

Abstract
The presynaptic, hemicholinium-3 sensitive, high-affinity choline transporter (CHT) supplies choline for acetylcholine (ACh) synthesis. In mice, a homozygous deletion of CHT (CHT−/−) leads to premature cessation of spontaneous or evoked neuromuscular signaling and is associated with perinatal cyanosis and lethality within 1 hr. Heterozygous (CHT+/−) mice exhibit diminished brain ACh levels and demonstrate an inability to sustain vigorous motor activity. We sought to explore the contribution of CHT gene dosage to motor function in greater detail using transgenic mice where CHT is expressed under control of the motor neuron promoter Hb9 (Hb9:CHT). On a CHT−/− background, the Hb9:CHT transgene conferred mice with the ability to move and breath for a postnatal period of ~24 hrs, thus increasing survival. Conversely, Hb9:CHT expression on a wild-type background (CHT+/+;Hb9:CHT) leads to an increased capacity for treadmill running compared to wild-type littermates. Analysis of the stimulated compound muscle action potential (CMAP) in these animals under basal conditions established that CHT+/+;Hb9:CHT mice display an unexpected, bidirectional change, producing either elevated or reduced CMAP amplitude, relative to CHT+/+ animals. To examine whether these two groups arise from underlying changes in synaptic properties, we used high-frequency stimulation of motor axons to assess CMAP recovery kinetics. Although CHT+/+;Hb9:CHT mice in the two groups display an equivalent, time-dependent reduction in CMAP amplitude, animals with a higher basal CMAP amplitude demonstrate a significantly enhanced rate of recovery. To explain our findings, we propose a model whereby CHT support for neuromuscular signaling involves contributions to ACh synthesis as well as cholinergic synaptic vesicle availability.
Keywords: choline, transport, acetylcholine, transgenic, synapse, neuromuscular
INTRODUCTION
Acetylcholine (ACh) serves as a neurotransmitter throughout the central and peripheral nervous systems and supports diverse physiological functions. In the brain, cholinergic terminals are present in nearly every brain region, even though the number of cholinergic nuclei and cells is relatively small (Woolf, 1991). Peripherally, ACh is utilized in both the sympathetic and parasympathetic branches of the autonomic nervous system, as well as the enteric nervous system. The most well-studied mammalian cholinergic synapse is the neuromuscular junction (NMJ), where release of ACh from motor neurons produces a depolarization of muscle fibers, triggering contraction of the muscle.
The presynaptic, high-affinity, choline transporter (CHT, SLC5A7) is essential for proper signaling at cholinergic synapses due to the transporter’s rate-limiting uptake of choline that is required to sustain ACh synthesis and release, particularly at high firing rates (Birks and MacIntosh, 1961, Haga, 1971, Barker and Mittag, 1975, Ferguson and Blakely, 2004). Interestingly, despite the known function of CHT to transport choline, the majority of CHT is present on synaptic vesicles (Nakata et al., 2004). In turn, multiple studies have documented that manipulation of CHT activity alters the strength of cholinergic signaling (Bazalakova and Blakely, 2006). Thus, genetically induced loss of CHT levels in either homozygous (Ferguson et al., 2004) or heterozygous (Bazalakova et al., 2006) knockout mice impairs cholinergic signaling, resulting in neonatal lethality or physical challenge-induced motor deficits respectively. Pharmacological manipulation of CHT also impacts cholinergic signaling; for example, treatment of NMJ synapses with the lethal CHT antagonist hemicholinium-3 (HC-3) reduces the level of ACh in NMJ presynaptic vesicles (Yu and Van der Kloot, 1991). CHT is also responsive to modulation of other elements that support cholinergic signaling. Reductions in the levels of choline acetyltransferase (ChAT) produce an increase in the level and activity of CHT (Brandon et al., 2004). Other models of impaired cholinergic function, such as acetylcholinesterase (AChE) transgenic overexpressing (Beeri et al., 1997) and knockout (Volpicelli-Daley et al., 2003) mice, demonstrate increased CHT levels that appear to represent a compensatory alteration to offset a loss of cholinergic signaling. Additionally, Krishnaswamy and Cooper recently demonstrated that presynaptic CHT downregulation is triggered by genetic elimination of nicotinic receptors on postganglionic neurons (Krishnaswamy and Cooper, 2009).
Pharmacological augmentation of cholinergic tone by enhancement of CHT expression and/or activity may be of benefit in disorders with cholinergic deficits, including myasthenia gravis, attention-deficit hyperactivity disorder (ADHD) and Alzheimer’s Disease. Currently, we lack small molecule, positive modulators of CHT that can inform as to the impact of elevated CHT expression or activity in vivo. We took advantage of the Hb9 promoter’s capability of driving motor neuron-specific gene expression (Arber et al., 1999, Thaler et al., 1999) to enhance CHT expression at NMJ synapses. In the CHT−/− mouse, that dies 30–60 min after birth, Hb9:CHT transgenic animals demonstrated significantly enhanced viability. In a CHT+/+ background, Hb9:CHT transgenic mice demonstrated altered physiological and behavioral motor phenotypes. Strikingly, studies of evoked responses of muscle fibers from the latter animals revealed a shift of excitatory events from a single population of summed action potential amplitudes to two populations, with either enhanced or diminished excitation. We discuss these findings with respect to a dual impact of overexpressed CHT to NMJ synaptic events, mediated either by an enhanced production of ACh, or a reduction in the readily-releasable pool of cholinergic synaptic vesicles.
EXPERIMENTAL PROCEDURES
Generation and genotyping of Hb9:CHT mice
All animal procedures were approved by the Vanderbilt University Institutional Care and Use Committee. pHb9-MCS-IRES-EGFP (Fig. 1A) was a kind gift of Dr. Thomas Jessell and contains a 9kb fragment of the Hb9 promoter, a 5′ splicing substrate, an internal ribosome entry sequence (IRES), enhanced green fluorescence protein (EGFP), and a bovine polyadenylation (polyA) signal (Arber et al., 1999, Wilson et al., 2005). BamHI/HindIII blunted cDNA encoding the mouse CHT (mCHT) (Apparsundaram et al., 2001) was cloned into the PmeI site, resulting in the expression construct pHb9:CHT-IRES-EGFP. DNA was sequenced using primer-directed Sanger sequencing in the DNA Core of the Vanderbilt Division of Human Genetics, Department of Medicine. To produce transgenic founders, the plasmid was first linearized and vector sequences removed using XhoI, and then purified and injected into the pronucleus of C57BL/6 single cell embryos in the ES/Transgenic Core of the Vanderbilt Stem Cell Center. Founders were screened by Southern blotting and PCR to identify carriers of exogenous mCHT cDNA or EGFP. DNA for genotyping was obtained by tail biopsy and processed using the RED Extract-n-Amp kit (Sigma; St. Louis, MO). Primers for PCR were 5′-ACACATGGGTTGGAGGAGG-3′ and 5′-CTAATGCAGAGAAAATTGC-3′, targeting the 5′ end of exon 3 and the 3′ end of exon 4 respectively. Products were analyzed using agarose gel electrophoresis. Quantitative PCR (qPCR) was performed using a LightCycler 480 (Roche) with SybrGreen fluorescence. Primers for qPCR were directed at 2 locations on the Hb9 promoter (5′-TGACCATCCACCAGGCTAAC-3′ with 5′-AGGGGCTCTTGATACAACCTC-3′ and 5′-GCGACAAAAATTGTCTGCCTA-3′ with 5′-TGATGCACACCAFCATCATA-3′) and 1 location in the mCHT cDNA in exon 3 (5′-GGGTTGGAGGAGGCTACATC-3′ with 5′-CTAGCCCAAGCTAGACCAC-3′). The reference primer was directed at a location 80kb upstream from genomic CHT (5′-CTGCTAACCTGGATGAAGCA-3′ with 5′-GCCTGTGAGAGCCTAGCTGA-3′).
