

Abstract
Structure-based drug design (SBDD) has emerged as a valuable pharmaceutical lead discovery tool, showing potential for accelerating the discovery process, while reducing developmental costs and boosting potencies of the drug that is ultimately selected. SBDD is a iterative, rational, lead compound sculpting process that involves both the synthesis of new derivatives and the evaluation of their binding to the target structure either through computational docking or elucidation of the target structure as a complex with the lead compound. This method heavily relies on the production of high-resolution (< 2Å) three-dimensional structures of the drug target, obtained through X-ray crystallographic analysis, in the presence or absence of the drug candidate. The lack of generalized methods for high quality crystal production is still a major bottleneck in the process of macromolecular crystallization. This review provides a brief introduction to SBDD and describes several macromolecular crystallization strategies, with an emphasis on advances and challenges facing researchers in the field today. Recent trends in the development of more universal macromolecular crystallization techniques, particularly nucleation-based techniques that are applicable to both soluble and integral membrane proteins, are also discussed.
1. Structure-Based Drug Design - From Lead Discovery to Drug Candidate Optimization
Identification of promising lead compounds early in the drug discovery process is critical for reducing the time required to optimize leads after the initial discovery of potential hits[1]. The time required to develop a drug lead from discovery to final form is typically between 6-12 years, resulting in total developmental costs exceeding $1 billion[2], emphasizing the need for new experimental approaches that will streamline this process. Although significant advances have been made using library-based screening strategies[3] for new lead identification, it has become increasingly clear that SBDD has even greater potential for accelerating the time to market for new pharmaceuticals entities and significantly reducing their cost of development.
The SBDD process is an iterative approach based on the three-dimensional structure of the target as a guide for the development of drug leads (Figure 1). This method utilizes computational analysis of target-lead complementarity using three-dimensional structures of the target or target-lead complex[1] so that both favorable and unfavorable lipophilic, hydrogen-bonding and polar interactions of the target-lead complex can be identified[4–6]. Structural refinements of the lead often emerge from target-lead complex crystals and analysis of the resulting high-resolution X-ray structures. An alternative approach, i.e., virtual screening, utilizes molecular docking simulations to screen libraries of potential small molecule leads to estimate the biological activity of successful drug candidates [7–10]. Regardless of the initial approach, the pharmaceutical properties of lead compounds (e.g., log P, bioavailability, metabolic toxicity, pharmacokinetics, structure-activity relationships, potency, etc) are continually optimized until a drug candidate is identified for clinical evaluation[4].
Figure 1. Schematic of drug discovery pipeline.
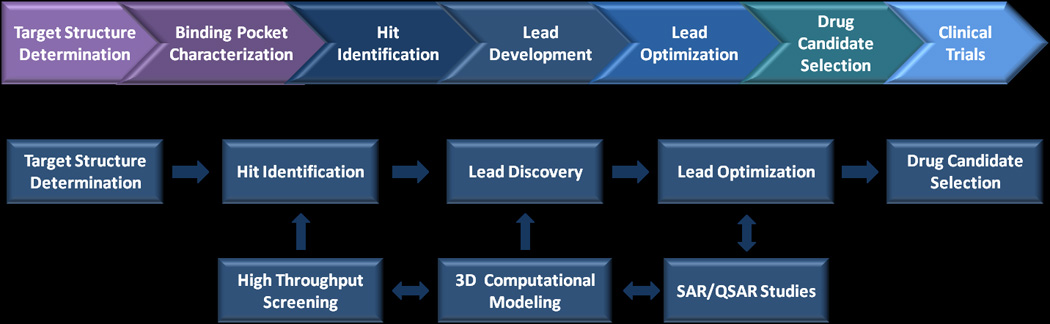
After identification of the drug target, its structure and drug-binding site is determined at the highest resolution possible. Atomically resolved structures enable more rapid refinements of lead compounds and optimization of the drug candidate.
