases as the Ccarbene–Cl separation shortens.), the lower limit of its activation barrier can be estimated by the corresponding reaction free energy, 44.1 kcal mol−1, the least endothermic among various Cu(II) species.19 Therefore, the calculations on the simplified model system favor a mechanism of Ccarbene–Cl reductive elimination from NHC Cu(III) chloride complexes for the oxidation of 1Cl by Selectfluor®.
Scheme 2.

The thermodynamically unfavorable Ccarbene–Cl reductive elimination from 9 is consistent with isolable NHC Cu(II) chloride complexes.20 Based on these results, we prefer a mechanism with two sequential inner- or outer-sphere 1e− oxidations followed by Ccarbene–Cl reductive elimination from NHC Cu(III) chloride complexes for the reaction with Cu(CF3SO3)2,21 although the detailed mechanism is still unclear. Furthermore, the quantitative formation of 2Cl instead of IPrCu(II) chloride complexes from the reaction of 1Cl and Selectfluor® suggests that Ccarbene–Cl reductive elimination from IPrCu(III) chloride complexes is much faster than reactions of 1Cl to form IPrCu(II) halide complexes. This reactivity is consistent with the calculated 3.5 kcal mol−1 activation barrier from 7 to 10. Such a low barrier is probably a consequence of the electrophilic nature of a Cu(III) center that renders the NHC Ccarbene susceptible to nucleophilic attack, as suggested by the remarkably close Ccarbene–Cl contacts (ca. 2.7 Å) in 5 and 7 (Figure 1) as well as the interactions between the chloride lone pair electrons and the Ccarbene pπ orbital (Figure 2). A similar interaction has been invoked to rationalize the short Ccarbene–Cl contact (ca. 2.85 Å) in NHC VV(O)Cl3.22 In contrast, no evidence of such an interaction exists for 8 (Figure 1).
Figure 1.

Optimized structures of 5, 7, and 8.
Figure 2.
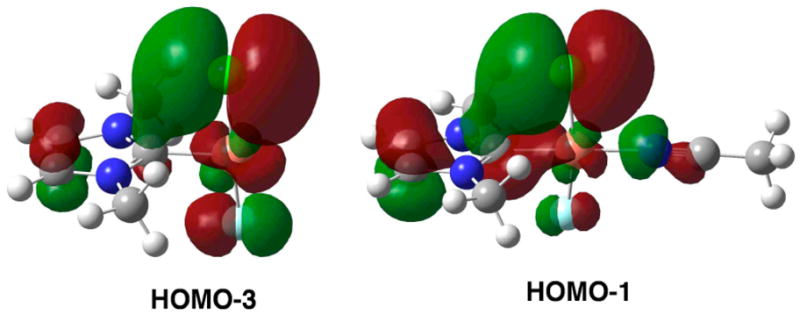
HOMO-3 of 5 and HOMO-1 of 7.
In summary, we have demonstrated that Ccarbene–halogen reductive eliminations readily occur from NHC copper halides at RT under oxidative conditions. These reactions provide new examples for the well-known oxidation-induced reductive eliminations. DFT calculations on a simplified model system suggest that the involvement of NHC Cu(III) halides is essential for these reactions and the reductive eliminations might be facilitated by the interaction between Ccarbene and the halogen lone pair. Given the ubiquity of NHC metal halide complexes, the facile Ccarbene–X reductive elimination reported here warrants consideration as a potential decomposition pathway in reactions involving NHC-supported high-valent metal complexes, especially with late transition metals under oxidative conditions.
Supplementary Material
Acknowledgments
This work was supported by NIH(GM50730) and ACS-PRF award. We thank Dr. Allen G. Oliver for the crystal structures, and Professor Xingwei Li and Dr. George S. Chen for their suggestions in the preparation of the manuscript.
Footnotes
Supporting Information Available: Detailed experimental and calculational data. This material is available free of charge at http://pubs.acs.org.
References
- 1.For reviews, see: Herrmann WA. Angew Chem Int Ed. 2002;41:1290–1309. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y.Kantchev EAB, O’Brien CJ, Organ MG. Angew Chem Int Ed. 2007;46:2768–2813. doi: 10.1002/anie.200601663.Díez-González S, Marion N, Nolan SP. Chem Rev. 2009;109:3612–3676. doi: 10.1021/cr900074m.Poyatos M, Mata JA, Peris E. Chem Rev. 2009;109:3677–3707. doi: 10.1021/cr800501s.Somojlowicz C, Bieniek M, Grela K. Chem Rev. 2009;109:3608–3742. doi: 10.1021/cr800524f.van Otterlo WAL, de Koning CB. Chem Rev. 2009;109:3743–3782. doi: 10.1021/cr900178p.Monfette S, Fogg DE. Chem Rev. 2009;109:3783–3816. doi: 10.1021/cr800541y.Alcaide B, Almendros P, Luna A. Chem Rev. 2009;109:3817–3858. doi: 10.1021/cr9001512.Vougioukalakis GC, Grubbs RH. Chem Rev. 2010;110:1746–1787. doi: 10.1021/cr9002424.