Fig. 1.

Generation of Hb9:CHT mice: (A) Schematic of Hb9:CHT transgene showing all elements that allow expression including a 5′ splice substrate (5′SS), CHT cDNA (mCHT), internal ribosome entry site (IRES), enhanced green fluorescent protein (EGFP), and a bovine polyadenylation signal (polyA). (B) PCR demonstrating presence of transgene. The 0.3kb band is derived from the cDNA for CHT and the 0.9kb band is derived from the genomic DNA for CHT. An aberrant 1.7kb band was also observed. (C) and (D) Growth curves for male (C) and female (D) mice (male: CHT+/+ n=12, Hb9:CHT n=4; female: CHT+/+ n=12, Hb9:CHT n=6, p>0.1 for genotype effect). (E–G) Hindbrain immunostaining for ChAT (E) and EGFP (F) with the merged image (G) showing colocalization in the soma of motor neurons from cranial nerve XII. Scale bar for (E–G) 20μm. (H–J) Midbrain immunostaining for ChAT (H) and EGFP (I) with the merged image (J) showing lack of EGFP in a non motor neuron population of cholinergic neurons in the lateral dorsal tegmentum. Scale bar for (H–J) is 50μm.
Mice, breeding strategy, and survival analysis
CHT+/− and CHT−/− mice have been previously described (Ferguson et al., 2004, Bazalakova et al., 2006). C57BL/6NHsd breeding females were obtained from Harlan (Harlan Laboratories; Indianapolis, IN). Mice were housed with 2 to 5 animals per cage on a 12-hour light/dark cycle with lights on at 0600h. Food and water were provided ad libitium (Purina Rodent Chow 5001). For staining and survival experiments, Hb9:CHT;CHT+/− males were bred to CHT+/− females. For treadmill and some CMAP experiments Hb9:CHT;CHT+/− males were bred to C57BL/6 females. For the remaining CMAP experiments, Hb9:CHT;CHT+/+ males were bred to C57BL/6 females. For neonatal experiments, multiple breeding triplets were established with the males removed after 5 days. Beginning on day 19 after the establishment of breedings, females were observed each hour beginning at 2200h, and if no pups were born, observation ended at 0900h the following day. If pups were born, litters were observed every 30 minutes for the first 6 hours and every hour subsequently up to 72 hours with the time of death recorded as the time the pup was found dead. All litters observed were completely born within 1 hour. For neonatal staining experiments, animals were taken 4 hours after birth or when observed as dead.
Tissue processing and immunofluorescence
Mice were deeply anaesthetized with Nembutal™ and euthanized by decapitation. For brain tissue, mice were perfused with 4% paraformaldehyde, brains removed and post-fixed for up to 1 week in 4% paraformaldehyde. Tissue was cryoprotected in 20% sucrose and sliced at 40μm using a freezing microtome. Pups were euthanized by decapitation if necessary. Diaphragms with attached ribs were pinned down at tension slightly above resting tension on Sylgard-lined (Ellsworth Adhesives; Germantown, WI ) plates and fixed for 30 minutes in 4% paraformaldehyde. NMJs were labeled with 1ug/mL alphaBungarotoxin-alexa488 (Invitrogen; Eugene, OR) for 30min. NMJs and floating brain sections were washed and then blocked with 3% normal donkey serum, 0.2% Triton X-100 in PBS for 1 hour. Primary antibodies (chick anti-Neurofilament-M, chick anti-Neurofilament-H, goat anti-ChAT (Millipore; Temecula, CA), rabbit anti-CHT (Ferguson et al., 2003)) were incubated overnight at 4°C in blocking medium. After washing, sections were stained with the appropriate, fluorescently labeled secondary antibodies, mounted with Aqua PolyMount, and imaged using a Zeiss LSM Meta 510 laser scanning confocal microscope.
NMJ area analysis
Flexor digitorum brevis (FDB) sections were processed for alpha bungarotoxin-alexa488 labeling of motor endplates. Projection images acquired on a Zeiss LSM510 Meta system were thresholded manually using the ImageJ software (NIH). Area of the image above threshold was then calculated based on the internal scaling from the microscope.
Treadmill testing
Mice between 8 and 11 weeks of age were examined for running endurance on a 6 lane motorized treadmill with an electric shock grid at one end (Columbus Instruments; Columbus OH). All experiments were conducted between 0900h and 1600h. For each mouse, training or trial sessions were ended when they reached a criterion of exhaustion, defined as willingness to receive 15s of shock in any 1 minute interval. On day 1 (training), mice were allowed to explore their lane for 5 minutes without shock followed by 5 minutes where the shock was turned on. All mice received a shock at least once during this stage. The treadmill was then started at 5m/min and accelerated to 15m/min at a rate of 1m/min/30sec. After 1 hour of running, the treadmill was then increased to 18m/min at 1m/min/30sec. Day 2 (test) began as described for day 1, but the treadmill was accelerated directly from 5m/min to 18m/min at the beginning. At 1 hour of running, the treadmill was accelerated to 20m/min. In these tests, endurance times for both the CHT+/+ (F(3,12)=15.28, P=0.0002, extra sum-of-squares F test), and the CHT+/+;Hb9:CHT (F(3,12)=9.650, P=0.0016, extra sum-of-squares F test) mice separated into bimodal distributions for endurance time. To reduce contributions in our data analysis from animals of either genotype with low performance values that likely fail to complete the task for reasons unrelated to physical endurance, we established a minimum criteria for performance for both genotypes. We averaged the minimum criteria for performance for each genotype (mean of the lower component of the bimodal distribution plus 3 standard deviations), and applied the result (28.74 min) to both genotypes.
Compound muscle action potential recording (CMAP)
All recordings were obtained between 1600h and 2330h. A separate cohort of mice from the treadmill between 8 and 11 weeks of age were anaesthetized with 3% isoflurane and maintained at 1.5% isoflurane on a heating pad at 37°C for the duration of the experiment. Experiments are based on protocols from Kaja et al. (Kaja et al., 2007) and Byun and Delpire (Byun and Delpire, 2007) with minor modifications. Data were collected and analyzed using a TECA Synergy portable electromyography system with platinum/iridium needle electrodes (Viasys Healthcare; Madison, WI). A bipolar stimulating electrode was inserted at the ankle to stimulate the tibial nerve, the recording electrode was inserted through the paw at the level of attachment for the first and fifth digits, and a reference electrode was inserted distally. A ground surface electrode was wrapped around the paw proximal to the recording electrode and affixed to a styrofoam surface. This allowed us to fix the position of the paw to mostly eliminate movement artifacts and record reliably before and after 50Hz stimulation although a small number of animals could not be analyzed due to the movement. Correct placement of the electrodes was determined by lack of noise >1microV and lack of an initial positive deflection upon stimulation. Stimulation intensity for each recording location was determined by increasing the stimulus intensity gradually during 0.5Hz stimulation until a plateau response was achieved. Suprathreshold stimulation was defined as 150% of the minimum stimulation needed to reach plateau response. This stimulation was recorded as the basal CMAP, and used for the duration of the experiment. High-frequency stimulation (50Hz, 1min) was then applied, immediately followed by test stimulations at 0.5Hz with data recorded every minute. All stimulations were delivered as 0.02ms square pulses and were reliably delivered at less than 4mA. The low-pass acquisition filter was 1Hz and the high-pass filter was 10kHz.
