

Abstract
BACKGROUND AND PURPOSE
Inflammatory pain is triggered by activation of pathways leading to the release of mediators such as bradykinin, prostaglandins, interleukins, ATP, growth factors and protons that sensitize peripheral nociceptors. The activation of acid-sensitive ion channels (ASICs) may have particular relevance in the development and maintenance of inflammatory pain. ASIC3 is of particular interest due to its restricted tissue distribution in the nociceptive primary afferent fibres and its high sensitivity to protons.
EXPERIMENTAL APPROACH
To examine the contribution of ASIC3 to the development and maintenance of muscle pain and inflammatory pain, we studied the in vivo efficacy of a selective ASIC3 inhibitor, APETx2, in rats.
KEY RESULTS
Administration of APETx2 into the gastrocnemius muscle prior to the administration of low pH saline prevented the development of mechanical hypersensitivity, whereas APETx2 administration following low-pH saline was ineffective in reversing hypersensitivity. The prevention of mechanical hypersensitivity produced by acid administration was observed whether APETx2 was applied via i.m. or i.t. routes. In the complete Freund's adjuvant (CFA) inflammatory pain model, local administration of APETx2 resulted in a potent and complete reversal of established mechanical hypersensitivity, whereas i.t. application of APETx2 was ineffective.
CONCLUSIONS AND IMPLICATIONS
ASIC3 contributed to the development of mechanical hypersensitivity in the acid-induced muscle pain model, whereas ASIC3 contributed to the maintenance of mechanical hypersensitivity in the CFA inflammatory pain model. The contribution of ASIC3 to established hypersensitivity associated with inflammation suggests that this channel may be an effective analgesic target for inflammatory pain states.
Keywords: inflammatory pain, acid-sensitive ion channels, amiloride, APETx2
Introduction
Inflammatory pain results from a cascade of cellular events initiated by tissue injury. Inflammatory mediators, including bradykinin, prostaglandins, growth factors, endothelin, interleukins, ATP and protons, are released at the site of injury and sensitize peripheral nociceptive neurons. The central projections of these peripheral nociceptors transmit this information to second-order neurons within the spinal cord (Julius and Basbaum, 2001). Ion channels expressed in both peripheral and central compartments of primary afferent nociceptors play an important role in pain signalling. Following tissue injury and inflammation, extracellular pH decreases due to the immune response, leak of intracellular components, hypoxic metabolism and related lactic acid production. Several studies have supported a direct link between tissue acidosis and the development of pain. In rats, a surgical incision resulted in a rapid drop of tissue pH. The development of acidosis was concomitant with observable pain behaviour, and this behaviour was diminished when pH returned to pre-incision value. (Woo et al., 2004). Repeated i.m. injections of acidified saline caused hyperalgesia that could last several days or weeks (Sluka et al., 2001). In human subjects, the direct subdermal injection of acidic saline resulted in development of localized pain (Ugawa et al., 2002). Activation of pain pathways by low pH is most likely receptor mediated, and several molecular targets for protons have been implicated to date, including ion channels belonging to three distinct classes: the acid-sensing ion channels (ASICs), vanilloid transient receptor potential channels (TRPVs) and TWIK-related potassium channels (TREKs) (channel nomenclature follows Alexander et al., 2009). Based on sequence identity, ASICs belong to the larger epithelial sodium channel/degenerin superfamily (Waldmann and Lazdunski, 1998). There are four mammalian ASIC genes (ASIC1–4) that have been identified, as well as several splice variants with varying sensitivities to pH and different expression profiles, reviewed in (Lingueglia, 2007).
Amiloride, a potassium-sparing diuretic, is a non-selective inhibitor of ASICs and has been used to evaluate the effects of pharmacological blockade of ASIC in rodent pain models. In the rat complete Freund's adjuvant (CFA) model of inflammatory pain, amiloride has been shown to significantly reduce both thermal and mechanical hypersensitivity (Dube et al., 2005; Kuduk et al., 2009). Amiloride was also reported to be effective in the rat formalin model, and prevented the development of mechanical hypersensitivity in the acid-induced muscle pain model (Rocha-Gonzalez et al., 2009). In addition, amiloride significantly reduced nociceptive behaviours induced by 5-HT, capsaicin or formalin applied in a low pH 6.2 solution (Rocha-Gonzalez et al., 2009). In humans, subdermal injection of amiloride attenuated acid-induced pain, whereas the TRPV1 antagonist, capsazepine, had no effect (Ugawa et al., 2002). Together, these studies suggest that amiloride-sensitive receptor(s), including ASICs, play a role in the development of inflammatory pain (Kellenberger and Schild, 2002).
In situ hybridization experiments have revealed that the ASIC1, ASIC2 and ASIC3 channel subtypes are expressed in peripheral neurons (Lingueglia et al., 1997; Waldmann et al., 1997; Chen et al., 1998). Disruption of the ACCN3 gene coding for ASIC3 resulted in reduced sensitivity to noxious stimuli, but increased sensitivity of mechanoreceptors detecting light touch (Price et al., 2001). Deletion of ACCN3, but not the ACCN1 gene (ASIC1), resulted in decreased muscle pain induced by repeated acid injection (Sluka et al., 2003). In addition, ACCN3 knock-out mice did not develop mechanical hypersensitivity after muscle inflammation when compared to wild-type mice (Sluka et al., 2007). These results support the role of ASIC3 in the development of inflammatory pain.
To further explore the role of ASIC3 in acid-induced muscle pain, as well as inflammatory pain, we utilized the ASIC3-selective peptide inhibitor, APETx2, isolated from sea anemone (Diochot et al., 2004). By comparing the efficacy of APETx2 following local versus i.t. administration, we investigated the role of ASIC3 in the development and maintenance of acid-induced muscle pain, and also evaluated the role of ASIC3 in the maintenance of CFA-induced inflammatory pain.
Methods
Synthesis and refolding of APETx2
APETx2 was synthesized using solid-phase methodology and a Boc protection method. Refolding of 0.02 mM linear APETx2 was performed in 1 mM guanidine HCl, 0.1 mM NH4OAc, pH 7.8, in the presence of 1.1 mM reduced glutathione, and 0.12 mM oxidized glutathione by 72 h air oxidation. The folding yield was ∼84%. Identity of refolded APETx2 was confirmed by electrospray mass spectroscopy. The inactive, linearized peptide was obtained by reducing APETx2 with dithiothreitol and blocking free sulphydryls with N-ethylmaleimide as previously described (Smyth et al., 1964) The large batch synthesis yielded ∼100 mg of APETx2, and this material was used in all in vitro and in vivo experiments.