A prime example of the SBDD process is the history of HIV-1 protease inhibitor development. HIV-1 protease is a critical enzyme in the life cycle of HIV, the retrovirus that causes autoimmune deficiency syndrome (AIDS). Since the elucidation of its structure in 1989, SBDD strategies have led to the introduction of three first-generation HIV protease inhibitors: Hoffman-La Roche’s Invirase™ (saquinavir), Abbott’s Norvir™ (ritonavir), and Merck Inc.’s Crixivan® (indinavir), as well as several more potent inhibitors in recent years[11–12]. For example, Mahalingam and coworkers developed a series of HIV-1 protease inhibitors, with activities that were up to 56 times more potent than current analogs[12]. The structure of the most potent inhibitor, co-crystallized with HIV-1 protease, is shown in Figure 2. This agent is considerably more potent (EC50 =7 nM) than indinavir (EC50= 50 nM) and is as potent as atazanavir (EC50=8 nM), the first protease inhibitor to be given on a once-a-day basis[13]. SBDD clearly has accelerated the process of drug development for HIV treatment since there have been more FDA approved drugs for its treatment than for all other viral infections combined[13]. Structure-based studies of other infectious diseases have resulted in the introduction of more than 200 active drugs on the market[4, 6, 14–15], including those for Severe Acute Respiratory Syndrome (SARS)[16–17] and influenza A virus[18–19].
Figure 2. Crystal structure of HIV-1 protease with bound inhibitor AHA599 (PDB accession number 2WKZ).
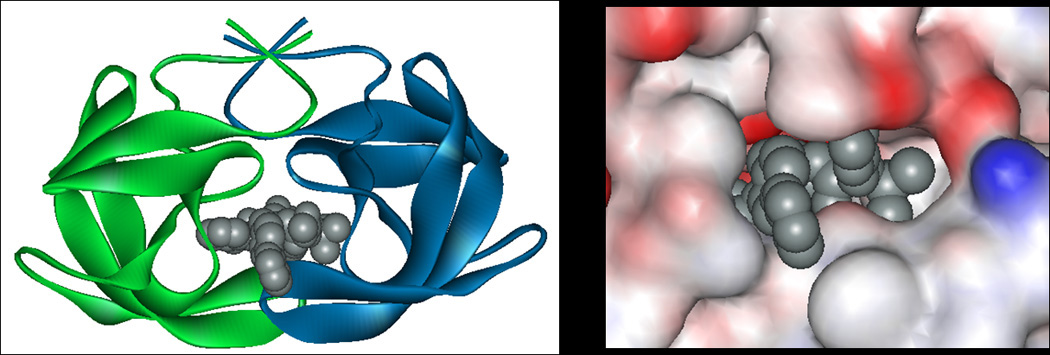
(left) Ribbon structure of HIV-1 protease that is complexed with AHA599, depicted as a space filling model. (right) Detail of the HIV-1 protease-inhibitor complex, illustrating important contacts and conformal fit between AHA599 and its protein target. This structure revealed that the binding interaction was dependent upon the homodimerization of the protein, providing insights into the design of next-generation inhibitors with enhanced binding interactions.
2. High Resolution Three Dimensional Structural Determination for SBDD
Since the SBDD process is most powerful when it begins with an atomically resolved structure of the target, it is essential to develop reliable, rapid and predictable methods for obtaining this information to enable the development of highly potent drug candidates. Structure elucidation of protein targets can be obtained from either X-ray crystallography or nuclear magnetic resonance (NMR) techniques; however, most protein targets used in SBDD have been obtained using X-ray crystallography. Despite their capacities to solve structures at atomic resolution, they both have practical limitations. X-ray crystallography is the most cost-effective and widely used tool; however, the structure determination rate and ultimate resolution are governed by the ability to grow large (> 10µm/side) diffraction quality crystals. This bottleneck illustrates another drawback of X-ray methods, i.e., the large amount of protein required to enable the screening of many crystallization conditions to discover those that are capable of producing high-quality single crystals. Structures of proteins up to approximately 30 kD can be determined in solution using techniques such as 2D nuclear Overhauser enhancement spectroscopy (NOESY) to observe interactions between hydrogen atoms that are in close proximity (< 5 Å apart) within a polypeptide backbone of known sequence. This process is labor-intensive since 15N- and 13C-enriched forms of the protein must be expressed in order to resolve overlapping peaks in 2D NOESY spectra.
Unfortunately, neither of these techniques have been highly effective for the elucidation of membrane protein structures thus far since less than 0.5% of non-redundant sequences deposited in the PDB as of June 2010 are membrane proteins. This has been especially problematic for the analysis of G protein-coupled receptors (GPCRs), a very important family of membrane proteins involved in cellular signaling that encode roughly 21% of the genes of known function[20–21] and represent 50–60% of current drug targets[22]. Drugs targeting GPCRs have been known for more than 50 years for cardiovascular, metabolic, neurodegenerative, psychiatric, and oncologic diseases[22], even though the molecular details of the GPCR family have been limited until elucidation of the human β2adrenergic receptor (β2AR) crystal structure in 2007[23–24]. This high-resolution structure enabled the rapid development of more potent β2AR-based therapeutics. In a recent study by Kolb and coworkers, over 1 million commercially available lead-like molecules were used in a docking screen with the β2AR. The most highly scored compounds in this screen were then tested, resulting in the discovery of six new inhibitors with binding affinities <4 µM, one with a Ki of 9 nM[25]. Thus, the high-resolution β2AR structure represents a significant advance in the understanding and utilization of structural information for GPCR drug discovery; however, there are many other members of this receptor family whose structures remain unsolved. This situation underscores the compelling need for new strategies aimed at membrane protein crystallization.
