

Abstract
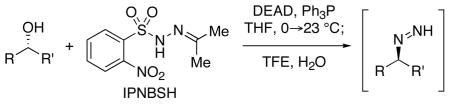
The reagent N-isopropylidene-N′-2-nitrobenzenesulfonyl hydrazine (IPNBSH) is used in the reduction of alcohols via the loss of dinitrogen from transiently formed monoalkyl diazene intermediates accessed by sequential Mitsunobu displacement, hydrolysis and fragmentation under mild reaction conditions.
The loss of dinitrogen from monoalkyl diazene intermediates is common in a wide range of transformations in organic chemistry.1,2 Several powerful methodologies for carbonyl reduction involve initial condensation with an arenesulfonyl hydrazine followed by reduction of the corresponding hydrazone leading to the loss of dinitrogen. In 1996, Myers reported a highly efficient, mild, and stereospecific conversion of a variety of propargylic alcohols to the corresponding allenes3a via a Mitsunobu4 reaction using the reagent 2-nitrobenzenesulfonyl hydrazide5 (NBSH). Subsequent reports discussed the use of NBSH in the reduction of allylic, benzylic, and saturated alcohols.3b–c This direct reduction of alcohols via the corresponding monoalkyl diazene intermediates under mild reaction conditions presents a highly versatile methodology for organic synthesis.6 We recently reported the use of the related reagent N-isopropylidene- N′-2-nitrobenzene-sulfonyl hydrazine (IPNBSH) for a difficult allylic reductive transposition step in our total syntheses of (−)-acylfulvene and (−)-irofulven.7 Herein we report our results on the general utility of IPNBSH, a complimentary reagent to NBSH, for conversion of alcohols to the corresponding monoalkyl diazene intermediates.
The stereospecific displacement of an alcohol by the reagent NBSH under the Mitsunobu reaction conditions affords the corresponding 1,1-disubstituted sulfonyl hydrazine 2 (Scheme 1).3
SCHEME 1.

Warming of the reaction mixture to ambient temperature provides the corresponding monoalkyl diazene 3 by elimination of 2-nitrobenzenesulfinic acid. Sigmatropic3a–b loss of dinitrogen from the unsaturated monoalkyl diazene, or expulsion of dinitrogen via a free-radical3c pathway from the saturated monoalkyl diazene, affords the corresponding reduction product. The thermal sensitivity of NBSH in solution and the corresponding Mitsunobu adduct 2 necessitate the execution of the initial step in these transformations at sub-ambient temperatures (−30 to −15 °C). Lower reaction temperatures obviate a competitive and undesired Mitsunobu reaction of the alcohol substrate with 2-nitrobenzenesulfinic acid, the thermal decomposition8 product of NBSH or adduct 2.3 For less reactive alcohols, the use of higher substrate concentrations and excess reagents in N-methyl morpholine has been described.3b–c
We recently found the use of the reagent IPNBSH to be advantageous over NBSH in a surprisingly difficult9 reductive allylic transposition reaction.7 In reduction of the substrate in entry 1 of Table 1, IPNBSH provided greater flexibility with respect to solvent choice, reaction temperature, order of addition, and concentration of substrate and reagents. As shown in Scheme 1, the Mitsunobu displacement of an alcohol with IPNBSH results in the stable and isolable arenesulfonyl hydrazone 4. We developed mild reaction conditions for in situ hydrolysis of hydrazone 4 to the same hydrazine 2 accessed using NBSH (Scheme 1).
TABLE 1.
Reduction of Alcohols using IPNBSHa
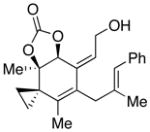

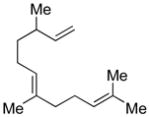
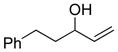
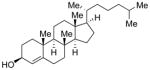
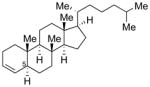
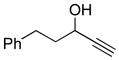
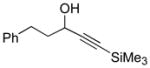
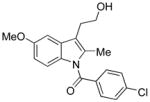
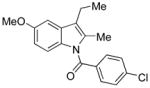
Conditions: Ph3P (1.2 equiv), DEAD (1.2 equiv), IPNBSH (1.2 equiv), THF [0.1M], 0→23°C, 2h; TFE–H2O (1:1), 2h.