Cloning rat ASIC3 and expression in Chinese hamster ovary (CHO) cells
The full-length rat ACCN3 gene was amplified from rat dorsal root ganglion total cDNA using Pfx polymerase, cloned into pENTR/D-TOPO entry vector (Invitrogen, Carlsbad, CA, USA) and confirmed by DNA sequencing. The expression construct was generated by performing LR recombination between the pENTR/D-TOPO entry clone containing the ACCN3 gene and the Gateway destination vector, pEF/FRT (Invitrogen). A stable CHO cell line was generated by co-transfection of ACCN3/pER/FRT and pOG44, and selection of hygromycin-resistant clones. Robust expression of the ASIC3 protein was confirmed by Western blot using the polyclonal anti-ASIC3 antibody (Alomone Laboratories Ltd, Jerusalem, Israel) (data not shown).
Patch clamp electrophysiology
ASIC3 currents were recorded using whole-cell voltage clamp techniques and an automated parallel patch clamp instrument (PatchXpress, Molecular Devices Corporation, Sunnyvale, CA, USA). From a holding potential of −60 mV, currents were activated by lowering pH in external solution containing (in mM): 150 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 12 dextrose, pH 7.4 (or 10 mM MES, pH 5.5). Intracellular patch clamp solution contained (in mM): 119 K gluconate, 15 KCl, 3.2 MgCl2, 5 EGTA, 5 HEPES, 5 K2ATP, pH 7.3; 0.1% BSA was added to APETx2 solutions.
Animals
All animal care and experimental procedures were approved by the Merck West Point Institutional Animal Care and Use Committee, and were performed in accordance with The Guide for the Care and Use of Laboratory Animals. Adult male Sprague Dawley rats (Taconic Farms, Germantown, NY, USA) weighing 200–300 g were used in all experiments, and the rats were maintained on a standard 12 h light–dark cycle where they had free access to food and water.
i.t. catheter implantation
For those studies in which APETx2 was injected i.t., rats received an indwelling i.t. catheter at least 5 days prior to nociceptive testing. The rats were anaesthetized using isoflurane (5%, inhalation), and using aseptic technique, a midline incision was made on the back of the neck to expose the atlanto-occipital membrane. The catheter was inserted into the spinal subarachnoid space by passing an 8.0 cm length of sterile polyurethane tubing (32-gauge; ReCath CS-1, Allison Park, PA, USA) through the membrane to the level of the rostral lumbar enlargement. The rostral end of the catheter was externalized and the incision was closed with 4-0 absorbable suture.
Acid-induced muscle pain model
Rats were placed on an elevated mesh galvanized steel platform in individual chambers, and mechanical sensitivity was determined by applying a series of calibrated von Frey filaments (0.25–15 g) to the plantar aspect of the left or right hind paw using the ‘up–down’ method to determine median withdrawal thresholds (Chaplan et al., 1994). Following determination of baseline mechanical sensitivity, the rats were briefly anaesthetized using isoflurane (5% to effect, by inhalation) and received an i.m. injection of saline (pH 4.0, 100 µL) into the left gastrocnemius muscle using a 27-gauge needle (Sluka et al., 2001). Five days following the i.m. saline injection, withdrawal thresholds were measured again and rats subsequently received a second i.m. injection of saline (pH 4.0, 100 µL) at the same site as the initial injection. Bilateral mechanical hypersensitivity was then measured following the second i.m. saline injection as described above.
CFA-induced inflammatory pain model
Rats were tested for baseline mechanical hind paw withdrawal thresholds by wrapping the rat in a towel and placing the hind paw (either left or right) in a modified Randall–Sellito paw pinch apparatus (Stoelting, Wood Dale, IL, USA). A plastic plinth was placed on the plantar aspect of the hind paw and an increasing force (measured in grams) was applied to the hind paw. The hind paw withdrawal threshold (g) was recorded when the rat either withdrew the hind paw from the stimulus or vocalized. The mechanical stimulus was applied to each hind paw three times at each testing time-point, and average mechanical hind paw withdrawal thresholds were determined for both the left and right hind paws. A maximal hind paw withdrawal threshold of 450 g was used to avoid tissue damage. Following determination of baseline thresholds, the rats received an intradermal injection of CFA (1 mg·mL−1, 100 µL) into the plantar aspect of the left hind paw, and were subsequently returned to their cages (Stein et al., 1988). Twenty-four hours following CFA injection, the rats were tested for mechanical hypersensitivity as described above.
Administration of APETx2
In those studies examining effects of APETx2 in the acid-induced pain model, APETx2 or vehicle was injected i.m. in a volume of 100 µL at the same site as the i.m. saline (pH 4.0) injection in the left gastrocnemius muscle using a 27-gauge needle. In those studies examining effects of APETx2 in the CFA inflammatory pain model, APETx2 was injected i.pl. into the left hind paw in a volume of 50 µL using a 27-gauge needle.
For i.t. administration, APETx2 was injected into the subarachnoid space at the level of the lumbar enlargement. A 5 µL volume was delivered over a 30 s period using a 33-gauge needle connected to a Hamilton 25 µL syringe using PE10 tubing. i.t. Catheter tip placement at the level of the lumbar enlargement was confirmed at the conclusion of the study by injecting 5 µL 4% lidocaine and examining temporary hind limb paralysis.
In all studies, APETx2 was administered at concentrations from 0.022 to 2.2 µM, which represented a range of concentrations from approximately the in vitro ASIC3 IC50 value (0.067 µM) to 33-fold over the IC50 value.
Data analysis
IC50 values were defined as the concentration of APETx2 that produced a 50% inhibition of hypersensitivity, and were calculated using a curve-fitting computer program (Tallarida and Murray, 1997). To determine IC50 values in the acid-induced pain model, the effects of APETx2 were expressed as per cent inhibition of hypersensitivity using the following equation: % inhibition = (post-drug threshold − mean post-vehicle threshold)/(baseline 15 g − mean post-vehicle threshold) × 100. To determine IC50 values in the CFA inflammatory pain model, the effects of APETx2 were expressed as per cent inhibition of hypersensitivity using the same equation by substituting the post-CFA threshold for mean post-vehicle threshold, and substituting the pre-CFA threshold for baseline 15 g threshold. Experimental groups consisted of n = 5–15/group depending on the experimental design, and the data were represented as mean ± SEM. Differences in responses from vehicle treatment were determined using two-way repeated measures anova with Tukey's post hoc test. SigmaStat was used for all statistical analyses, and significance was defined as P < 0.05.