Muscle and muscle fiber cross-sectional analysis
Mice were euthanized by rapid cervical dislocation. The gastrocnemius was rapidly dissected with the distal tendon cut and left attached at the proximal end. The muscle was held at resting length and frozen for 20 seconds in 2-methylbutane cooled in liquid nitrogen. Muscles were then mounted for cryostat sectioning at 20μm and thaw mounted onto superfrost slides (Fisher). Muscle sections were dried for at least 30min, then stained for succinate dehydrogenase activity as previously described (De Paepe et al., 2009). Briefly, slides were incubated for 30 minutes at 37°C in a reaction buffer containing 100mM Na-succinate and 1.2mM nitroblue tetrazolium in 0.2M Na-phosphate buffer (pH=7.4). Slides were then rinsed and cleared in ascending and descending acetone solutions followed by washes with water. Slides were mounted with Aqua PolyMount and imaged using a Zeiss Discovery V.12 stereo microscope (for whole muscle cross-sectional area) or a Zeiss Axiophot upright microscope (for muscle fiber cross-sectional area).
AChE and ChAT enzyme activities
Gastrocnemius muscles were dissected, frozen on dry ice, and stored at −80°C until use. Muscles were weighed and then placed into 3mL of PBS to be homogenized using a polytron. 3mL of PBS was then added to the initial homogenate and then further homogenized using a dounce homogenizer. A portion of the dounce homogenized tissue was then taken and Triton X-100 was added to a final concentration of 0.5% and frozen at −20°C for later ChAT assays. AChE was determined using the Amplex Red kit (Invitrogen). ChAT was determined by the synthesis of [14C]-ACh from [14C]-Acetyl-CoA using the method of Frick et al (Frick et al., 2002).
Statistical Analysis
Statistics were computed using Prism 5.0 (GraphPad). Survival curves were analyzed first for all genotypes and, if significant, reanalyzed for the experimental pair using the Mantel-Cox Log-Rank test. To assess the bimodality of the CMAP experiment, CMAPs were binned into 0.75mV intervals and the resulting histogram used to calculate probability density functions. Comparison between models using a single normal distribution or a sum of two normal distributions was performed using the extra sum-of-squares F test. Data for the recovery curves were analyzed by normalizing data to findings obtained from 0 minutes to 5 minutes following stimulation (0% and 100% respectively). A single exponential function was fitted constraining Y0 to 0 and plateau to 100 and the K values of the following equation were compared: Y=Y0 + (Plateau−Y0)*(1−exp(−K*x)).
RESULTS
Motor neuron-specific expression of mCHT transgene
Of five founder animals generated following pronuclear injection of a pHb9:mCHT-IRES-EGFP construct (Fig 1A), three animals transmitted the Hb9:CHT transgene to offspring. The presence of the transgene was examined by presence of the CHT cDNA (Fig. 1B). One line did not achieve stable transmission to successive generations. Of the two remaining lines, we selected the line (Tg2) that exhibited the highest copy number as determined by qPCR (10 copies versus 7 copies in the other line, Tg9) for analysis. Furthermore, Tg9 animals displayed lower NMJ CHT expression on a CHT −/− background than Tg2 animals, as determined by fewer terminals with CHT and no visible CHT in incoming fibers (data not shown). They also did not display rescue from neonatal lethality, even when possessing the transgene on both chromosomes, suggesting overall low CHT protein expression. As shown in Fig 1C and D, both male and female Hb9:CHT transgenic mice from the Tg2 line grew normally (Fig 1C and D), and the transgene segregated randomly from CHT with no evidence for lethality (n=80, chi2=7.6, df=5, P>0.1). [Approximate location of Figure 1]
As the mCHT protein expressed from our transgene does not differ in sequence from the native CHT, we utilized the coexpressed EGFP to examine expression of the transporter in somatic motor neurons. Although the Tg2 line carries multiple copies of the Hb9:CHT transgene, direct EGFP fluorescence signal was not apparent. We therefore utilized anti-EGFP antibody-based detection to examine transgene expression. In Fig 1E–F, we demonstrate colocalization of anti-EGFP immunoreactivity with ChAT in the large majority of hypoglossal nucleus motor neurons (Fig. 1E). Consistent with the specificity of the Hb9 promoter, EGFP immunoreactivity was absent from neurons of the cholinergic lateral dorsal tegmental nucleus (Fig. 1H–J). Western blotting of brainstem extracts however did not reveal an elevation in CHT levels (data not shown), possibly because of efficient export of transporters to motor neuron terminals. As with others (Wessler and Kilbinger, 1986, Wessler and Sandmann, 1987), we have to date been unsuccessful in immunoblotting or in monitoring CHT activity at the NMJ, most likely due to the limited density of neuronal elements in muscle preparations and the presence of a non-CHT choline accumulation mechanism in muscle tissue. Nonetheless, on the CHT−/− background, we could readily detect CHT expression by immunofluorescence (Fig 2A–I) in CHT−/−;Hb9:CHT diaphragms. [Approximate location of Figure 2] In the diaphragm of neonatal, CHT+/+ animals, CHT immunoreactivity colocalized as previously described (Ferguson et al., 2004) with alpha bungarotoxin-labeled nicotinic receptors (Fig. 2A to C). Specificity of labeling for CHT was evident, as seen in a parallel colocalization series produced with nontransgenic CHT −/− mice (Fig 2D–F). In Hb9:CHT transgenic mice on the CHT−/− background (Fig 2G–I), CHT labeling was evident, though weaker than observed in CHT+/+ preparations (compare 2B and 2H) and, as in CHT+/+ specimens, was colocalized with alpha bungarotoxin. Interestingly, the lateral distribution of endplates across the diaphragm of both CHT −/− and Hb9:CHT transgenic mice on the CHT−/− background was more irregular than seen with sections from CHT+/+ animals, and not all terminals labeled for the CHT transgene opposed dense clusters of nicotinic receptors. These findings indicate that although the CHT transgene is present in multiple copies, expression of CHT protein does not reach that of the transporter at the wild-type NMJ.
Fig. 2.

Expression and function of CHT from HB9:CHT demonstrated in Hb9:CHT;CHT−/− neonatal mice: (A–I) Immunostaining for alpha Bungarotoxin and neurofilaments (A, D, G) and CHT (B, E, H) with the merged images (C, F, I) in CHT+/+ (A–C), CHT−/− (D–F) and Hb9:CHT;CHT−/− (G–I) mice. Although weaker than CHT+/+, CHT immunoreactivity is present opposed to α-bungarotoxin staining in CHT−/−;Hb9:CHT diaphragms. Scale bar is 50μm. (J) Survival time from birth for mice of all six possible genotypes (CHT+/+ n=5, CHT+/− n=22, CHT−/− n=5, Hb9:CHT;CHT+/+ n=5, Hb9:CHT;CHT+/− n=22, Hb9:CHT;CHT−/− n=3). CHT+/+ open triangles; CHT+/− not shown, all survived to 4320 min; CHT−/− open circles; Hb9:CHT;CHT+/+ filled triangles; Hb9:CHT;CHT+/− not shown, all survived to 4320 min; Hb9:CHT;CHT−/− filled circles. (K–M) Photographs of mice at 4 hrs after birth. CHT−/−;Hb9:CHT mice have similar color to CHT+/+ mice and occasionally have a milk-spot (indicated by arrows) that is noticeably smaller in this pup.