Average of two experiments.
Reference 7.
Hydrolysis step required 3 h.
The Mitsunobu reaction was complete in 15 min.
E:Z = 93:7.
2.0 equiv of reagents was used; contains <8% of C5-diastereomer.
1.6 equiv of reagents was used; PhNHNH2 (5.0 equiv) was used in place of TFE–H2O.
 |
(1) |
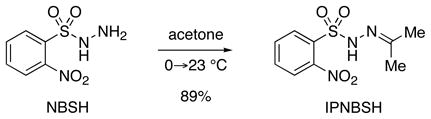
IPNBSH can be prepared by dissolution of the readily available NBSH5 in acetone (eq 1). For optimal results, IPNBSH is triturated with hexanes to give the desired reagent as a white solid. Comparison of the X-ray structure of IPNBSH10 with that of the closely related acetone p-toluenesulfonyl hydrazone11 reveals longer C–S (1.774 vs. 1.753 Å) and slightly shorter N(2)–S (1.636 vs. 1.637 Å) bonds consistent with greater interaction between the nitrogen(2) and sulfur in IPNBSH. The smaller N–N–S bond angle found in IPNBSH (112.4° vs. 114.1°) was expected to reduce the steric interaction of the syn-coplanar N–H and C-Me groups.
Significantly, the greater thermal stability of IPNBSH compared to NBSH was expected to provide flexibility for the Mitsunobu reaction while offering equally rapid thermal fragmentation after hydrolysis of the adduct 4 (Scheme 1). For comparison, heating a solution of IPNBSH in DMSO-d6 [0.02M] at 50 °C for 30 minutes did not result in decomposition while a significant quantity of NBSH (~60%) was found to fragment under the same conditions. Solutions of IPNBSH as described above did not decompose at 75 or 100 °C after 30 min whereas considerable decomposition was observed at 150 °C within minutes. The greater thermal stability of IPNBSH allows it to be stored at room temperature for several months.5b–c, 12
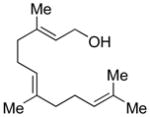
The utility of IPNBSH in the challenging7 reductive allylic transposition mentioned above prompted our evaluation of this reagent’s broader utility for organic synthesis. Addition of diethylazadicarboxylate (DEAD, 1.20 equiv) to a solution of trans,trans-farnesol (1a, 1 equiv, Scheme 2), IPNBSH (1.21 equiv), and triphenylphosphine (Ph3P, 1.21 equiv) in THF (9.0 mL) rapidly (15 min) provided the desired Mitsunobu adduct 4a in 86% isolated yield. The isolation of 4a is not necessary and its direct hydrolysis under optimal conditions was achieved by introduction of trifluoroethanol–water (TFE–H2O, 1:1) upon completion of the Mitsunobu reaction.13 The hydrolysis step results in fragmentation of 2-nitrobenzenesulfinic acid followed by sigmatropic loss of dinitrogen to give the triene 5a in 87% isolated yield. The ease of hydrolysis of these hydrazones under mild conditions is likely due to the rapid fragmentation of the corresponding sulfonyl hydrazine derivatives at room temperature.14
SCHEME 2.

A uniform set of reaction conditions proved effective for a single-step reduction of a range of alcohols (Table 1). Allylic alcohols (Table 1, entries 1–5) and propargylic alcohols (Table 1, entries 6–7) provided the desired reduction products via the expected sigmatropic loss of dinitrogen from the intermediate monoalkyl diazenes generated by in situ hydrolysis. For unhindered primary alcohols the reaction could be conducted at even lower concentration (0.05M) and sub-ambient temperatures with equal efficiency. The use of IPNBSH allowed the use of other solvents (e.g., toluene, fluorobenzene, and chlorobenzene) in place of THF with similar results.