Results
In vitro electrophysiological evaluation of synthetic APETx2
ASIC3 current was recorded in CHO cells stably expressing rat ASIC3 in a whole-cell voltage clamp configuration. The rapid change in pH from 7.4 to 6.0 resulted in a large (6 ± 1 nA), rapidly desensitizing inward current (Figure 1A). Pre-incubation of cells with 1 µM of folded, synthetic APETx2 resulted in almost complete block of the proton-evoked current (Figure 1A). The effect of APETx2 was fully reversible, as the ASIC3 current was restored to control levels after wash (not shown). APETx2 inhibited ASIC3 current in a dose-dependent manner, and a non-linear regression analysis of concentration–response curve determined an IC50 of 67 nM (Figure 1B). This value obtained for the synthetic APETx2 peptide agrees well with the IC50 of 63 nM reported for native APETx2 toxin purified from Anthopleura elegantissima (Diochot et al., 2004). The inactive, linearized peptide was obtained by reducing the disulphide bridges of APETx2 with dithiothreitol and blocking the free sulphydryls with N-ethylmaleimide. This linearized APETx2 peptide failed to block ASIC3 currents (Figure 1A) and was included as a control in selected in vivo experiments.
Figure 1.
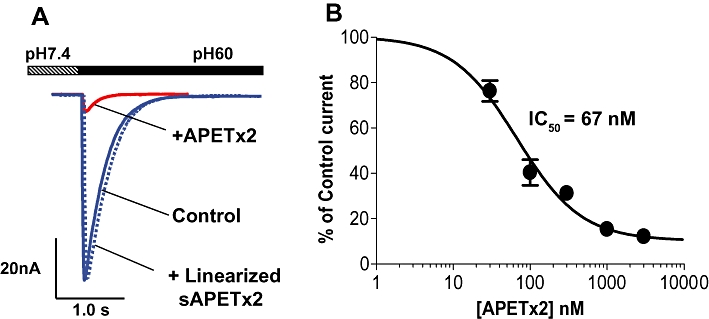
Effects of synthetic APETx2 on ASIC3 current recorded in CHO cells. ASIC3 currents were recorded using whole-cell voltage clamp techniques. From a holding potential of −60 mV, currents were activated by lowering external pH to 5.5 in the absence or presence of 3 µM APETx2 peptide, or 3 µM of the control, linearized APETx2 peptide (A). Concentration–response curve for block of ASIC3 currents by APETx2 (B). Results were plotted as mean ± SD (n = 3).
Effect of i.m. administration of APETx2 in the acid-induced muscle pain model
The effects of APETx2 were studied in the acid-induced pain model (Sluka et al., 2001). In this model, two injections of acidified saline (pH 4.0) into the left gastrocnemius muscle of rats 5 days apart produced long-lasting, bilateral mechanical hypersensitivity in the hind paw (Figure 2A,B). To examine the role of peripheral ASIC3 receptors in the development of acid-induced pain, APETx2 was administered by i.m. injection prior to the second injection of acidified saline. This treatment resulted in a dose-dependent inhibition of acid-induced mechanical hypersensitivity in both the ipsilateral and contralateral hind paws with IC50 values of ∼0.7 and ∼0.4 µM, respectively (Figure 2A,B). At the highest concentration tested (∼2 µM), APETx2 inhibited mechanical hypersensitivity in the ipsilateral and contralateral hind paws by 65 and 87% (Figure 2C). In contrast, APETx2 applied following the second injection of acid saline produced no visible change in hypersensitivity (Figure 2D; only data for ipsilateral hind paw shown).
Figure 2.
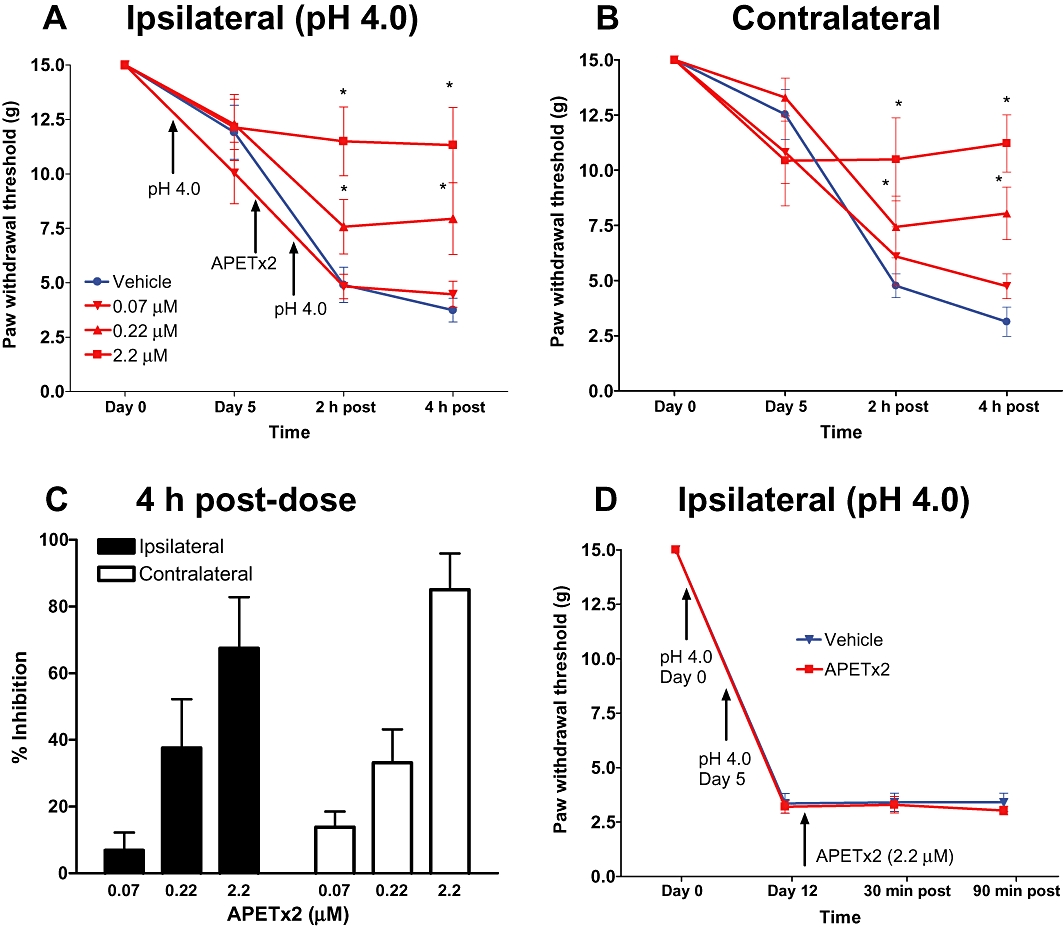
Effects of i.m. administration of APETx2 on mechanical hypersensitivity in the acid-induced muscle pain model. Acidic saline (pH 4, 100 µL) was injected into the gastrocnemius muscle of the rat twice, 5 days apart. APETx2 was administered prior to the second injection of acid saline. Mechanical hypersensitivity was assessed by measuring withdrawal thresholds to von Frey filament stimulation for the ipsilateral (A, C) and contralateral (B) hind paws. Data are expressed as mean ± SEM. Change in threshold relative to pretreatment levels was calculated and plotted as a function of the dose of APETx2 (C). APETx2 was also applied after second i.m. injection of acid saline (D). *P < 0.05 versus vehicle group.