To explore the capacity of the Hb9:CHT transgene to rescue CHT −/− mice from neonatal lethality (Ferguson et al., 2004), we monitored the survival of CHT+/+, CHT+/−, and CHT−/−, with or without the Hb9:CHT transgene, all derived from a cross of CHT+/−;Hb9:CHT with CHT+/− parents. As previously observed (Ferguson et al., 2004), CHT−/− mice died within 30min after birth, becoming progressively paralyzed and cyanotic, presumably due to an inability to contract the diaphragm. As expected, CHT +/− and CHT+/+ littermates exhibited normal growth and survival. Strikingly, CHT−/−;Hb9:CHT mice survived up up to 24 hours (Fig. 2J), exhibiting normal coloration in the first hour after birth (Fig. 2M), grossly normal movements and evidence of nursing behavior.
Hb9:CHT transgenic mice on a CHT+/+ background display enhanced endurance on a treadmill
Our Hb9-based transgenic strategy was designed not only to assess the capacity of motor neuron-specific expression of CHT to rescue lethality of CHT−/− mice, but also to permit evaluation of the functional consequences of supranormal CHT expression when the transgene is placed on a CHT+/+ background. To explore the latter opportunity, CHT+/+ and CHT+/+;Hb9:CHT transgenic mice were assessed for endurance in a paradigm of forced exercise on a motorized treadmill. We have previously shown that CHT+/− mice display an inability to achieve the speeds, or exhibit the endurance, of CHT+/+ mice (Bazalakova et al., 2006). The distributions of time until exhaustion for CHT+/+ and CHT+/+;Hb9:CHT mice that met performance criteria for inclusion are shown in Fig 3A and B. The mean time until exhaustion for the CHT+/+;Hb9:CHT mice was significantly greater than that of CHT+/+ littermates (two-tailed t-test, P=0.0179) (Fig 3C). [Approximate location of Figure 3]
Fig. 3.
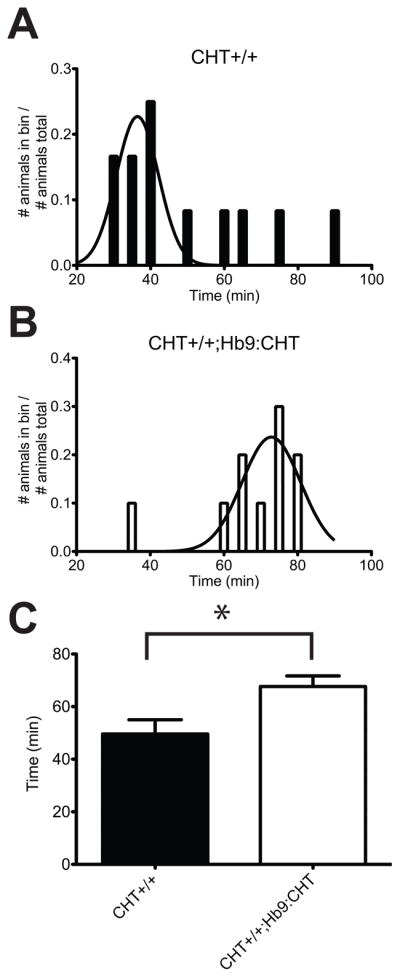
Endurance of Hb9:CHT mice during a forced treadmill exercise paradigm. (A and B) Distributions for running times of CHT+/+ (A) and CHT+/+;Hb9:CHT (B) mice that met performance criteria. The fitted curve is the probability density function for a Gaussian distribution of the performing mice. (C) Comparison of running times for each genotype. CHT+/+;Hb9:CHT mice are significantly elevated (two-tailed t-test, P=0.0179).
Basal CMAP and CMAP recovery from high frequency stimulation are altered in Hb9:CHT transgenic mice
With evidence of enhanced endurance in the CHT+/+;Hb9:CHT mice, we sought evidence of altered neuromuscular signaling that might contribute to our behavioral observations by examining evoked compound muscle action potentials (CMAP). We observed no genotype effect on the amplitude of the baseline evoked CMAP (Mann-Whitney U test, P=0.8294) (Fig. 4A). However, recordings from the CHT+/+;Hb9:CHT mice revealed a clear shift from the unimodal distribution of values evident in CHT+/+ animals (Fig. 4B) to a bimodal distribution (P=.0023, F(3,5)=23.04, extra sum-of-squares F-test) (Fig. 4C). Both the low and high amplitude Hb9:CHT;CHT+/+ groups were significantly different than the CHT+/+ group P<0.001, ANOVA with Bonferroni post-test). [Approximate location of Figure 4]
Fig. 4.

CMAP analysis of the hindlimb paw muscles: (A) Maximal stimulated recorded compound muscle action potential (CMAP) prior to high frequency stimulation (CHT+/+ n=11, CHT+/+Hb9:CHT n=12). Histogram of CMAP amplitudes for CHT+/+ (B) and CHT+/+;Hb9:CHT (C) mice and the resulting probability density functions.
To further explore the properties of the high and low amplitude groups described above, we examined the CMAP recovery rates following prolonged 50Hz stimulation. CHT+/+, low amplitude CHT+/+;Hb9:CHT and high amplitude CHT+/+;Hb9:CHT mice exhibited the same percentage CMAP reduction (~80%) following high frequency stimulation (P=0.6047) (Fig. 5A). Additionally, the kinetics for CMAP recovery for the low amplitude CHT+/+;Hb9:CHT group was indistinguishable from the CMAP recovery rate of CHT +/+ mice (CHT+/+ K=0.71+/−0.04; CHT+/+;Hb9:CHT(Low) K=0.79 +/− 0.08, P=0.3304, F(1,64)=0.9168, extra sum-of-squares F-test) (Fig 5C). In contrast, the high CMAP amplitude, CHT+/+;Hb9:CHT group displayed a significantly faster recovery rate (CHT+/+ K=0.71+/−0.04; CHT+/+;Hb9:CHT(High) K=0.95+/− 0.06, P=0.0017, F(1,88)=10.54, extra sum-of-squares F-test) (Fig 5B). [Approximate location of Figure 5]
Fig. 5.

Recovery of stimulated CMAP after 60s of 50Hz stimulation: (A) % reduction of CMAP from basal to first stimulation after high frequency stimulation. (B) and (C) Normalized recovery curves comparing CHT+/+ (n=8) mice against the high (n=7) (B) and low (n=3) (C) CMAP components of CHT+/+;Hb9:CHT mice.
Physical characteristics and enzyme activities of muscles are not changed by the Hb9:CHT transgene
To examine possible structural and compensatory changes related to altered cholinergic signaling, we examined the size of the NMJ and muscles and the activities of enzymes involved in ACh synthesis and degradation. For analysis of NMJ area (Fig. 6A–C), we used the Flexor digitorum brevis (FDB) near the body of the third metatarsal as this muscle was the predominant muscle for CMAP recording. The NMJ areas of this muscle were not changed between genotypes (two-tailed t-test, P=0.2705) (Fig 6C). [approximate location of figure 6]
Fig. 6.