Interestingly, while the Mitsunobu reaction with the primary alcohol in entry 8 of Table 1 proceeded smoothly under standard conditions (adduct isolated in 93% yield), the hydrolysis of the corresponding hydrazone adduct 4 under the conditions used for propargylic and allylic substrates proved ineffective, affording less than 20% yield of the desired reduction product.15
We reasoned that while slow hydrolysis and generation of unsaturated (propargylic and allylic) monoalkyl diazenes led to an efficient sigmatropic loss of dinitrogen, the loss of dinitrogen from saturated monoalkyl diazenes would be optimal at higher concentrations of the diazene intermediate.16 The replacement of the hydrolysis step with a hydrazone exchange reaction (phenylhydrazine), reduced the time for the complete consumption of the Mitsunobu adduct from 4h to 30 min (TLC analysis).17 The limitation of IPNBSH is its greater sensitivity toward sterically hindered substrates as compared to NBSH. For example, the Mitsunobu reaction with the saturated secondary alcohol in entry 9 of Table 1 proved difficult as compared to the reaction of unsaturated secondary alcohols (Table 1, entries 4–7).18 The use of more forcing reaction conditions with the alcohol in entry 9 of Table 1 did not increase the isolated yield of the desired product and returned the starting alcohol.19
The thermal stability of IPNBSH allows for unique transformations such as the one shown in equation 2. N-Alkylation of the corresponding sodium sulfonamide of IPNBSH with allylic bromide 6 followed by in situ hydrolysis afforded the desired terminal alkene 5a via sigmatropic loss of dinitrogen. This single-step N-alkylation–reduction strategy offers a valuable alternative to the approach based on the Mitsunobu reaction.9a
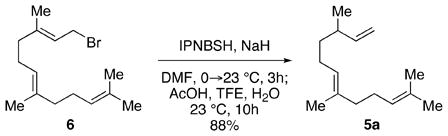 |
(2) |
In addition to IPNBSH, we examined a series of other hydrazone derivatives as potential reagents for the conversion of alcohols to the corresponding monoalkyl diazenes. These included the 2-nitrobenzenesulfonyl hydrazones of trifluoroacetone, dichloroacetone, cyclobutanone, benzaldehyde, acetaldehyde, and trimethylacetaldehyde, in addition to methanesulfonyl hydrazones of acetone and benzaldehyde. IPNBSH was identified as the best reagent based on optimal reactivity in the Mitsunobu reaction and ease of hydrolysis of the corresponding adduct. Interestingly, while the Mitsunobu reaction of the aldehyde hydrazone derivatives proceeded with equal efficiency as IPNBSH their hydrolysis gave the corresponding carbonyl hydrazide and not the expected reduction product (Scheme 3). This is likely due to isomerization20 of transient α-hydroxyalkyldiazene intermediates 10a–b.
SCHEME 3.

In conclusion, the reagent IPNBSH serves as a complementary reagent to NBSH for conversion of alcohols to the corresponding reduction products via monoalkyl diazene intermediates. Attractive features of this reagent include ease of preparation, storage, and use due to excellent thermal stability. IPNBSH offers flexibility with respect to choice of reaction solvent, reaction temperature, order of addition, and concentration of substrate and reagents in the Mitsunobu reaction. We expect IPNBSH will find applications in unique transformations such as that illustrated in equation 2.