Effect of i.t. administration of APETx2 in the acid-induced muscle pain model
To determine the contribution of spinal ASIC3 receptors to the development and maintenance of acid-induced muscle pain, APETx2 was administered by i.t. injection. The administration of APETx2 prior to the second i.m. injection of acid saline resulted in a dose-dependent inhibition of mechanical hypersensitivity in both the ipsilateral and contralateral hind paws (Figure 3A,B). At the highest concentration tested (0.44 µM), APETx2 inhibited mechanical hypersensitivity in the ipsilateral and contralateral hind paws by 84 ± 11% and 87 ± 15%, respectively (Figure 3C). i.t. Injection of the inactive, linearized APETx2 peptide produced no significant effect on the development of mechanical hypersensitivity (Figure 3A–C). To examine the role of spinal ASIC3 receptors in the maintenance of acid-induced muscle pain, APETx2 was injected after the second injection of acid saline when the hyperalgesia was fully developed. Single-dose administration of 0.44 µM of APETx2 produced a small increase in withdrawal thresholds in both the ipsilateral (% inhibition = 14 ± 4 at 90 min) and contralateral (% inhibition = 7 ± 2 at 90 min) hind paws; however, these effects failed to reach statistical significance (Figure 3D).
Figure 3.
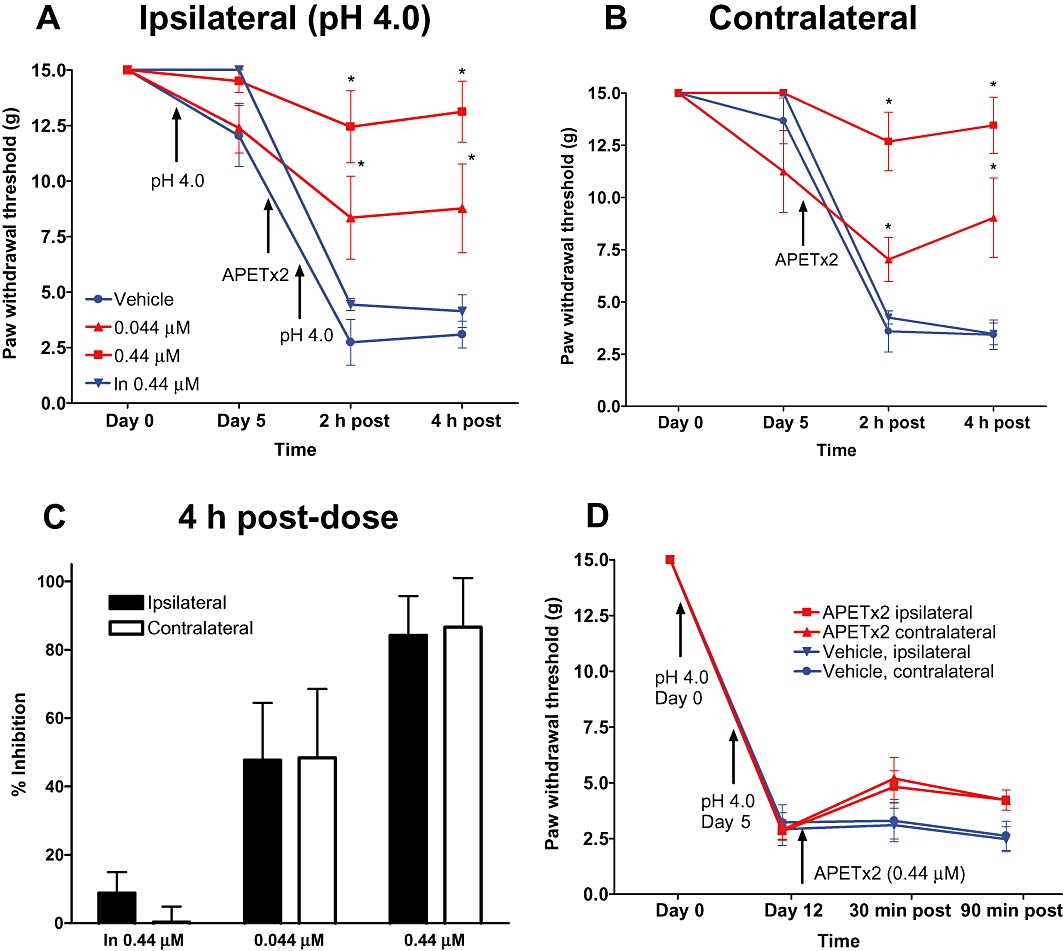
Effects of i.t. administration of APETx2 on mechanical hypersensitivity in the acid-induced muscle pain model. APETx2 or inactive, linearized APETx2 was administered via i.t. administration prior to the second i.m. injection of acid saline (A, B) or following the second acid saline injection after hypersensitivity was established (D). Data are expressed as mean ± SEM. *P < 0.05 versus vehicle group.