Physical characteristics of NMJ and muscle fibers of Hb9:CHT mice. (A and B) Sample NMJ of CHT+/+ (A) and CHT+/+;Hb9:CHT (B) from the Flexor digitorum brevis (FDB). (C) The NMJ area is not different between genotypes (CHT+/+ n=4, Hb9:CHT n=5; two-tailed t-test, P>0.1). (D and E) Sample images of cross-sections of gastrocnemius muscles stained for SDH. (F) Cross-sectional area of each fiber type is not different between genotypes (CHT+/+ n=3, Hb9:CHT n=5; two-way ANOVA, P>0.1). (G) Cross-sectional area of the entire gastrocnemius is also not different between genotypes (CHT+/+ n=4, Hb9:CHT n=5; two-tailed t-test, P>0.1).
In order to compare the physical aspects of the muscle and enzyme activities, it was necessary to use the gastrocnemius due to its larger size and the ability to generate a complete cross section of the muscle (not possible for FDB). The cross-sectional area of the gastrocnemius was not changed in CHT+/+;Hb9:CHT mice (two-tailed t-test, P=0.8573) (Fig 6G). We also analyzed the size of both Type I/IIa fibers (dark SDH staining) and Type IIb fibers (light SDH staining). In both cases, there was not change in the CHT+/+;Hb9:CHT mice (two-way ANOVA, P=0.7951 for genotype effect) (Fig 6F). Furthermore, there was no change in the activity of either AChE or ChAT (Fig 7) [approximate location of figure 7].
Fig. 7.

ACh metabolizing enzyme activities of Hb9:CHT mice. (A) ChAT activity is not affected by the Hb9:CHT transgene (CHT+/+ n=3, Hb9:CHT n=4; two-tailed t-test, P>0.1). (B) AChE activity is also not affected (CHT+/+ n=3, Hb9:CHT n=5; two-tailed t-test, P>0.1).
DISCUSSION
Our results provide evidence of successful motor neuron and NMJ expression of CHT in Hb9:CHT transgenic mice. Due to the limitations of measuring CHT levels and activity in motor neurons (Wessler and Kilbinger, 1986, Wessler and Sandmann, 1987), we verified CHT expression of our Hb9:CHT transgene via visualization of transgene expression on a CHT−/− background. Although we detected CHT at the NMJ, staining levels did not reach the levels observed in CHT +/+ mice. We suspect that although we integrated multiple copies of the Hb9:CHT transgene into the genome of these mice, the Hb9 promoter may be intrinsically weaker than the native CHT promoter, or be limited by the site of integration. Qualitative observation of the NMJ region of these mice also displayed morphological characteristics (wider band of NMJ across diaphragm, as seen in Fig. 2A–I) seen in mice lacking ACh synthesis (Brandon et al., 2003) or no CHT expression (Ferguson et al., 2004), including less compact synaptic zones. Lack of diaphragm function from insufficient ACh release has been shown as the cause of death in mice with severely compromised cholinergic synapses (Misgeld et al., 2002), including our CHT−/− mice (Ferguson et al., 2004). Although a full rescue of CHT was not achieved, we did observe a significant extension of lifespan in CHT−/−;Hb9:CHT mice. Pursuit of the basis for limited longevity in these mice is beyond the scope of the current study but may arise from deficits in autonomic signaling supporting normal cardiovascular function (Vaseghi and Shivkumar, Schwartz et al., 1988). Additionally, central cholinergic control of motor neuron activity, such as control of respiratory motor neurons by pontine cholinergic neurons (Shao and Feldman, 2005), may be compromised since transgene expression is limited to brainstem and spinal motor neurons (Fig. 1E–J).
One of the most striking phenotypes observed in CHT+/− mice was reduced endurance during forced treadmill exercise. We were therefore eager to determine whether the opposite phenotype would emerge when CHT expression was elevated in motor neurons of wild-type animals. Our treadmill paradigm employed for this assessment was designed to ameliorate some anticipated confounds. Specifically, we used a very low amount of training so that we could better identify increases in running time caused by elevated CHT and minimize any training induced plasticity (Massett and Berk, 2005). We also used a lower speed that would be more likely to elicit a challenge-induced phenotype (Bazalakova et al., 2006). Other groups have used longer training regimens (Lerman et al., 2002) and more intense testing protocols (Lightfoot et al., 2001). Our demonstration of enhanced endurance in the Hb9:CHT mice supports the capacity of elevated CHT expression to positively contribute to exercise endurance. Since the haploinsufficiency of our CHT+/− mice is constitutive, we were unable to determine previously (Bazalakova et al., 2006) whether the reduced endurance of these animals was caused by deficits in neuromuscular, autonomic, or central cholinergic signaling. The elevated endurance apparent in the Hb9:CHT mice provides strong evidence that modulation of CHT expression/activity at the NMJ can both decrease or increase endurance. Whether this effect arises as a result of developmental changes in the strength and stability of NMJ signaling or is a feature of ongoing NMJ activity during the challenge protocol is unknown, but should be amenable to study in transgenic models where CHT expression is controlled in an inducible manner. If the latter explanation holds, pharmacological induction of increased CHT activity could represent a novel therapeutic strategy to ameliorate deficits in NMJ signaling, as seen in myasthenic syndromes (Schwab et al., 1957, Sussman et al., 2008).
To complement the behavioral challenge of the treadmill and to monitor physiological features of motor neuron signaling arising from a neuromuscular challenge, we assessed the magnitude of evoked stimulation and the rate of recovery from depression that arises from high frequency (50 Hz) stimulation. As CMAP measures the summation of action potentials of the muscle rather than the signal from individual nAChRs, we did not expect to see differences between genotypes in the basal CMAP amplitude. Our amplitudes were evoked using supramaximal stimulation, reducing concerns that results derived from variations in initiation of axonal action potentials. However, we observed a striking bimodal distribution in the basal CMAP amplitude, and the predicted enhanced recovery phenotype was only observed in the “high-CMAP” group. Given that the tibial nerve was supramaximally stimulated and muscles are thought to have a strong safety factor (Wood and Slater, 2001), it was surprising to see a population with increased CMAP amplitude. An elevated CMAP amplitude, caused by increased fidelity of neuromuscular transmission, would be expected if the safety factor of the studied muscle in CHT+/+ mice is low enough such that stimulation of all motor axons does not necessarily result in action potentials of all muscle fibers. Our recording setup primarily recorded from the FDB, which has been suggested to have a lower safety factor compared to the more extensively studied diaphragm (Molenaar, 2008). However, increased CMAP amplitude is usually associated with pathological conditions that reduce ACh release where CMAP can be elevated by repetitive nerve stimulation (i.e. Lambert-Eaton myasthenic syndrome). In non-pathological conditions, increased CMAP amplitude has been best described only under a few conditions, but all relate to periods of increased use. During timescales on the order of minutes, exercise has been shown to increase CMAP amplitude in response to exercise (Lentz and Nielsen, 2002). Several explanations have been offered for the increase in CMAP including increased muscle excitability (Hicks et al., 1989), increased number of muscle action potentials (Lee et al., 1977), and increased synchronization of muscle action potentials (Asawa et al., 2004). As an alternative hypothesis to improved neuromuscular transmission, increased CHT could alter the excitability of the muscle. For example, Fong and colleagues recently demonstrated that increased quantal size at the NMJ can alter the steady-state electrical properties of the muscle membrane (Fong et al., 2010). As the expected mechanism for increased cholinergic signaling with elevated CHT is an increase in quantal size (Fig 8), a progressive change in muscle exciteability may explain increased CMAP amplitude.
Fig. 8.