- 2.For examples of Ccarbene–C couplings, see: McGuinness DS, Cavell KJ, Yates BF, Skelton BW, White AH. J Am Chem Soc. 2001;123:8317–8328. doi: 10.1021/ja010628p.Marshall WJ, Grushin VV. Organometallics. 2003;22:1591–1593.Bacciu D, Cavell KJ, Fallis IA, Ooi LL. Angew Chem Int Ed. 2005;44:5282–5284. doi: 10.1002/anie.200500884.Becker E, Stingl V, Dazinger G, Puchberger M, Mereiter K, Kirchner K. J Am Chem Soc. 2006;128:6572–6573. doi: 10.1021/ja061454k.Shih WC, Wang CH, Chang YT, Yap GPA, Ong TG. Organometallics. 2009;28:1060–1067.Steinke T, Shaw BK, Jong H, Patrick BO, Fryzuk MD, Green JC. J Am Chem Soc. 2009;131:10461–10466. doi: 10.1021/ja901346g.Romain C, Miqueu K, Sotiropoulos JM, Bellemin-Laponnaz S, Dagorne S. Angew Chem Int Ed. 2010;49:2198–2201. doi: 10.1002/anie.200906702.
- 3.For examples of NHC ligand displacement/dissociation, see: Ku RZ, Huang JC, Cho JY, Kiang FM, Reddy R, Chen YC, Lee KJ, Lee JH, Lee GH, Peng SM, Liu ST. Organometallics. 1998;28:863–870.Titcomb LR, Caddick S, Cloke FGN, Wilsona DJ, McKerrecher D. Chem Comm. 2001:1388–1389.Simms RW, Drewitt MJ, Baird MC. Organometallics. 2002;21:2958–2963.de K Lewis AK, Caddick S, Cloke FGN, Billingham NC, Hitchcock PB, Leonard J. J Am Chem Soc. 2003;125:10066–10073. doi: 10.1021/ja035565k.Dorta R, Stevens ED, Hoff CD, Nolan SP. J Am Chem Soc. 2003;125:10490–10491. doi: 10.1021/ja0362151.Torres O, Martín M, Sola E. Organometallics. 2009;28:863–870.
- 4.For other reactions involving the cleavage of the M–Ccarbene bond: Turner ZR, Bellabarba R, Tooze RP, Arnold PL. J Am Chem Soc. 2010;132:4050–4051. doi: 10.1021/ja910673q.
- 5.For reviews, see: Vigalok A. Chem Eur J. 2008;14:5102–5108. doi: 10.1002/chem.200701738.Sheppard TD. Org Biomol Chem. 2009;7:1043–1052. doi: 10.1039/b818155a.
- 6.Halogenolysis of the metal–Ccarbene bonds to form Ccarbene–X bonds does occur, presumably through either direct electrophilic cleavage of metal–Ccarbene bonds by X2 or oxidative addition of X2 followed by Ccarbene–X reductive elimination. DFT calculations support a direct electrophilic cleavage of Pd– Ccarbene bond by I2.9fLappert MF, Pye PL. J Chem Soc, Dalton Trans. 1977:1283–1291.Liu ST, Ku RZ, Liu CY, Kiang FM. J Organomet Chem. 1997;543:249–250.Cole ML, Davies AJ, Jones C. J Chem Soc, Dalton Trans. 2001:2451–2452.Fooladi E, Dalhus B, Tilset M. Dalton Trans. 2004:3909–3917. doi: 10.1039/b408750j.Heckenroth M, Neels A, Garnier MG, Aebi P, Ehlers AW, Albercht M. Chem Eur J. 2009;15:9375–9386. doi: 10.1002/chem.200900249.Lee E. Ph D Dissertation. Stanford University; Stanford, CA: 2009.
- 7.For examples, see: Ribas X, Jackson DA, Donnadieu B, Mahía J, Parella T, Xifra R, Hedman B, Hodgson KO, Llobet A, Stack TDP. Angew Chem Int Ed. 2002;41:2991–2994. doi: 10.1002/1521-3773(20020816)41:16<2991::AID-ANIE2991>3.0.CO;2-6.Mirica LM, Ribas X, Stack TDP. Chem Rev. 2004;104:1013–1046. doi: 10.1021/cr020632z.Mirica LM, Vance M, Rudd DJ, Hedman B, Hodgson KO, Solomon EI, Stack TDP. Science. 2005;308:1890–1892. doi: 10.1126/science.1112081.Bertz SH, Cope S, Murphy M, Ogle CA, Taylor BJ. J Am Chem Soc. 2007;129:7208–7209. doi: 10.1021/ja067533d.Hu H, Snyder JP. J Am Chem Soc. 2007;129:7210–7211. doi: 10.1021/ja0675346.Gärtner T, Henze W, Gschwind RM. J Am Chem Soc. 2007;129:11362–11363. doi: 10.1021/ja074788y.Storr T, Verma P, Pratt RC, Wasinger EC, Shimazaki Y, Stack TDP. J Am Chem Soc. 2008;130:15448–15459. doi: 10.1021/ja804339m.Herres-Pawlis S, Verma P, Haase R, Kang P, Lyons CT, Wasinger EC, Flörke U, Henkel G, Stack TDP. J Am Chem Soc. 2009;131:1154–1169. doi: 10.1021/ja807809x.Huffman LM, Stahl SS. J Am Chem Soc. 2008;130:9196–9197. doi: 10.1021/ja802123p.