Effect of i.pl. administration of APETx2 in the CFA inflammatory pain model
To examine the contribution of ASIC3 in inflammatory pain, we used the rat CFA-induced inflammation model. CFA-treated rats developed mechanical hypersensitivity in the ipsilateral hind paw 24 h following injection (post-CFA) measured as a decrease in paw withdrawal thresholds to mechanical stimulation (Figure 4A). i.pl. Injection of APETx2 into the inflamed hind paw resulted in a complete reversal of mechanical hypersensitivity to the levels measured prior to CFA treatment. The reversal of hypersensitivity reached maximal efficacy at 30 min after the administration of APETx2 and was dose dependent (EC50 = 0.34 µM). At the highest concentration evaluated (2.2 µM), the reversal of hypersensitivity by APETx2 was almost complete (98 ± 16% at 30 min) (Figure 4C). No effect of APETx2 was observed in the contralateral hind paw (Figure 4B). In addition, i.pl. injection of the inactive, linearized APETx2 peptide (2.2 µM) had no effect on mechanical hypersensitivity (Figure 4C). Furthermore, i.pl. injection of APETx2 had no effect on mechanical sensitivity in naïve animals (Figure 4D).
Figure 4.
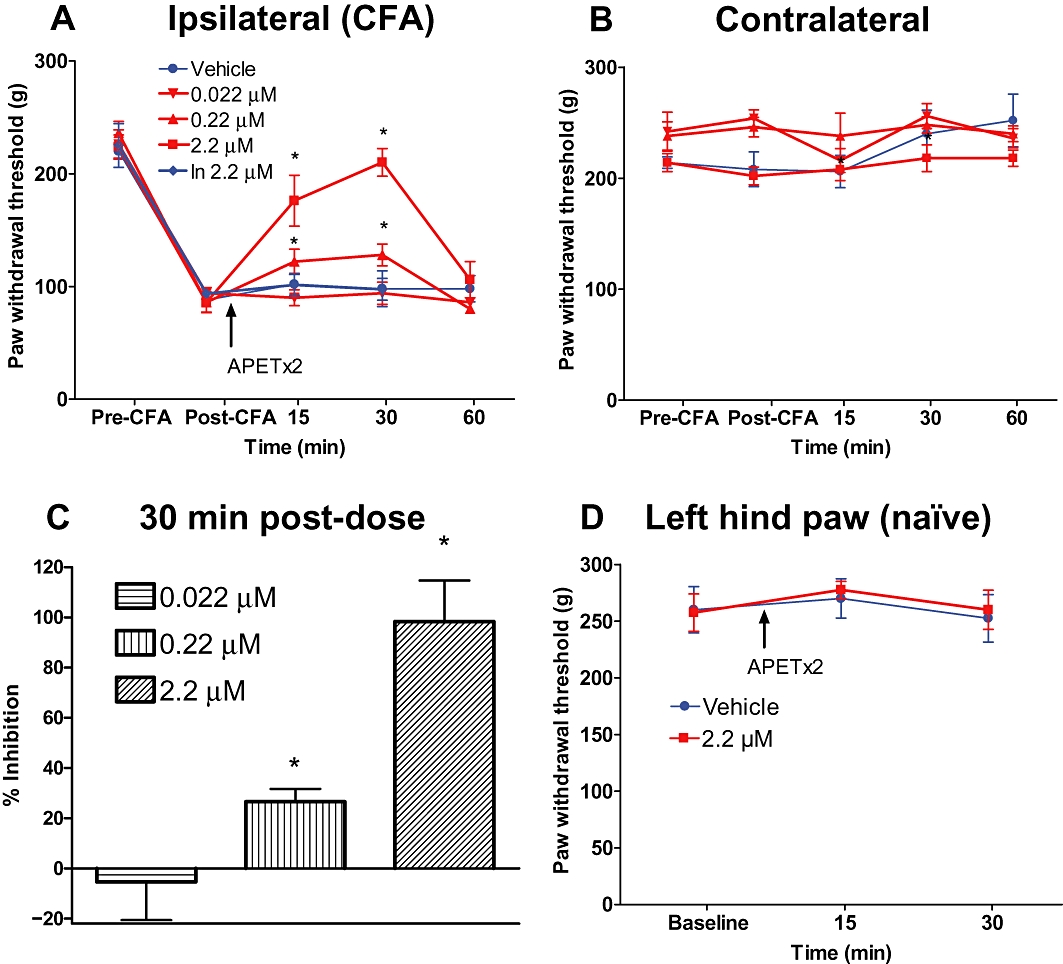
Effects of i.pl. administration of APETx2 in the CFA inflammatory pain model. Mechanical hypersensitivity was produced by injection of CFA into the left hind paw. Twenty-four hours after establishment of inflammation and mechanical hypersensitivity, APETx2 was administered into the inflamed hind paw, and paw withdrawal thresholds were measured at different time intervals for the injected paw (A) and uninjected paw (B). Data from (A) were plotted as % reversal compared to pre-CFA controls (C). APETx2 was administered i.pl. to naïve (non-CFA) animals (D). Data are expressed as mean ± SEM. *P < 0.05 versus vehicle group.
Effect of i.t. administration of APETx2 in the CFA inflammatory pain model
The potential contribution of spinal ASIC3 receptors to the maintenance of inflammatory pain was examined by i.t. administration of APETx2 in the CFA model. Twenty-four hours following CFA injection into the left hind paw, rats developed mechanical hypersensitivity in the ipsilateral hind paw that was unaffected by subsequent i.t. injection of APETx2 (1.3 µM; Figure 5A). Additionally, the i.t. injection of APETx2 had no effect on mechanical paw withdrawal thresholds in the contralateral hind paw (Figure 5B).
Figure 5.
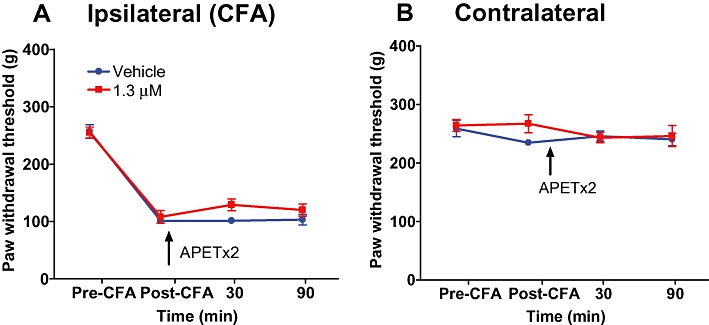
Effects of i.t. administration of APETx2 in the CFA inflammatory pain model. Mechanical hypersensitivity was produced by injection of CFA into the left hind paw. Twenty-four hours after establishment of inflammation and hypersensitivity, APETx2 was administered by i.t. injection, and paw withdrawal thresholds were measured at different time intervals for the inflamed (A) and non-inflamed hind paw (B). Data are expressed as mean ± SEM.