Experimental Section
(E)-3,7,11-Trimethyldodeca-1,6,10-triene (5a, Table 1, entry 3)
DEAD (74 μL, 0.47 mmol, 1.2 equiv) was added drop-wise to a solution of IPNBSH (122 mg, 0.474 mmol, 1.21 equiv), trans,trans-farnesol (1a, 0.100 mL, 0.393 mmol, 1 equiv), and triphenylphosphine (124 mg, 0.473 mmol, 1.21 equiv) in anhydrous THF (9.0 mL) at 0 °C under an argon atmosphere. After 5 min, the reaction mixture was allowed to warm to 23 °C. After 20 min, a mixture of trifluoroethanol and water (1:1, 4.5 mL) was added to the reaction mixture to enable formation of the allylic diazene intermediate. After 3 h, the reaction mixture was partitioned between diethyl ether (25 mL) and water (25 mL), and the aqueous layer was extracted with diethyl ether (2 × 25 mL). The combined organic layers were dried over anhydrous sodium sulfate, were filtered, and were concentrated. The residue was purified by flash column chromatography on silica gel (100% npentane) to give triene 5a (71 mg, 87%). All spectroscopic data were in agreement with literature.21 TLC (40% ethyl acetate in hexanes), Rf: 0.8 (CAM, KMnO4). 1H NMR (400 MHz, CDCl3, 20 °C): 5.25–5.15 (m, 1H), 5.13–5.10 (m, 2H), 4.99 (t, 1H, J = 2.0 Hz), 4.94–4.91 (m, 2H), 2.14–1.98 (m, 7H), 1.69 (s, 3H), 1.61 (s, 3H), 1.60 (s, 3H), 1.36–1.33 (m, 2H), 0.99 (d, 3H, J = 8 Hz). 13C NMR (500 MHz, CDCl3, 20 °C): 145.0, 135.1, 131.5, 124.7, 124.6, 112.7, 39.9, 37.5, 36.9, 26.9, 25.9, 25.8, 20.4, 20.4, 17.9, 16.2. MS (m/z) C15H26 [M]+: 206. FTIR (thin film): 3077 (m), 2966 (s), 2915 (s), 2856 (s), 1640 (m), 1453 (s), 1376 (s).
5-Phenylpenta-1,2-diene (Table 1, Entry 6)
DEAD (67.5 μL, 0.429 mmol, 1.20 equiv) was added drop-wise to a solution of IPNBSH (110 mg, 0.429 mmol, 1.20 equiv), 5-phenylpent-1-yn-3-ol (57 mg, 0.36 mmol, 1 equiv), and triphenylphosphine (112 mg, 0.428 mmol, 1.20 equiv) in anhydrous THF (3.5 mL) at 0 °C under an argon atmosphere. After 5 min, the reaction mixture was allowed to warm to 23 °C. After 2 h, a mixture of trifluroethanol and water (1:1, 1.7 mL) was added to the reaction mixture to enable the formation of the propargylic diazene intermediate. After 2 h, the reaction mixture was partitioned between npentane (25 mL) and water (25 mL). The organic layer was washed with water (2 × 25mL), was dried over anhydrous sodium sulfate, was filtered, and was concentrated. The residue was purified by flash column chromatography on silica gel (100% npentane) to give the allene (36 mg, 70%). All spectroscopic data were in agreement with the literature.22 1H NMR (400 MHz, CDCl3, 20°C): 7.31–7.28 (m, 2H), 7.22–7.21 (m, 3H), 5.17 (tt, 1H, J = 13.2, 6.8 Hz), 4.69 (dt, 2H, J = 6.8, 3.2 Hz), 2.75 (t, 2H, J = 7.6 Hz), 2.35–2.32 (m, 2H). 13C NMR (500 MHz, CDCl3, 20°C): 208.8, 142.0, 128.7, 128.5, 126.1, 89.6, 75.3, 35.6, 30.2. MS (m/z) C11H12 [M]+: 144. FTIR (thin film): 3027 (s), 2922 (s), 2855 (s), 1955 (s), 1495 (m), 1453 (m).