3. Current Strategies and Obstacles in Macromolecular Crystallization
The methods most often employed for macromolecular crystallization are either high-throughput plate-based screening techniques or microfluidics-type platforms. Using automated high-throughput methods, it is currently possible to test thousands of potential crystallization conditions by setting up and monitoring trials using sub-microliter volumes of protein solution[23, 26–27]. This miniaturization of experiments reduces the labor burden devoted to protein expression and purification, thus enabling the evaluation of up to 105 crystallization trials per day[28]. This can be extremely beneficial for protein targets that are labile and/or are only available in very small quantities. Microfluidic platforms have also been used to further reduce the volumes needed down to ≤ 10 nL per crystallization condition[29]. Other strategies used include determination of the “crystallization slot”, where osmotic second virial coefficients (B22) lying in a range between −8 × 10−4 and −2 × 10−4 mol/g2 are experimentally determined [30], the “relative crystallizability”, a parameter that describes the percentage of crystal nucleation phase area within the protein and precipitating agent concentration ranges[31], or the crystallization coefficient (ξc, which is proportional to the ratio between the volume diffusion rate and the surface integration rate) to determine the “kinetic crystallization window” where 1 < ξc < 8 for crystals grown at the air-water interface[32].
The methods above have resulted in the crystallization of thousands of soluble proteins; however, these methods are most often not amenable to membrane protein crystallization for reasons that are briefly described below. These proteins are difficult to obtain in highly pure form due to proteolytic degradation and the presence of occult impurities, in spite of advances made to increase the yields from membrane protein expression in both pro- and eukaryotic systems[33–34]. Membrane proteins also have a high degree of conformational flexibility that often produces misfolded states, a situation that is exacerbated by the high detergent concentrations that are used during their extraction and purification. In addition, they display complex phase diagrams that are further convoluted by the presence of detergent and endogenous membrane lipids, are highly sensitive to solution conditions (e.g., pH, salinity, temperature, etc), are difficult to concentrate under mild conditions, and typically yield crystals that are fragile due to their high water content. Figure 3 illustrates the different phase diagram zones encountered in the crystallization process. The information from this diagram can be used to drive the transition into the nucleation zone where crystal growth can occur; however, the phase diagram for most proteins is unknown. This often leads to the production of amorphous aggregates or crystalline showers instead of high quality single crystals. In addition, since most crystallization processes are batch procedures, growth of large high quality crystals is challenging because the protein concentration is constantly changing as growth ensues. Thus, the production of high quality crystals remains as the major stumbling block in the drug discovery process for membrane protein targets of SBDD.
Figure 3. Conceptual illustration of protein solution phase diagram that identifies key regions important for controlling nucleation and crystallization.
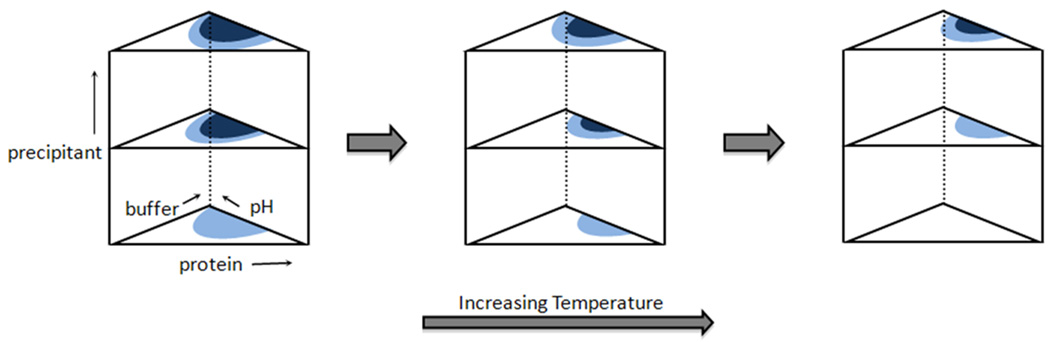
Reliable methods for mapping the transition from the supersaturation zone (dark blue), where no further nucleation occurs, into the metastable zone (light blue), where nucleation can begin, are typically unknown for most SBDD targets. In addition to protein solution composition, physical factors such as sample temperature can influence the available phase space for crystal nucleation and growth.