Discussion
In the present study, local i.m. or spinal administration of the ASIC3 inhibitor APETx2 was found to prevent the development of mechanical hypersensitivity in a model of muscle pain produced by i.m. injection of acidic saline. In contrast, i.m. or spinal administration of APETx2 following the development of acid-induced mechanical hypersensitivity did not affect established hypersensitivity. In the CFA model of inflammatory pain, local i.pl. injection of APETx2 was found to reverse CFA-induced mechanical hypersensitivity while spinal administration was ineffective. These data support a role for ASIC3 in the development, but not maintenance, of mechanical hypersensitivity in a model of acid-induced muscle pain. Additionally, these data suggest that peripheral ASIC3 contributes to the maintenance of mechanical hypersensitivity in the CFA inflammatory pain model.
To examine the contribution of ASIC3 to mechanical hypersensitivity in models of acid-induced muscle pain and inflammatory pain, we used a potent peptide ASIC3 channel inhibitor, APETx2 (Diochot et al., 2004). In-house whole-cell voltage clamp studies using CHO cells stably expressing rat ASIC3 revealed that this peptide is a potent ASIC3 inhibitor with IC50 = 67 nM. While APETx2 was utilized as a potent ASIC3 inhibitor for the in vivo studies, we cannot completely rule out the possibility that observed in vivo effects of APETx2 may involve additional mechanisms independent of ASIC3 blockade. The potency and selectivity of APETx2, however, suggest that this is unlikely. APETx2 does not inhibit closely related ASICs, including ASIC1 and ASIC2 (Diochot et al., 2004). Additionally, it has been reported that APETx2 does not inhibit several other ion channels from a diverse repertoire, including the potassium channels Kv1.4, hERG, Kv2.2, Kv3.1, Kv4.2, Kv4.3 (Diochot et al., 2004). Moreover, in the present study, the in vivo anti-nociceptive actions of APETx2 were found to be specific for mechanical hypersensitivity and do not reflect a general anti-nociceptive action, because i.pl. injection of APETx2 into the hind paw of naïve (non-inflamed) rats did not affect mechanical withdrawal thresholds. Thus, the totality of these results suggests that APETx2 is likely to be selective for ASIC3, and that this selectivity may occur as a consequence of its complex three-dimensional structure and its binding to ASIC3 at several structural clusters, rather than at a single site.
In the present study, peripheral i.m. injection or spinal administration of APETx2 prevented the development of mechanical hypersensitivity in the acid-induced muscle pain model, supporting a role for peripheral ASIC3 in the development of acid-induced pain. ASIC3 is expressed throughout the periphery in sensory neurons (Waldmann et al., 1997; Molliver et al., 2005), taste cells (Richter et al., 2004), retina (Maubaret et al., 2002), testes (Babinski et al., 2000) and bone (Jahr et al., 2005), reviewed in (Lingueglia, 2007). A role for peripheral ASIC3 in acid-induced pain was previously demonstrated by the observation that APETx2 was found to block acid-induced currents and action potentials in peripheral sensory fibres, and inhibit acid-induced pain behaviours following peripheral administration (Deval et al., 2008). Additionally, global disruption of the ACCN3 gene encoding ASIC3 was reported to result in diminished pain responses in a murine model of acid-induced muscle pain (Sluka et al., 2003) In ASIC3–/– mice, local injection of an ASIC3-encoding herpes virus into muscle resulted in the expression of functionally active ASIC3 and re-establishment of acid-induced hypersensitivity, strongly supporting a role for peripheral ASIC3 in acid-induced muscle pain (Sluka et al., 2007). In the present study, we also found that spinal administration of APETx2 produced a similar inhibition of the development of acid-induced mechanical hypersensitivity, suggesting a role for spinal ASIC3 in this phenomenon. While activation of peripheral ASIC3 via lowered local pH is a reasonable explanation for the role of peripheral ASIC3 in the acid-induced muscle pain model, it is less obvious how central, spinal ASIC3 could be contributing to hypersensitivity produced by lowered pH in peripheral muscle. While the central mechanisms responsible for activation of spinal ASIC3 following i.m. acid injection would be speculative, it is important to note that a role of other ASICs in the spinal cord has been reported earlier, in persistent pain models. For example, both peripheral and spinal TRPV1 receptors contribute to CFA-induced inflammatory pain (Cui et al., 2006). Although the endogenous mediators responsible for spinal TRPV1 activation are unknown, these results are similar to our findings in that either lowered pH in the spinal cord or an as yet unidentified mechanism(s) is responsible for activation of spinal ASICs in the presence of a peripheral insult.
In contrast to their role in the development of acid-induced hypersensitivity, ASIC3 does not appear to be involved in the maintenance of this hypersensitivity, because peripheral or spinal administration of APETx2 was found to be ineffective when injected following the development of mechanical hypersensitivity. It is well accepted that initial hyperexcitability of peripheral sensory afferent fibres results in plasticity in the CNS (spinal cord and supraspinal sites) resulting in subsequent central sensitization characterized by persistent hyperexcitability of spinal dorsal horn sensory neurons (Latremoliere and Woolf, 2009). In the original study describing the acid-induced muscle pain model (Sluka et al., 2001), the authors noted that the bilateral mechanical hypersensitivity must involve central sensitization processes, because peripheral sensitization mechanisms would be likely only to result in unilateral hypersensitivity. To further examine central sensitization mechanisms involved in this model, a subsequent paper was published by Tillu et al. (2008) demonstrating that descending pain facilitatory systems from the rostral ventromedial medulla contribute to bilateral hypersensitivity. The results from the present study are consistent with the notion that ASIC3 appears to play a direct role in peripheral sensitization, which subsequently results in the development of central sensitization and persistent hypersensitivity mediated by other pro-nociceptive mechanisms including descending pain facilitatory systems from supraspinal sites and excitatory amino acid receptors (Skyba et al., 2002).