Supplementary Material
Acknowledgments
M.M. is a Dale F. and Betty Ann Frey Damon Runyon Scholar supported by the Damon Runyon Cancer Research Foundation (DRS-39-04). We thank Mr. Michael A. Schmidt and Dr. Peter Mueller for X-ray crystallographic analysis of IPNBSH. We acknowledge financial support by NIH-NIGMS (GM074825), Paul M. Cook Fund, Firmenich, Sigma–Aldrich, Amgen, and Boehringer Ingelheim Pharmaceutical Inc.
Footnotes
Supporting Information Available: Experimental procedures, spectroscopic data for all new compounds, and the X-ray structure of IPNBSH. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.For representative examples, see: Szmant HH, Harnsberger HF, Butler TJ, Barie WP. J Org Chem. 1952;74:2724.Nickon A, Hill AS. J Am Chem Soc. 1964;86:1152.Corey EJ, Cane DE, Libit L. J Am Chem Soc. 1971;93:7016.Hutchins RO, Kacher M, Rua L. J Org Chem. 1975;40:923.Kabalka GW, Chandler JH. Synth Commun. 1979;9:275.Corey EJ, Wess G, Xiang YB, Singh AK. J Am Chem Soc. 1987;109:4717.Myers AG, Kukkola PJ. J Am Chem Soc. 1990;112:8208.Myers AG, Finney NS. J Am Chem Soc. 1990;112:9641.Guziec FS, Jr, Wei D. J Org Chem. 1992;57:3772.Wood JL, Porco JA, Jr, Taunton J, Lee AY, Clardy J, Schreiber SL. J Am Chem Soc. 1992;114:5898.Bregant TM, Groppe J, Little RD. J Am Chem Soc. 1994;116:3635.Ott GR, Heathcock CH. Org Lett. 1999;1:1475. doi: 10.1021/ol990281j.Chai Y, Vicic DA, McIntosh MC. Org Lett. 2003;5:1039. doi: 10.1021/ol034052f.Sammis GM, Flamme EM, Xie H, Ho DM, Sorensen EJ. J Am Chem Soc. 2005;127:8612. doi: 10.1021/ja052383c.
- 2.(a) Kosower EM. Acc Chem Res. 1971;4:193. [Google Scholar]; (b) Tsuji T, Kosower EM. J Am Chem Soc. 1971;93:1992. [Google Scholar]
- 3.(a) Myers AG, Zheng B. J Am Chem Soc. 1996;118:4492. [Google Scholar]; (b) Myers AG, Zheng B. Tetrahedron Lett. 1996;37:4841. [Google Scholar]; (c) Myers AG, Movassaghi M, Zheng B. J Am Chem Soc. 1997;119:8572. [Google Scholar]
- 4.(a) Mitsunobu O. Synthesis. 1981:1. [Google Scholar]; (b) Hughes DL. Org React. 1982;42:335. [Google Scholar]
- 5.(a) Dann AT, Davies W. J Chem Soc. 1929:1050. [Google Scholar]; (b) Myers AG, Zheng B, Movassaghi M. J Org Chem. 1997;62:7507. doi: 10.1021/jo9710137. [DOI] [PubMed] [Google Scholar]; (c) Myers AG, Movassaghi M. In: e- Encyclopedia of Reagents for Organic Synthesis. Paquette LA, editor. John Wiley & Sons; 2003. [Google Scholar]
- 6.For examples in the utility of NBSH in synthesis, see: Shepard MS, Carreira EM. J Am Chem Soc. 1997;119:2597.Corey EJ, Huang AX. J Am Chem Soc. 1999;121:710.Arredondo VM, Tian S, McDonald FE, Marks TJ. J Am Chem Soc. 1999;121:3633.Kozmin SA, Rawal VH. J Am Chem Soc. 1999;121:9562.Charette AB, Jolicoeur E, Bydlinski GAS. Org Lett. 2001;3:3293. doi: 10.1021/ol0165140.Haukaas MH, O’Doherty GA. Org Lett. 2002;4:1771. doi: 10.1021/ol025844x.Regás D, Afonso MM, Rodríguez ML, Palenzuela JA. J Org Chem. 2003;68:7845. doi: 10.1021/jo034480z.McGrath MJ, Fletcher MT, König WA, Moore CJ, Cribb BW, Allsopp PG, Kitching W. J Org Chem. 2003;68:3739. doi: 10.1021/jo026213j.Ng SS, Jamison TF. Tetrahedron. 2005;61:11405.Michael FE, Duncan AP, Sweeney ZK, Bergman RG. J Am Chem Soc. 2005;127:1752. doi: 10.1021/ja045607k.Charest MG, Lerner CD, Brubaker JD, Siegel DR, Myers AG. Science. 2005;308:395. doi: 10.1126/science.1109755.