Proposed model for bimodal distribution of CMAP data. In a wild-type synapse, CHT imports choline that is rapidly synthesized into ACh and packaged into vesicles for release and postsynaptic effect. In CHT+/+;Hb9:CHT mice with an elevated basal response, increased plasma membrane CHT increases both choline transport and ACh synthesis. The distribution of vesicles in the elevated condition is minimally affected, if at all, but the amount of ACh per vesicle is substantially elevated. Conversely, in the CHT+/+;Hb9:CHT mice with reduced response, ACh release is reduced by the lack of readily releasable or recycling vesicles. Vesicles are instead shunted to the reserve pool by the presence of CHT, probably by the interaction of CHT with an unknown adaptor protein.
Previous studies have documented that altered ACh vesicular content can alter the amount of ACh released (Yu and Van der Kloot, 1991, Gras et al., 2008). Extended periods of stimulation, even at frequencies as low as 10Hz, lead to the emergence of larger quantal sizes (Yu and Van der Kloot, 1991), suggesting that elevating quantal size could be dependent on the history of motor activity of each mouse. As these increases were shown to be HC-3 sensitive, it is reasonable to speculate that increased CHT could increase the variability between animals by increasing the effect of animal experience on measured neuromuscular function. As noted above, we recognize that the effects we observed could be due either to a developmental effect of elevated ACh synthesis and release or elevated ACh signaling at the time of stimulation. Further studies are needed to address this issue that utilize temporally controlled CHT expression.
[Approximate location of Figure 8]The generation of a population of animals with low basal CMAP amplitudes was surprising given the expectation of a gain of function. Similar amplitudes obtained from recordings at multiple sites in the same animal convinced us that the changes observed are reflections of stable physiological characteristics as opposed to experimental artifacts. The vesicular localization of CHT at the NMJ and other cholinergic synapses has been postulated to relate to the functionally defined reserve pool of cholinergic synaptic vesicles (Ferguson et al., 2003, Ferguson and Blakely, 2004, Nakata et al., 2004). Potentially, elevated CHT could negatively impact the size of CMAPs if elevated CHT increases the abundance of reserve pool vesicles at the expense of the readily releasable pool (Fig 6). In preliminary studies examining the NMJs of nematodes genetically deprived of the CHT homolog CHO-1, we have observed a loss of synaptic vesicles within 100nm of the plasma membrane (D.S. Matthies and R.D. Blakely, unpublished findings). Increasing the ratio of reserve pool to releasable pool vesicles would reduce the amount of ACh available for release during lighter periods of stimulation and thereby reduce overall stimulation if the mobilization of reserve pool vesicles is too slow to compensate. The two hypotheses we present to explain high and low CMAP values may interrelate as newly synthesized ACh is thought to preferentially load more rapidly recycling vesicles (Gracz et al., 1988, Bonzelius and Zimmermann, 1990). Possibly, only in mice where there is a sufficiently large population of recycling vesicles can elevated ACh synthesis “win out” and result in an elevation in cholinergic signaling. Although we did not observe a group of CHT+/+;Hb9:CHT mice with reduced endurance to match our findings of reduced CMAP levels, we utilized a cutoff for non-performance in this task that could have masked such an effect. Indeed, retrospective analysis of treadmill times for the nonperforming group reveals a broader distribution of running times than seen in the CHT+/+ mice that might reflect incorporation of mice with a negative shift in endurance. Future experiments will directly assess the relationship between changes in treadmill running time and changes in CMAP amplitude.
An alternative hypothesis for the bimodal distribution of the CMAP is a potential compensatory effect occurring at the neuromuscular junction. In CHT+/− mice, M1 and M2 muscarinic ACh receptor levels are reduced in a brain region-specific manner (Bazalakova et al., 2006), and these animals display a reduced locomotor sensitivity to the muscarinic antagonist scopolamine. Both M1 and M2 receptors have been identified on motor neuron presynaptic terminals, where they may also be influenced by elevations in CHT expression. At the NMJ, M1 facilitates ACh release whereas M2 inhibits ACh release and functional interactions between these receptors have been described (Oliveira and Correia-de-Sa, 2006). At low frequencies of stimulation, M1 stimulation of release predominates whereas at higher stimulation frequencies, M2-mediated suppression is dominant. If prior experience can alter the balance between M1 and M2 signaling, then the effects of altered CHT expression should lead to enhanced or suppressed neuromuscular transmission. It is unlikely that the transgene initiates any RNA level feedback regulation as our construct uses the CHT cDNA and therefore lacks potential 3′-untranslated region regulatory elements associated with cholinergic signaling. Finally, differences in the ratio of ACh and ATP storage and release could drive signaling toward enhanced or diminished signaling.
If the two populations identified in CMAP responses derive from contributions of CHT to fundamentally different components of presynaptic signaling capacity (e.g. ACh synthesis and alterations in availability of releasable versus reserve vesicle pools), we hypothesized that these differences might be observable in the kinetic properties of evoked responses. In support of this idea, CHT+/+ and CHT−/− mice display differences in the kinetics of rundown of both spontaneous and evoked ACh release (Ferguson et al., 2004). In the context of our manipulations, we reasoned that if the high-CMAP group derives from NMJ signaling that is dominated by elevated ACh synthesis and release, recycling vesicles might be more rapidly refilled after acute depletion, leading to a more rapid recovery. In contrast, if the low-CMAP group derives from a reduction in the availability of readily releasable vesicles, the kinetics of recovery might not be impacted by CHT overexpression. Indeed, we found a statistically significant enhanced rate of recovery in the high-, but not the low-CMAP group. Importantly, this difference was observed in the context of equivalent percentage suppression of CMAP in both groups induced by high frequency stimulation. Altered recovery kinetics of heart rate (HR) in the cardiovascular system have been associated with changes in mortality (Cole et al., 1999) and we have demonstrated reductions in HR recovery in CHT+/− mice (English et al., 2010). As with our discussion of the origins of differences in basal CMAP values, we recognize that at present, there are likely other explanations for the kinetic differences we observed. Two approaches are currently being pursued to gain finer resolution of the effects of CHT overexpression. One is a quantitative, optical evaluation of presynaptic vesicle cycling rates where CHT overexpression is achieved in NMJ synaptopHluorin mice (Wyatt and Balice-Gordon, 2008, Gaffield et al., 2009). These studies offer an opportunity to assess the impact of CHT on the segregation of synaptic vesicle pools. The second is a quantal analysis of miniature end-plate potentials (Everett and Ernst, 2004) that can report on the ACh content of single synaptic vesicles. Given the range of disorders that display reduced cholinergic signaling, ranging from neuromuscular disorders to Alzheimer’s Disease, the results of these studies should not only clarify molecular contributions to cholinergic signaling but also lay a foundation for new, cholinomimetic therapeutics.