The results from the present study also support a role for peripheral ASIC3 in the maintenance of mechanical hypersensitivity in the CFA-induced inflammatory pain model, as i.pl. injection of APETx2 dose-dependently reversed mechanical hypersensitivity. These data are consistent with those of Deval et al. (2008), who reported that i.pl. injection of APETx2 inhibited CFA-induced heat hypersensitivity, supporting a role for peripheral ASIC3 in inflammation-induced hypersensitivity associated with both mechanical and heat stimuli. That peripheral administration of APETx2 was found to block CFA-induced hypersensitivity in the present study is not surprising, because local pH in inflamed tissue is lowered, which can consequently activate ASIC3 on peripheral terminals of primary sensory afferents innervating the area of inflammation in the hind paw (Deval et al., 2008). Additionally, other ASICs, such as TRPV1, have also been shown to contribute to CFA-induced hypersensitivity through a peripheral site of action (Cui et al., 2006). Interestingly, Deval et al. (2008) also found that chronic i.t. administration of siRNA targeting ASIC3 prior to CFA inflammation prevented the development of CFA-induced thermal hypersensitivity. Given that i.t. administration of APETx2 was found to be ineffective in reversing ongoing CFA-induced hypersensitivity in the present study, the data would perhaps suggest a greater role for spinal ASIC3 in the development, rather than maintenance, of hypersensitivity, or differing roles of spinal ASIC3 in mechanical or heat hypersensitivity. Additionally, while i.t. APETx2 administration was likely to be localized to the spinal cord in the present study, it is not clear where (peripheral or spinal) the ASIC3 was down-regulated by the siRNA in the study by Deval et al. (2008). Nevertheless, the data from the present study strongly support a role for peripheral ASIC3 in the maintenance of inflammation-induced hypersensitivity, which is consistent with a number of other studies demonstrating a role for ASICs in inflammatory pain using small molecule inhibitors and knock-out mice (Dube et al., 2005; Sluka et al., 2007; Ikeuchi et al., 2008; Kuduk et al., 2009; Walder et al., 2010).
The differential role of ASIC3 in the development versus maintenance of mechanical hypersensitivity in the acid-induced muscle pain model and CFA inflammatory pain model may be attributed to several fundamental characteristics which are inherently different in these models. The most likely explanation, as outlined above, is that established secondary hypersensitivity in the acid-induced muscle pain model is likely to involve mechanisms associated with central sensitization (i.e. non-ASIC3) to a greater extent than primary hypersensitivity measured in the CFA inflammatory pain model. Additionally, ASIC3 may have a differential role in peripheral sensitization induced by low pH compared to CFA-induced infiltration of inflammatory mediators. Lastly, in the acid-induced muscle pain model, mechanical hypersensitivity was measured using a phasic punctuate mechanical stimulus (the von Frey filaments), whereas in the CFA model, hypersensitivity was measured using a graded mechanical pressure stimulus (the Randall–Sellito test). Nevertheless, the results from the present study demonstrate a different role for ASIC3 in the development and maintenance of mechanical hypersensitivity in the acid-induced muscle pain model, and CFA inflammatory pain model respectively.
In summary, a potent peptide ASIC3 inhibitor, APETx2, was used in the present study to further support a role for ASIC3 in both acid-induced muscle pain and inflammatory pain. The data suggest that ASIC3 has a more prominent role in the development of mechanical hypersensitivity in a model of acid-induced muscle pain. On the other hand, the results from the present study demonstrating a prominent role for peripheral ASIC3 in ongoing inflammatory pain support further investigation of ASIC3 as a potential analgesic target for the treatment of inflammatory pain conditions.
Acknowledgments
We would like to thank the Laboratory Animal Resources Department at Merck Research Laboratories West Point for providing surgical support.
Glossary
Abbreviations
- ASIC
acid-sensitive ion channel
- CFA
complete Freund's adjuvant
- TREK
TWIK-related potassium channel
- TRP
transient receptor potential channel
Conflict of interest
The authors state no conflict of interest.
Supplemental material
References
- Alexander SP, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edition. Br J Pharmacol. 2009;158(Suppl. 1):S1–254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babinski K, Catarsi S, Biagini G, Seguela P. Mammalian ASIC2a and ASIC3 subunits co-assemble into heteromeric proton-gated channels sensitive to Gd3+ J Biol Chem. 2000;275:28519–28525. doi: 10.1074/jbc.M004114200. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Chen CC, England S, Akopian AN, Wood JN. A sensory neuron-specific, proton-gated ion channel. Proc Natl Acad Sci U S A. 1998;95:10240–10245. doi: 10.1073/pnas.95.17.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui M, Honore P, Zhong C, Gauvin D, Mikusa J, Hernandez G, et al. TRPV1 receptors in the CNS play a key role in broad-spectrum analgesia of TRPV1 antagonists. J Neurosci. 2006;26:9385–9393. doi: 10.1523/JNEUROSCI.1246-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deval E, Noel J, Lay N, Alloui A, Diochot S, Friend V, et al. ASIC3, a sensor of acidic and primary inflammatory pain. Embo J. 2008;27:3047–3055. doi: 10.1038/emboj.2008.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diochot S, Baron A, Rash LD, Deval E, Escoubas P, Scarzello S, et al. A new sea anemone peptide, APETx2, inhibits ASIC3, a major acid-sensitive channel in sensory neurons. Embo J. 2004;23:1516–1525. doi: 10.1038/sj.emboj.7600177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube GR, Lehto SG, Breese NM, Baker SJ, Wang X, Matulenko MA, et al. Electrophysiological and in vivo characterization of A-317567, a novel blocker of acid sensing ion channels. Pain. 2005;117:88–96. doi: 10.1016/j.pain.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Ikeuchi M, Kolker SJ, Burnes LA, Walder RY, Sluka KA. Role of ASIC3 in the primary and secondary hyperalgesia produced by joint inflammation in mice. Pain. 2008;137:662–669. doi: 10.1016/j.pain.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahr H, van Driel M, van Osch GJ, Weinans H, van Leeuwen JP. Identification of acid-sensing ion channels in bone. Biochem Biophys Res Commun. 2005;337:349–354. doi: 10.1016/j.bbrc.2005.09.054. [DOI] [PubMed] [Google Scholar]