- 7.Movassaghi M, Piizzi G, Siegel DS, Piersanti G. Angew Chem Int Ed. 2006;45:5859. doi: 10.1002/anie.200602011. [DOI] [PubMed] [Google Scholar]
- 8.Hünig S, Müller HR, Thier W. Angew Chem, Int Ed Engl. 1965;4:271. [Google Scholar]
- 9.For other examples, see: Ott GR, Heathcock CH. Org Lett. 1999;1:1475. doi: 10.1021/ol990281j.The use of high reaction temperatures described in Vostrikov NS, Vasikov VZ, Miftakhov MS. Russ J Org Chem. 2005;41:967.likely cause decomposition of NBSH.
- 10.See the Supporting Information for the crystal structure of IPNBSH.
- 11.Ojala CR, Ojala WH, Pennamon SY, Gleason WB. Acta Cryst. 1998;C54:57. [Google Scholar]
- 12.Due to potential toxicity and flammability of IPNBSH similar to related arenesulfonylhydrazides (e.g., p-toluenesulfonylhydrazide), prudent experimental practices should be followed in handling of IPNBSH with gloves under inert atmosphere. See Paquette LA. Encyclopedia of Reagents for Organic Synthesis. Vol. 7. John Wiley & Sons; 1995. p. 4953.
- 13.These conditions for the hydrolysis step were found to be superior to others explored including the use of substoichiometric quantities of acetic acid, Sc(OTf)3, or use of other co-solvents (MeOH and EtOH).
- 14.Consistent with this hypothesis, hydrolysis of the Mitsunobu adducts of secondary alcohols is faster than those derived from primary alcohols.
- 15.Addition of acetic acid, 2,6-lutidine, or 1,4-cyclohexadiene, and the use of ethanedithiol instead of water did not improve the yield of the reduction product.
- 16.A faster release of the sulfonyl hydrazine derivative 2 and formation of the diazene intermediate 3 could provide a more efficient free-radical loss of dinitrogen. Please see reference 2 and Myers AG, Movassaghi M, Zheng B. Tetrahedron Lett. 1997;38:6569.
- 17.The addition of phenylhydrazine was only necessary in the deoxygenation of saturated alcohols.
- 18.For comparison, the use of NBSH in the reduction of the saturated secondary alcohol in entry 9 of Table 1 under optimum reaction conditions provided the desired deoxygenated product in 80% yield. See reference 3c.
- 19.Unfortunately, various procedural changes described in a more recent and related report Keith JM, Gomez L. J Org Chem. 2006;71:7113. doi: 10.1021/jo061185g. did not provide any advantage over our original reaction conditions in reference 7 for the use of IPNBSH in the Mitsunobu reaction.
- 20.Tezuka T, Otsuka T, Wang PC, Murata M. Tetrahedron Lett. 1986;27:3627. [Google Scholar]
- 21.Saplay KM, Sahni R, Damodaran NP, Dev S. Tetrahedron. 1980;36:1455. [Google Scholar]
- 22.Ohno H, Miyamura K, Tanaka T. J Org Chem. 2002;67:1359. doi: 10.1021/jo016320y. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.