4. Recent Advances and Opportunities in Macromolecular Crystallization
In spite of the recent advances that have been made to accelerate the rate of protein structure determination with the approaches mentioned above, major impediments remain. Since nucleation is the first step in the crystallization process, the development of general methods to control protein nucleation could help to reduce the bottleneck in the protein crystallization process[35]. Some of the most promising approaches are briefly described below.
An important method based on the bicontinuous cubic phases formed by monoolein-rich dispersions has led to the successful crystallization of several membrane proteins, including the β2AR system noted above. Initially reported by Landau and Rosenbusch[36] for the crystallization of bacteriorhodopsin, the method for growing crystals simply involves mixing two parts solubilized protein with three parts monoolein to produce a homogeneous solution. The lipid cubic phase forms spontaneously at 20°C within hours to weeks, occasionally requiring the addition of precipitant. An attractive feature of the cubic phase method is its capacity to retain the original phase state, even in the presence of a wide array of polar, apolar and amphiphilic additives. Caffrey and coworkers[37–38] have developed a family of single chain lipids that enable tailoring of the cubic phase microstructure to suit the requirements of the target protein; however, in most cases, this information is not known precisely in advance. Application of this method was instrumental in growing crystals of β2AR for the high-resolution structure described above.
Sligar and coworkers have reported the use of nanodiscs comprised of bilayer membrane bounded by a ring of peptidic amphiphiles as a means of solubilizing and stabilizing membrane proteins for solution characterization[39]. These dispersions offer potential for assembling nanodisc-solubilized membrane protein into ordered arrays suitable for structural analysis. This method offers advantages over liposomes or detergent micelles due to their stability, small size, ability to add genetically-modifiable features to the nanodisc structure, and accessiblilty to both sides of the phospholipid bilayer domain[40]. Nanodiscs have been used to successfully reconstitute membrane proteins for structural determination by high resolution solution NMR[41] and cryoelectron microscopy for 3D particle reconstruction[42]. Although the potential for use of nanodics for protein crystallization applications is still unknown, the rapid advances made thus far with nanodisc approaches suggests that they have tremendous potential for advancing the field of membrane protein structure determination for SBDD.
A microfluidics-based approach developed by Ismagilov and coworkers creates supersaturated protein solutions within convergent microfluidic channels that controllably mix buffer, protein, precipitant, and a carrier fluid[43]. Isolated protein droplets are transported through microfluidics channels to produce mixing that induces protein nucleation[43–44]. An advantage of this approach is the ability to directly load the crystals grown within the microfluidic channels into x-ray capillaries for diffraction analysis. Crystals exceeding 50 µm in length have been grown in this manner from solutions of thaumatin, bovine liver catalase, and glucose isomerase[45]. Another microfluidics-based approach developed by Kenis and coworkers provides control over the extent and rate of protein supersaturation by varying the dimensions of the protein and evaporation chambers in a multi-well format[46]. This approach consumes less than 20 nL of material and has been successfully applied to the crystallization of bacteriorhodopsin. A similar approach developed by Ismagilov and coworkers utilizes fluorinated nitriliotriacetic acid (NTA) amphiphiles for the specific capture of histidine-tagged proteins within microfluidic channels[47], a strategy that can be extended to other histidine-tagged proteins. Microfluidic devices present a potential problem; however, especially for solutions containing detergents and other amphiphiles for the crystallization of membrane proteins. If the surface properties of the device are not carefully controlled, surfactant molecules could be inadvertently stripped from the membrane protein dispersion, thereby altering its stability and crystallization capacity.[48] These devices also require integrated imaging systems capable of monitoring the crystallization conditions and database management systems for storing the information collected[49].
A novel seeding technique was recently introduced by Villasenor and coworkers that uses acoustic waves to deliver seed suspensions into protein drops[50]. The technique improved upon other sparse-matrix screening approaches by reducing the chemical bias introduced into the protein droplet by reducing the volume of the seed suspension to nanoliter volumes. The authors also discovered that this seeding technique was more effective at increasing the number of sparse-matrix conditions that yield protein crystals compared to buffer additives alone. A particular challenge in this approach is that seed-induced crystallization may occur using conditions that without seed had been deemed unfavorable, suggesting that there is no clear correlation between unproductive protein solutions and good seeding conditions. Although this uncertainty makes it challenging to know when one is close to successful crystallization conditions, this seeding method clearly demonstrates the potential for improving the yield of sparse matrix conditions for crystal growth.