- 8.Connelly NG, Geiger WE. Chem Rev. 1996;96:877–910. doi: 10.1021/cr940053x. [DOI] [PubMed] [Google Scholar]
- 9.Inamo M, Kumagai H, Harada U, Itoh S, Iwatsuki S, Ishihara K, Takagi HD. Dalton Trans. 2004:1703–1707. doi: 10.1039/b403760j. [DOI] [PubMed] [Google Scholar]
- 10.2Cl was characterized by NMR (1H, 13C, and 19F), mass spectroscopy, and X-ray crystallography (2Cl·SbF6·CH2Cl2).19 Reactions with lesser amounts of oxidants led to unreacted 1Cl and lower yields of 2Cl. The amounts of oxidant and the color of the reaction solution suggest that the side product is Cu(II) (blue) and Cu(I) (colorless) species for the reaction with Selectfluor® and CuII(CF3SO3)2, respectively.
- 11.2Br and 2I were characterized by NMR (1H, 13C, and 19F), mass spectroscopy, and X-ray crystallography (2Br·CF3SO3·CH2Cl2 and 2I· I3).19 The reaction of IPrCuIF with CuII(CF3SO3)2 led to the formation of IPrCuI(CF3SO3) (a crystal structure was obtained),19 while the reaction with Selectfluor® afforded a species with broad 1H NMR signals similar to that of a reported NHC-ligated Cu(II) dimer20b, suggesting that C-F formation is slower than the comproportionation between Cu(I) and Cu(III).
- 12.Wong CL, Kochi JK. J Am Chem Soc. 1979;101:5593–5603. [Google Scholar]
- 13.Protonolysis of the Cu-Ccarbene bond was also observed, leading to ca. 20 % yield of the corresponding imidazolium. In addition, no well-defined oxidation occurs with 1Cl by cyclic voltammetry at low temperatures (ca. −40 °C).
- 14.All calculations were performed at the level of B3LYP/6-311+G** by Gaussian 03 program. The solvation effect was estimated by PCM/UA0 model. Relative free energies are used in the computational discussion. See supporting information for more detailed descriptions of the computational methods. (a) Pople, J. A. et al. Gaussian03, Revision E.01, Gaussian, Inc., Wallingford, CT, 2004. (b) The gas phase entropies were converted to corresponding entropies19 (1 M in CH3CN) according to the method in Wertz DH. J Am Chem Soc. 1980;102:5316–5322.
- 15.A 12.8 kcal mol−1 activation free energy was calculated for this step (fluorine transfer) at a lowerlevel method (B3LYP/6-311+G**//B3LYP/LANL2DZ).19
- 16.6 is proposed as an intermediate in the reaction. It might ligate to copper in the final uncharacterized Cu(II) product. Coordination of MeCN to 3 might facilitate the oxidation. However, the corresponding 3-coordinate species could not be located computationally.
- 17.η2-Arene-coordination intermediates have been reported in a related computational study for oxidative addition of Ph–Br to various CuI species. ( Zhang SL, Liu L, Fu Y, Guo QX. Organometallics. 2007;26:4546–4554.) By contrast, no analogous intermediate was located computationally for our system.
- 18.These include two isomers of 7, one 5-coordinate Cu(III) species, and a complex with 6 coordinated to 5.19
- 19.See supporting information.
- 20.(a) Arnold PL, Rodden M, Davis KM, Scarisbrick AC, Blake AJ, Wilson C. Chem Comm. 2004:1612–1613. doi: 10.1039/b404614e. [DOI] [PubMed] [Google Scholar]; (b) Larsen AO, Leu W, Oberhuber CN, Campbell JE, Hoveyda AH. J Am Chem Soc. 2004;126:11130–11131. doi: 10.1021/ja046245j. [DOI] [PubMed] [Google Scholar]
- 21.Previous work indicated that AgSbF6 (1.35 V vs NHE)8 can oxidize CuII-Salen to [CuIIISalen](SbF6) in CH2Cl2.7d Therefore, the oxidation of 1Cl to Cu(III) by CuII(CF3SO3)2 (1.30 V vs NHE)9 seems possible.
- 22.Abernethy CD, Codd GM, Spicer MD, Taylor MK. J Am Chem Soc. 2003;125:1128–1129. doi: 10.1021/ja0276321. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.