Acknowledgments
The authors thank Chris Svitek, Qiao Han, Angela Steele, Tracy Moore-Jarret and Kathryn Lindler for excellent laboratory support. The authors would also like to like to thank Dr. David Robertson and Dr. Martin Appalsamy for training and equipment related to the CMAP experiments and Dr. James Sutlcliffe and Dr. Sabata Lund for assistance with copy number determination. Treadmill experiments were performed through the use of the Murine Neurobehavioral Core lab at the Vanderbilt University Medical Center, managed by Dr. John D. Allison. Imaging experiments, analysis, and presentation of confocal images were performed largely through the use of the Vanderbilt University Medical Center Cell Imaging Shared Resource. This work was supported by NIH award MH073159 to RDB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apparsundaram S, Ferguson SM, Blakely RD. Molecular cloning and characterization of a murine hemicholinium-3-sensitive choline transporter. Biochem Soc Trans. 2001;29:711–716. doi: 10.1042/0300-5127:0290711. [DOI] [PubMed] [Google Scholar]
- Arber S, Han B, Mendelsohn M, Smith M, Jessell TM, Sockanathan S. Requirement for the homeobox gene Hb9 in the consolidation of motor neuron identity. Neuron. 1999;23:659–674. doi: 10.1016/s0896-6273(01)80026-x. [DOI] [PubMed] [Google Scholar]
- Asawa T, Shindo M, Momoi H. Compound muscle action potentials during repetitive nerve stimulation. Muscle Nerve. 2004;29:724–728. doi: 10.1002/mus.20037. [DOI] [PubMed] [Google Scholar]
- Barker LA, Mittag TW. Comparative studies of substrates and inhibitors of choline transport and choline acetyltransferase. J Pharmacol Exp Ther. 1975;192:86–94. [PubMed] [Google Scholar]
- Bazalakova MH, Blakely RD. The high-affinity choline transporter: a critical protein for sustaining cholinergic signaling as revealed in studies of genetically altered mice. Handb Exp Pharmacol. 2006:525–544. doi: 10.1007/3-540-29784-7_21. [DOI] [PubMed] [Google Scholar]
- Bazalakova MH, Wright J, Schneble EJ, McDonald MP, Heilman CJ, Levey AI, Blakely RD. Deficits in acetylcholine homeostasis, receptors and behaviors in choline transporter heterozygous mice. Genes Brain Behav. 2006 doi: 10.1111/j.1601-183X.2006.00269.x. [DOI] [PubMed] [Google Scholar]
- Beeri R, Le Novere N, Mervis R, Huberman T, Grauer E, Changeux JP, Soreq H. Enhanced hemicholinium binding and attenuated dendrite branching in cognitively impaired acetylcholinesterase-transgenic mice. J Neurochem. 1997;69:2441–2451. doi: 10.1046/j.1471-4159.1997.69062441.x. [DOI] [PubMed] [Google Scholar]
- Birks R, MacIntosh F. Acetylcholine metabolism of a sympathetic ganglion. Can J Physiol Pharmacol. 1961;39:787–827. [Google Scholar]
- Bonzelius F, Zimmermann H. Recycled synaptic vesicles contain vesicle but not plasma membrane marker, newly synthesized acetylcholine, and a sample of extracellular medium. J Neurochem. 1990;55:1266–1273. doi: 10.1111/j.1471-4159.1990.tb03134.x. [DOI] [PubMed] [Google Scholar]
- Brandon EP, Lin W, D’Amour KA, Pizzo DP, Dominguez B, Sugiura Y, Thode S, Ko CP, Thal LJ, Gage FH, Lee KF. Aberrant patterning of neuromuscular synapses in choline acetyltransferase-deficient mice. J Neurosci. 2003;23:539–549. doi: 10.1523/JNEUROSCI.23-02-00539.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandon EP, Mellott T, Pizzo DP, Coufal N, D’Amour KA, Gobeske K, Lortie M, Lopez-Coviella I, Berse B, Thal LJ, Gage FH, Blusztajn JK. Choline transporter 1 maintains cholinergic function in choline acetyltransferase haploinsufficiency. J Neurosci. 2004;24:5459–5466. doi: 10.1523/JNEUROSCI.1106-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byun N, Delpire E. Axonal and periaxonal swelling precede peripheral neurodegeneration in KCC3 knockout mice. Neurobiol Dis. 2007;28:39–51. doi: 10.1016/j.nbd.2007.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole CR, Blackstone EH, Pashkow FJ, Snader CE, Lauer MS. Heart-rate recovery immediately after exercise as a predictor of mortality. N Engl J Med. 1999;341:1351–1357. doi: 10.1056/NEJM199910283411804. [DOI] [PubMed] [Google Scholar]
- De Paepe B, De Bleecker JL, Van Coster R. Histochemical methods for the diagnosis of mitochondrial diseases. Curr Protoc Hum Genet. 2009;Chapter 19(Unit19):12. doi: 10.1002/0471142905.hg1902s63. [DOI] [PubMed] [Google Scholar]
- English BA, Appalsamy M, Diedrich A, Ruggiero AM, Lund D, Wright J, Keller NR, Louderback KM, Robertson D, Blakely RD. Tachycardia, reduced vagal capacity, and age-dependent ventricular dysfunction arising from diminished expression of the presynaptic choline transporter. Am J Physiol Heart Circ Physiol. 2010;299:H799–810. doi: 10.1152/ajpheart.00170.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett AW, Ernst EJ. Increased quantal size in transmission at slow but not fast neuromuscular synapses of apolipoprotein E deficient mice. Exp Neurol. 2004;185:290–296. doi: 10.1016/j.expneurol.2003.10.006. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Bazalakova M, Savchenko V, Tapia JC, Wright J, Blakely RD. Lethal impairment of cholinergic neurotransmission in hemicholinium-3-sensitive choline transporter knockout mice. Proc Natl Acad Sci U S A. 2004;101:8762–8767. doi: 10.1073/pnas.0401667101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson SM, Blakely RD. The choline transporter resurfaces: new roles for synaptic vesicles? Mol Interv. 2004;4:22–37. doi: 10.1124/mi.4.1.22. [DOI] [PubMed] [Google Scholar]
- Ferguson SM, Savchenko V, Apparsundaram S, Zwick M, Wright J, Heilman CJ, Yi H, Levey AI, Blakely RD. Vesicular Localization and Activity-Dependent Trafficking of Presynaptic Choline Transporters. J Neurosci. 2003;23:9697–9709. doi: 10.1523/JNEUROSCI.23-30-09697.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong SW, McLennan IS, McIntyre A, Reid J, Shennan KI, Bewick GS. TGF-beta2 alters the characteristics of the neuromuscular junction by regulating presynaptic quantal size. Proc Natl Acad Sci U S A. 2010;107:13515–13519. doi: 10.1073/pnas.1001695107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frick KM, Burlingame LA, Delaney SS, Berger-Sweeney J. Sex differences in neurochemical markers that correlate with behavior in aging mice. Neurobiol Aging. 2002;23:145–158. doi: 10.1016/s0197-4580(01)00237-8. [DOI] [PubMed] [Google Scholar]
- Gaffield MA, Tabares L, Betz WJ. The spatial pattern of exocytosis and post-exocytic mobility of synaptopHluorin in mouse motor nerve terminals. J Physiol. 2009;587:1187–1200. doi: 10.1113/jphysiol.2008.166728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gracz LM, Wang WC, Parsons SM. Cholinergic synaptic vesicle heterogeneity: evidence for regulation of acetylcholine transport. Biochemistry. 1988;27:5268–5274. doi: 10.1021/bi00414a048. [DOI] [PubMed] [Google Scholar]