Interfacial templating strategies using various organic (e.g., lipid monolayers[51–54], lipid tubules[55] or cubic phases[36]) and inorganic substrates (e.g., gold or mica) have been developed for controlled nucleation of two-dimensional protein crystals (for a review of this and other interfacial approaches see ref.[54]). Mirkin and coworkers have demonstrated the directed growth of polypeptide single crystals using dip pen nanolithography (DPN)[56]. Their findings suggests that protein crystal growth may be controlled by utilizing highly periodic substrates as nucleation templates and laminar flow of the protein target on the template to control crystal growth rates. Thompson and coworkers have also developed a noncovalent, cyclodextrin-based template which utilizes reversible host:guest chemistry between the cyclodextrin template and NTA-modified guest ligands that capture histidine-tagged proteins[53]. Among the advantages of this technique are the orientation of the protein with respect to the template, concentration at the interface to achieve local supersaturation, reversible, non-covalent binding at the interface, and pseudo-epitaxial nucleation of crystals at the highly structured surface.
5. Expert Opinion
The small, but growing number of proteins that have been crystallized using the interfacial templating approach suggests that this method is still largely untapped for the structure elucidation of SBDD targets. Interfacial templates and other site-specific nucleation strategies can emerge as very powerful tools for high-throughput structure determination, particularly if universal, rapid and predictable methods for controlled crystal growth can be merged with these techniques in stepwise fashion. The most verdant area for new methods development is in the creation of controlled nucleation and crystallization techniques for membrane proteins for SBDD. Unfortunately, the biochemical and colloid science challenges noted above have greatly impeded progress in this field by limiting the range of innovative new strategies that have been attempted. This is understandable on one hand, because efforts that are tightly focused on structure elucidation with a very small batch of newly-isolated functional and purified membrane protein are unlikely to be spent exploring novel techniques due to the high value of the target. Nonetheless, the slow progress in this field can be directly attributed to this overly narrow focus. Consequently, there is still a great need for the design and development of new materials and processes that embody as many of the features outlined below as possible:
Utilization of in vitro expression systems to minimize the complexity of the sample by removing as many non-target protein contaminants as possible;
Development of improved detergents whose phase diagrams have been mapped with respect to surfactant concentration, biomembrane components, ionic strength, pH, temperature and commonly used precipitants;
Non-aggressive, detergent stripping methods that enable gradual removal of the amphiphile to avoid commonly encountered problems like precipitation and crystal showers;
Experimentally-controllable sites and rates of nucleation to guide the formation of single, large crystals on diffraction-compatible substrates rather than showers of small crystals due to heterogeneous nucleation events;
Migration of crystallization schemes from batch-wise processes to flow-based strategies to more precisely transition protein solutions into supersaturation regimes. This will enable crystal growth rate control and maintainence of solution compositions to avoid concentration-limited growth of large crystals for diffraction analysis;
Continued emphasis on sample volume reduction to relieve the burden on expression and purification systems to both expand the number of membrane protein targets that are amenable to X-ray diffraction structure elucidation and increase the acceptance rate of novel methodological approaches by structural biologists.
Realization of designs that address these constraints are destined to accelerate the drug discovery process by improving the efficiency and universality of macromolecular crystallization techniques, particularly for membrane proteins, leading to faster structural determinations of potential drug targets for SBDD.
Acknowledgments
The authors were supported by the National Institute of Health (Grant No. GM079058) and the Purdue Research Foundation.