- Julius D, Basbaum AI. Molecular mechanisms of nociception. Nature. 2001;413:203–210. doi: 10.1038/35093019. [DOI] [PubMed] [Google Scholar]
- Kellenberger S, Schild L. Epithelial sodium channel/degenerin family of ion channels: a variety of functions for a shared structure. Physiol Rev. 2002;82:735–767. doi: 10.1152/physrev.00007.2002. [DOI] [PubMed] [Google Scholar]
- Kuduk SD, Di Marco CN, Chang RK, Dipardo RM, Cook SP, Cato MJ, et al. Amiloride derived inhibitors of acid-sensing ion channel-3 (ASIC3) Bioorg Med Chem Lett. 2009;19:2514–2518. doi: 10.1016/j.bmcl.2009.03.029. [DOI] [PubMed] [Google Scholar]
- Latremoliere A, Woolf CJ. Central sensitization: a generator of pain hypersensitivity by central neural plasticity. J Pain. 2009;10:895–926. doi: 10.1016/j.jpain.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingueglia E. Acid-sensing ion channels in sensory perception. J Biol Chem. 2007;282:17325–17329. doi: 10.1074/jbc.R700011200. [DOI] [PubMed] [Google Scholar]
- Lingueglia E, de Weille JR, Bassilana F, Heurteaux C, Sakai H, Waldmann R, et al. A modulatory subunit of acid sensing ion channels in brain and dorsal root ganglion cells. J Biol Chem. 1997;272:29778–29783. doi: 10.1074/jbc.272.47.29778. [DOI] [PubMed] [Google Scholar]
- Maubaret C, Delettre C, Sola S, Hamel CP. Identification of preferentially expressed mRNAs in retina and cochlea. DNA Cell Biol. 2002;21:781–791. doi: 10.1089/104454902320908432. [DOI] [PubMed] [Google Scholar]
- Molliver DC, Immke DC, Fierro L, Pare M, Rice FL, McCleskey EW. ASIC3, an acid-sensing ion channel, is expressed in metaboreceptive sensory neurons. Mol Pain. 2005;1:35. doi: 10.1186/1744-8069-1-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MP, McIlwrath SL, Xie J, Cheng C, Qiao J, Tarr DE, et al. The DRASIC cation channel contributes to the detection of cutaneous touch and acid stimuli in mice. Neuron. 2001;32:1071–1083. doi: 10.1016/s0896-6273(01)00547-5. [DOI] [PubMed] [Google Scholar]
- Richter TA, Dvoryanchikov GA, Roper SD, Chaudhari N. Acid-sensing ion channel-2 is not necessary for sour taste in mice. J Neurosci. 2004;24:4088–4091. doi: 10.1523/JNEUROSCI.0653-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha-Gonzalez HI, Herrejon-Abreu EB, Lopez-Santillan FJ, Garcia-Lopez BE, Murbartian J, Granados-Soto V. Acid increases inflammatory pain in rats: effect of local peripheral ASICs inhibitors. Eur J Pharmacol. 2009;603:56–61. doi: 10.1016/j.ejphar.2008.12.017. [DOI] [PubMed] [Google Scholar]
- Skyba DA, King EW, Sluka KA. Effects of NMDA and non-NMDA ionotropic glutamate receptor antagonists on the development and maintenance of hyperalgesia induced by repeated intramuscular injection of acidic saline. Pain. 2002;98:69–78. doi: 10.1016/s0304-3959(01)00471-7. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Kalra A, Moore SA. Unilateral intramuscular injections of acidic saline produce a bilateral, long-lasting hyperalgesia. Muscle Nerve. 2001;24:37–46. doi: 10.1002/1097-4598(200101)24:1<37::aid-mus4>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Price MP, Breese NM, Stucky CL, Wemmie JA, Welsh MJ. Chronic hyperalgesia induced by repeated acid injections in muscle is abolished by the loss of ASIC3, but not ASIC1. Pain. 2003;106:229–239. doi: 10.1016/S0304-3959(03)00269-0. [DOI] [PubMed] [Google Scholar]
- Sluka KA, Radhakrishnan R, Benson CJ, Eshcol JO, Price MP, Babinski K, et al. ASIC3 in muscle mediates mechanical, but not heat, hyperalgesia associated with muscle inflammation. Pain. 2007;129:102–112. doi: 10.1016/j.pain.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth DG, Blumenfeld OO, Konigsberg W. Reactions of N-ethylmaleimide with peptides and amino acids. Biochem J. 1964;91:589–595. doi: 10.1042/bj0910589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Millan MJ, Herz A. Unilateral inflammation of the hindpaw in rats as a model of prolonged noxious stimulation: alterations in behavior and nociceptive thresholds. Pharmacol Biochem Behav. 1988;31:455–451. doi: 10.1016/0091-3057(88)90372-3. [DOI] [PubMed] [Google Scholar]
- Tallarida R, Murray R. Manual of Pharmacologic Calculations with Computer Programs. New York: Springer-Verlag; 1997. [Google Scholar]
- Tillu DV, Gebhart GF, Sluka KA. Descending facilitatory pathways from the RVM initiate and maintain bilateral hyperalgesia after muscle insult. Pain. 2008;136:331–339. doi: 10.1016/j.pain.2007.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugawa S, Ueda T, Ishida Y, Nishigaki M, Shibata Y, Shimada S. Amiloride-blockable acid-sensing ion channels are leading acid sensors expressed in human nociceptors. J Clin Invest. 2002;110:1185–1190. doi: 10.1172/JCI15709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walder RY, Rasmussen LA, Rainier JD, Light AR, Wemmie JA, Sluka KA. ASIC1 and ASIC3 play different roles in the development of hyperalgesia after inflammatory muscle injury. J Pain. 2010;11:210–218. doi: 10.1016/j.jpain.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldmann R, Lazdunski M. H(+)-gated cation channels: neuronal acid sensors in the NaC/DEG family of ion channels. Curr Opin Neurobiol. 1998;8:418–424. doi: 10.1016/s0959-4388(98)80070-6. [DOI] [PubMed] [Google Scholar]
- Waldmann R, Bassilana F, de Weille J, Champigny G, Heurteaux C, Lazdunski M. Molecular cloning of a non-inactivating proton-gated Na+ channel specific for sensory neurons. J Biol Chem. 1997;272:20975–20978. doi: 10.1074/jbc.272.34.20975. [DOI] [PubMed] [Google Scholar]
- Woo YC, Park SS, Subieta AR, Brennan TJ. Changes in tissue pH and temperature after incision indicate acidosis may contribute to postoperative pain. Anesthesiology. 2004;101:468–475. doi: 10.1097/00000542-200408000-00029. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.