- Gras C, Amilhon B, Lepicard EM, Poirel O, Vinatier J, Herbin M, Dumas S, Tzavara ET, Wade MR, Nomikos GG, Hanoun N, Saurini F, Kemel ML, Gasnier B, Giros B, El Mestikawy S. The vesicular glutamate transporter VGLUT3 synergizes striatal acetylcholine tone. Nat Neurosci. 2008;11:292–300. doi: 10.1038/nn2052. [DOI] [PubMed] [Google Scholar]
- Haga T. Synthesis and release of ( 14 C)acetylcholine in synaptosomes. J Neurochem. 1971;18:781–798. doi: 10.1111/j.1471-4159.1971.tb12008.x. [DOI] [PubMed] [Google Scholar]
- Hicks A, Fenton J, Garner S, McComas AJ. M wave potentiation during and after muscle activity. J Appl Physiol. 1989;66:2606–2610. doi: 10.1152/jappl.1989.66.6.2606. [DOI] [PubMed] [Google Scholar]
- Kaja S, van de Ven RC, van Dijk JG, Verschuuren JJ, Arahata K, Frants RR, Ferrari MD, van den Maagdenberg AM, Plomp JJ. Severely impaired neuromuscular synaptic transmission causes muscle weakness in the Cacna1a-mutant mouse rolling Nagoya. Eur J Neurosci. 2007;25:2009–2020. doi: 10.1111/j.1460-9568.2007.05438.x. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy A, Cooper E. An activity-dependent retrograde signal induces the expression of the high-affinity choline transporter in cholinergic neurons. Neuron. 2009;61:272–286. doi: 10.1016/j.neuron.2008.11.025. [DOI] [PubMed] [Google Scholar]
- Lee C, Barnes A, Yang E, Katz RL. Neuromuscular facilitation during train-of-four and tetanic stimulation in healthy volunteers: observations with half-refractory paired responses. Br J Anaesth. 1977;49:555–560. doi: 10.1093/bja/49.6.555. [DOI] [PubMed] [Google Scholar]
- Lentz M, Nielsen JF. Post-exercise facilitation and depression of M wave and motor evoked potentials in healthy subjects. Clin Neurophysiol. 2002;113:1092–1098. doi: 10.1016/s1388-2457(02)00031-7. [DOI] [PubMed] [Google Scholar]
- Lerman I, Harrison BC, Freeman K, Hewett TE, Allen DL, Robbins J, Leinwand LA. Genetic variability in forced and voluntary endurance exercise performance in seven inbred mouse strains. J Appl Physiol. 2002;92:2245–2255. doi: 10.1152/japplphysiol.01045.2001. [DOI] [PubMed] [Google Scholar]
- Lightfoot JT, Turner MJ, Debate KA, Kleeberger SR. Interstrain variation in murine aerobic capacity. Med Sci Sports Exerc. 2001;33:2053–2057. doi: 10.1097/00005768-200112000-00012. [DOI] [PubMed] [Google Scholar]
- Massett MP, Berk BC. Strain-dependent differences in responses to exercise training in inbred and hybrid mice. Am J Physiol Regul Integr Comp Physiol. 2005;288:R1006–1013. doi: 10.1152/ajpregu.00476.2004. [DOI] [PubMed] [Google Scholar]
- Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR. Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron. 2002;36:635–648. doi: 10.1016/s0896-6273(02)01020-6. [DOI] [PubMed] [Google Scholar]
- Molenaar PC. A relative weak leg muscle in the rolling Nagoya mouse as a model for Lambert-Eaton myasthenic syndrome. J Neuroimmunol. 2008;201–202:166–171. doi: 10.1016/j.jneuroim.2008.05.027. [DOI] [PubMed] [Google Scholar]
- Nakata K, Okuda T, Misawa H. Ultrastructural localization of high-affinity choline transporter in the rat neuromuscular junction: enrichment on synaptic vesicles. Synapse. 2004;53:53–56. doi: 10.1002/syn.20029. [DOI] [PubMed] [Google Scholar]
- Oliveira L, Correia-de-Sa P. Dissociation between M1-facilitation of acetylcholine release and crosstalk with A2A- and M2-receptors on rat motoneurons. Signal Transduct. 2006;6:19–31. [Google Scholar]
- Schwab RS, Osserman KE, Tether JE. Treatment of myasthenia gravis; prolonged action with multiple-dose tablets of neostigmine bromide and mestinon bromide. J Am Med Assoc. 1957;165:671–674. doi: 10.1001/jama.1957.02980240029008. [DOI] [PubMed] [Google Scholar]
- Schwartz P, Vanoli E, Stramba-Badiale M, De Ferrari G, Billman G, Foreman R. Autonomic mechanisms and sudden death. New insights from analysis of baroreceptor reflexes in conscious dogs with and without a myocardial infarction. Circulation. 1988;78:969–979. doi: 10.1161/01.cir.78.4.969. [DOI] [PubMed] [Google Scholar]
- Shao XM, Feldman JL. Cholinergic neurotransmission in the preBotzinger Complex modulates excitability of inspiratory neurons and regulates respiratory rhythm. Neuroscience. 2005;130:1069–1081. doi: 10.1016/j.neuroscience.2004.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman JD, Argov Z, McKee D, Hazum E, Brawer S, Soreq H. Antisense treatment for myasthenia gravis: experience with monarsen. Ann N Y Acad Sci. 2008;1132:283–290. doi: 10.1196/annals.1405.022. [DOI] [PubMed] [Google Scholar]
- Thaler J, Harrison K, Sharma K, Lettieri K, Kehrl J, Pfaff SL. Active suppression of interneuron programs within developing motor neurons revealed by analysis of homeodomain factor HB9. Neuron. 1999;23:675–687. doi: 10.1016/s0896-6273(01)80027-1. [DOI] [PubMed] [Google Scholar]
- Vaseghi M, Shivkumar K. The Role of the Autonomic Nervous System in Sudden Cardiac Death. Prog Cardiovasc Dis. 50:404–419. doi: 10.1016/j.pcad.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Hrabovska A, Duysen EG, Ferguson SM, Blakely RD, Lockridge O, Levey AI. Altered striatal function and muscarinic cholinergic receptors in acetylcholinesterase knockout mice. Mol Pharmacol. 2003;64:1309–1316. doi: 10.1124/mol.64.6.1309. [DOI] [PubMed] [Google Scholar]
- Wessler I, Kilbinger H. Release of [3H]acetylcholine from a modified rat phrenic nerve-hemidiaphragm preparation. Naunyn Schmiedebergs Arch Pharmacol. 1986;334:357–364. doi: 10.1007/BF00569370. [DOI] [PubMed] [Google Scholar]
- Wessler I, Sandmann J. Uptake and metabolism of [3H]choline by the rat phrenic nerve-hemidiaphragm preparation. Naunyn Schmiedebergs Arch Pharmacol. 1987;335:231–237. doi: 10.1007/BF00172789. [DOI] [PubMed] [Google Scholar]
- Wilson JM, Hartley R, Maxwell DJ, Todd AJ, Lieberam I, Kaltschmidt JA, Yoshida Y, Jessell TM, Brownstone RM. Conditional rhythmicity of ventral spinal interneurons defined by expression of the Hb9 homeodomain protein. J Neurosci. 2005;25:5710–5719. doi: 10.1523/JNEUROSCI.0274-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SJ, Slater CR. Safety factor at the neuromuscular junction. Prog Neurobiol. 2001;64:393–429. doi: 10.1016/s0301-0082(00)00055-1. [DOI] [PubMed] [Google Scholar]
- Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37:475–524. doi: 10.1016/0301-0082(91)90006-m. [DOI] [PubMed] [Google Scholar]
- Wyatt RM, Balice-Gordon RJ. Heterogeneity in synaptic vesicle release at neuromuscular synapses of mice expressing synaptopHluorin. J Neurosci. 2008;28:325–335. doi: 10.1523/JNEUROSCI.3544-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SP, Van der Kloot W. Increasing quantal size at the mouse neuromuscular junction and the role of choline. J Physiol. 1991;433:677–704. doi: 10.1113/jphysiol.1991.sp018450. [DOI] [PMC free article] [PubMed] [Google Scholar]