List of Abbreviations
- SBDD
structure-based drug design
- HIV
human immunodeficiency syndrome
- AIDS
acquired immunodeficiency syndrome
- NTA
nitrilotriacetic acid
- NMR
nuclear magnetic resonance
- GPCR
G-protein coupled receptor
- SARS
severe acute respiratory syndrome
- SAR
structure activity relationship
- NOESY
nuclear Overhauser effect spectroscopy
- DPN
dip-pen nanolithography
Footnotes
Declaration of Interest
References
- 1.Andricopulo AD, Salum LB, Abraham DJ. Structure-based drug design strategies in medicinal chemistry. Curr Top Med Chem. 2009;9:771–790. doi: 10.2174/156802609789207127. [DOI] [PubMed] [Google Scholar]
- 2.Kuntz ID. Structure-based strategies for drug design and discovery. Science. 1992;257:1078–1082. doi: 10.1126/science.257.5073.1078. [DOI] [PubMed] [Google Scholar]
- 3.Ghosh S, Nie A, An J, Huang Z. Structure-based virtual screening of chemical libraries for drug discovery. Curr Opin Chem Biol. 2006;10(3):194–202. doi: 10.1016/j.cbpa.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 4.Leach AR, Gillet VJ, Lewis RA, Taylor R. Three-dimensional pharmacophore methods in drug discovery. J Med Chem. 2010;53:539–558. doi: 10.1021/jm900817u. [DOI] [PubMed] [Google Scholar]
- 5.Guido RV, Oliva G, Andricopulo AD. Virtual screening and its integration with modern drug design technologies. Curr Med Chem. 2008;15:37–46. doi: 10.2174/092986708783330683. [DOI] [PubMed] [Google Scholar]
- 6.Scapin G. Structural biology and drug discovery. Curr Pharm Des. 2006;12:2087–2097. doi: 10.2174/138161206777585201. [DOI] [PubMed] [Google Scholar]
- 7.Kolb P, Ferreira RS, Irwin JJ, Shoichet BK. Docking and chemoinformatic screens for new ligands and targets. Curr Opin Biotech. 2009 Aug;20(4):429–436. doi: 10.1016/j.copbio.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolb P, Irwin JJ. Docking screens: right for the right reasons? Curr Top Med Chem. 2009;9(9):755–770. doi: 10.2174/156802609789207091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Azevedo WF., Jr Structure-based virtual screening. Current Drug Targets. 2010;11(3):261–263. doi: 10.2174/138945010790711969. [DOI] [PubMed] [Google Scholar]
- 10.De Azevedo WF., Jr MolDock applied to structure-based virtual screening. Current Drug Targets. 2010;11(3):327–334. doi: 10.2174/138945010790711941. [DOI] [PubMed] [Google Scholar]
- 11.Navia MA, Fitzgerald PM, McKeever BM, Leu CT, Heimbach JC, Herber WK, et al. Three-dimensional structure of aspartyl protease from human immunodeficiency virus HIV-1. Nature. 1989 Feb 16;337(6208):615–620. doi: 10.1038/337615a0. [DOI] [PubMed] [Google Scholar]
- 12.Mahalingam AK, Axelsson L, Ekegren JK, Wannberg J, Kihlstrom J, Unge T, et al. HIV-1 protease inhibitors with a transition-state mimic comprising a tertiary alcohol: improved antiviral activity in cells. J Med Chem. 2010;53:607–615. doi: 10.1021/jm901165g. [DOI] [PubMed] [Google Scholar]
- 13.Mehellou Y, De Clercq E. Twenty-six years of anti-HIV drug discovery: where do we stand and where do we go? J Med Chem. 2010;53:521–538. doi: 10.1021/jm900492g. [DOI] [PubMed] [Google Scholar]
- 14.Arinaminpathy Y, Khurana E, Engelman DM, Gerstein MB. Computational analysis of membrane proteins: the largest class of drug targets. Drug Disc Today. 2009;14:1130–1135. doi: 10.1016/j.drudis.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lundstrom K. Structural genomics for membrane proteins. Cell Mol Life Sci. 2006;63:2597–2607. doi: 10.1007/s00018-006-6252-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ghosh AK, Xi K, Johnson ME, Baker SC, Mesecar AD. Progress in anti-SARS coronavirus chemistry, biology and chemotherapy. Annu Rep Med Chem. 2007;41:183–196. doi: 10.1016/S0065-7743(06)41011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ghosh AK, Takayama J, Aubin Y, Ratia K, Chaudhuri R, Baez Y, et al. Structure-based design, synthesis, and biological evaluation of a series of novel and reversible inhibitors for the severe acute respiratory syndrome-coronavirus papain-like protease. J Med Chem. 2009 doi: 10.1021/jm900611t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.von Itzstein M. The war against influenza: discovery and development of sialidase inhibitors. Nat Rev Drug Disc. 2007;6:967–974. doi: 10.1038/nrd2400. [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Zhang J, Xu W. Recent progress in rational drug design of neuraminidase inhibitors. Curr Med Chem. 2007;14:2872–2891. doi: 10.2174/092986707782360024. [DOI] [PubMed] [Google Scholar]
- 20.Roth BL. Receptor systems: will mining the receptorome yield novel targets for pharmacotherapy? Pharmacol Ther. 2005;108:59–64. doi: 10.1016/j.pharmthera.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 21.Schwartz TW, Hubbell WL. Structural biology: A moving story of receptors. Nature. 2008;455:473–474. doi: 10.1038/455473a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lundstrom K. An overview on GPCRs and drug discovery: structure-based drug design and structural biology on GPCRs. Methods Mol Biol. 2009;552:51–66. doi: 10.1007/978-1-60327-317-6_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cherezov V, Rosenbaum DM, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. High-resolution crystal structure of an engineered human beta2-adrenergic G protein-coupled receptor. Science. 2007;318:1258–1265. doi: 10.1126/science.1150577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rosenbaum DM, Cherezov V, Hanson MA, Rasmussen SG, Thian FS, Kobilka TS, et al. GPCR engineering yields high-resolution structural insights into beta2-adrenergic receptor function. Science. 2007;318:1266–1273. doi: 10.1126/science.1150609. [DOI] [PubMed] [Google Scholar]
- 25.Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK. Structure-based discovery of beta2-adrenergic receptor ligands. Proc Natl Acad Sci USA. 2009;106:6843–6848. doi: 10.1073/pnas.0812657106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wan H, Bergstrom F. High-throughput screening of drug-protein binding in drug discovery. J, Liq Chromatog Rel Tech. 2007;30:681–700. [Google Scholar]
- 27.Chayen NE. Optimization techniques for automation and high throughput. Methods Mol Biol. 2007;363:175–190. doi: 10.1007/978-1-59745-209-0_9. [DOI] [PubMed] [Google Scholar]
- 28.Bolanos-Garcia VM, Chayen NE. New directions in conventional methods of protein crystallization. Prog Biophys Mol Biol. 2009 doi: 10.1016/j.pbiomolbio.2009.12.006. [DOI] [PubMed] [Google Scholar]
- 29.Gerdts C, Nollert P. Advances in microfluidic membrane protein crystallization techniques. Membrane Protein Crystallization. 2009:179–189. [Google Scholar]
- 30.Narayanan J, Liu XY. Biophys J. 2003;84:523. doi: 10.1016/S0006-3495(03)74871-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu DW, Garneau A, Mazumdar M, Zhou M, Xu GJ, Lin SX. Attempts to rationalize protein crystallization using relative crystallizability. J Struct Biol. 2006;154:297–302. doi: 10.1016/j.jsb.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 32.Jia Y, Liu XY. From surface self-assembly to crystallization: prediction of protein crystallization conditions. J Phys Chem B. 2006;110:6949–6955. doi: 10.1021/jp0536089. [DOI] [PubMed] [Google Scholar]
- 33.Ito K, Sugawara T, Shiroishi M, Tokuda N, Kurokawa A, Misaka T, et al. Advanced method for high-throughput expression of mutated eukaryotic membrane proteins in Saccharomyces cerevisiae. Biochem Biophys Res Comm. 2008;371:841–845. doi: 10.1016/j.bbrc.2008.04.182. [DOI] [PubMed] [Google Scholar]
- 34.Forstner M, Leder L, Mayr LM. Optimization of protein expression systems for modern drug discovery. Expert Rev Proteomics. 2007;4:67–78. doi: 10.1586/14789450.4.1.67. [DOI] [PubMed] [Google Scholar]
- 35.Saridakis E, Chayen NE. Towards a 'universal' nucleant for protein crystallization. Trends Biotechnol. 2009;27:99–106. doi: 10.1016/j.tibtech.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 36.Landau EM, Rosenbusch JP. Lipidic cubic phases: a novel concept for the crystallization of membrane proteins. Proc Natl Acad Sci USA. 1996;93:14532–14535. doi: 10.1073/pnas.93.25.14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Caffrey M. Membrane protein crystallization. J Struct Biol. 2003;142:108–132. doi: 10.1016/s1047-8477(03)00043-1. [DOI] [PubMed] [Google Scholar]
- 38.Misquitta Y, Cherezov V, Havas F, Patterson S, Mohan JM, Wells AJ, et al. Rational design of lipid for membrane protein crystallization. J Struct Biol. 2004;148:169–175. doi: 10.1016/j.jsb.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 39.Civjan NR, Bayburt TH, Schuler MA, Sligar SG. Direct solubilization of heterologously expressed membrane proteins by incorporation into nanoscale lipid bilayers. BioTechniques. 2003;35:556–563. doi: 10.2144/03353rr02. [DOI] [PubMed] [Google Scholar]
- 40.Bayburt TH, Sligar SG. Membrane protein assembly into Nanodiscs. FEBS letters. 2010 May 3;584(9):1721–1727. doi: 10.1016/j.febslet.2009.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shenkarev ZO, Lyukmanova EN, Solozhenkin OI, Gagnidze IE, Nekrasova OV, Chupin VV, et al. Lipid-protein nanodiscs: possible application in high-resolution NMR investigations of membrane proteins and membrane-active peptides. Biochem (Mosc) 2009 Jul;74(7):756–765. doi: 10.1134/s0006297909070086. [DOI] [PubMed] [Google Scholar]
- 42.Lyukmanova EN, Shenkarev ZO, Paramonov AS, Sobol AG, Ovchinnikova TV, Chupin VV, et al. Lipid-protein nanoscale bilayers: a versatile medium for NMR investigations of membrane proteins and membrane-active peptides. J Amer Chem Soc. 2008 Feb 20;130(7):2140–2141. doi: 10.1021/ja0777988. [DOI] [PubMed] [Google Scholar]
- 43.Zheng B, Gerdts CJ, Ismagilov RF. Using nanoliter plugs in microfluidics to facilitate and understand protein crystallization. Curr Opin Struct Biol. 2005;15:548–555. doi: 10.1016/j.sbi.2005.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song H, Chen DL, Ismagilov RF. Reactions in droplets in microfluidic channels. Angew Chem Int Ed. 2006;45:7336–7356. doi: 10.1002/anie.200601554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng B, Roach LS, Ismagilov RF. Screening of protein crystallization conditions on a microfluidic chip using nanoliter-size droplets. J Amer Chem Soc. 2003 Sep 17;125(37):11170–11171. doi: 10.1021/ja037166v. [DOI] [PubMed] [Google Scholar]
- 46.Perry SL, Roberts GW, Tice JD, Gennis RB, Kenis PJA. Microfluidic generation of lipidic mesophases for membrane protein crystallization. Cryst Growth Des. 2009;9:2566–2569. doi: 10.1021/cg900289d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kreutz JE, Li L, Roach LS, Hatakeyama T, Ismagilov RF. Laterally mobile, functionalized self-assembled monolayers at the fluorous-aqueous interface in a plug-based microfluidic system: characterization and testing with membrane protein crystallization. J Am Chem Soc. 2009;131:6042–6043. doi: 10.1021/ja808697e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li L, Ismagilov RF. Protein crystallization using microfluidic technologies based on valves, droplets, and SlipChip. Annu Rev Biophys. 2010 Feb 1; doi: 10.1146/annurev.biophys.050708.133630. [DOI] [PubMed] [Google Scholar]
- 49.Price WN, Chen Y, Handelman SK, Neely H, Manor P, Karlin R, et al. Understanding the physical properties that control protein crystallization by analysis of large-scale experimental data. Nat Biotech. 2009 Jan;27(1):51–57. doi: 10.1038/nbt.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Villasenor AG, Wong A, Shao A, Garg A, Kuglstatter A, Harris SF. Acoustic matrix microseeding: improving protein crystal growth with minimal chemical bias. Acta Cryst. 2010 May;66(Pt 5):568–576. doi: 10.1107/S0907444910005512. [DOI] [PubMed] [Google Scholar]
- 51.Barklis E, McDermott J, Wilkens S, Schabtach E, Schmid J, Fuller S, et al. Structural analysis of membrane-bound retrovirus capsid proteins. EMBO J. 1997;16:1199–1213. doi: 10.1093/emboj/16.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barklis E, McDermott J, Wilkens S, Fuller S, Thompson DH. Organization of HIV-1 capsid proteins on a lipid monolayer. J Biol Chem. 1998;273:7177–7180. doi: 10.1074/jbc.273.13.7177. [DOI] [PubMed] [Google Scholar]
- 53.Zhou M, Haldar S, Franses J, Kim J-M, Thompson DH. Synthesis and self-assembly properties of acylated cyclodextrins and nitrilotriacetic acid (NTA)-modified inclusion ligands for interfacial protein crystallization. Supramol Chem. 2005;17:101–111. [Google Scholar]
- 54.Thompson DH, Zhou MK, Grey J, Kim H-K. Design, synthesis, and performance of NTA-modified lipids as templates for histidine-tagged protein crystallization. Chem Lett. 2007;36:956–975. [Google Scholar]
- 55.Wilson-Kubalek EM, Brown RE, Celia H, Milligan RA. Proc Natl Acad Sci USA. 1998;95:8040. doi: 10.1073/pnas.95.14.8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu X, Zhang Y, Goswami DK, Okasinski JS, Salaita K, Sun P, et al. The controlled evolution of a polymer single crystal. Science. 2005;307:1763–1766. doi: 10.1126/science.1109487. [DOI] [PubMed] [Google Scholar]