

Summary
The y-linked autoimmune accelerating (Yaa) locus drives the transition to fatal lupus nephritis when combined with B6.Sle1 in our B6-congenic model of systemic autoimmunity. We and others recently demonstrated that the translocation of a cluster of X-linked genes onto the Y chromosome is the genetic lesion underlying Yaa (Subramanian, S. et al., Proc Natl Acad Sci USA 2006. 103: 9970–9975; Pisitkun, P. et al., Science 2006. 312: 1669–1672). In male mice carrying Yaa, the transcription of several genes within the translocated segment is increased roughly 2-fold. Although the translocated X chromosome segment in Yaa may contain as many as 16 genes, the major candidate gene for causation of the Yaa-associated autoimmune phenotypes has been TLR7. To confirm the role of TLR7 in Yaa-mediated autoimmune phenotypes, we introgressed a targeted disruption of TLR7 (TLR7−) onto B6.Sle1Yaa to produce B6.Sle1YaaTLR7− and examined evidence of disease at 6 and 9 months of age. Our results demonstrate that the upregulation of TLR7 in the B6.Sle1Yaa strain is responsible for splenomegaly, glomerular nephritis and the majority of the cellular abnormalities of B, T and myeloid cells. The upregulation of TLR7 was also responsible for driving the infiltration and activation of leukocytes into the kidney, in which activated T cellswere a primary component. However, the resolution of TLR7 upregulation did not eliminate the enhanced humoral autoimmunity observed in B6.SleYaa, suggesting that additional elements in the translocation may contribute to the disease phenotype.
Keywords: Autoimmunity, TLR7, genetics, SLE, congenic
Introduction
Systemic Lupus Erythematosus (SLE) is an autoimmune disease classically associated with a loss in immune tolerance that leads to the production of a complex array of autoantibodies that predominantly recognize nuclear material[1]. In severe forms of disease, immune complex deposition can affect multiple end organs and initiate a chronic inflammation that can culminate in severe pathology and fatal disease, as exemplified in lupus nephritis. Genetic predisposition plays a dominant role in SLE susceptibility and a variety of genetic studies in both humans and animal models have identified multiple susceptibility alleles that interact to cause a profound dysregulation of the immune system.
We have utilized a congenic dissection strategy to characterize the individual genetic components that interact to drive the development of severe pathology in murine models of SLE.[2] Our initial studies focused on the NZM2410 strain, which is a congenic recombinant inbred strain derived from a backcross of NZB onto NZW.[3, 4] In an extensive series of investigations, we produced a collection of B6-congenic strains carrying individual susceptibility loci derived from NZM2410 (reviewed in ref [2]). Detailed analyses of the component phenotypes expressed in these strains have defined three separate pathways that interact to mediate severe disease (reviewed in ref [1]). Extensive characterizations of the B6.Sle1strain have identified polymorphisms in the SLAM/CD2 gene cluster as causative for a loss in immune tolerance in both B and T lymphocytes.[5] When isolated on the B6 background, Sle1 confers a mild autoimmune phenotype, with antinuclear antibodies developing as early as 3 months of age, together with hypergammaglobinemia and a mild splenomegaly chararacterised by an expansion of the CD4 T and B cell populations [6, 7]. However, the addition of a second susceptibility locus, such as Sle3 or Yaa, causes the transition of this benign autoimmune phenotype into severe disease [8, 9]. Thus, our congenic dissection strategy has provided a model system with which to characterize the genetic interactions that drive the development of severe disease pathology.
The Y-linked autoimmune accelerating (Yaa) locus is derived from the lupus-prone BXSB mouse[10]. Although Yaa mediates only minimal autoimmune consequences in isolation on the normal B6 background, it is highly epistatic with the Sle1 region, accelerating the rapid development of a highly penetrant and fatal form of disease at a relatively young age[11]. We and others recently demonstrated that the genetic lesion underlying Yaa is a X to Y translocation in which a telomeric segment (> 1 mBase) of the X chromosome is translocated onto the Y chromosome. This translocation results in a duplication of about 16 genes in male mice carrying Yaa, which leads to a roughly 2-fold increase in the transcription of several of the translocated genes. Within the cluster of translocated genes, the major candidate gene for causation of the Yaa-associated autoimmune phenotypes has been TLR7.
There is burgeoning evidence for a role of Toll-like receptors in autoimmune disease (TLR). Most interest has focused on TLR3, TLR7 and TLR9, which recognize dsRNA, ssRNA and dsDNA respectively. The association of these genes with lupus arises predominantly from murine models, where stimulation of any of these receptors in vivo augments nephritis [12–17]. Furthermore, more recently, it has been demonstrated that inhibitors of TLR7 reduce a number of lupus associated phenotypes, in the MRLlpr and NZB/W strains [18, 19]. Taken together, these studies provide a compelling argument for the involvement of toll receptors in the development of systemic autoimmunity.
In the present study, we have introgressed a TLR7-deficient allele (TLR7−) onto B6.Sle1Yaa to produce B6.Sle1YaaTLR7−. The expression of TLR7 in this strain is limited to the single copy of TLR7 translocated onto the Y chromosome. Expression analyses demonstrate that TLR7 transcription in B6.Sle1YaaTLR7− mice is indistinguishable from normal B6 levels, although other genes within the translocated segment on Yaa are still over-expressed. Examination of a cohort of B6.Sle1YaaTLR7−mice aged 9 months revealed that gross splenic pathology, GN and most cellular phenotypes observed in the B6.Sle1Yaa strain have returned to phenotypes characteristic of B6.Sle1. Surprisingly, the increased levels of humoral autoimmunity in B6.Sle1Yaa were not fully resolved in B6.Sle1YaaTLR7−. Nonetheless, these results indicate that the majority of the autoimmune phenotypes associated with the Yaa translocation are conferred by the 2-fold increase in TLR7 expression.
Results
Transition to severe pathology in B6.Sle1Yaa is dependent on dysregulation of TLR7
We produced male cohorts of B6, B6.Sle1, B6.Yaa, B6.Sle1Yaa, B6.YaaTLR7−, and B6.Sle1YaaTLR7− mice and assessed the expression of TLR7. The upregulation of TLR7 expression observed in purified B cells from Yaa-bearing males was eliminated with deletion of the X-derived copy of TLR7, although other translocated genes, such as Prsp2, continued to be expressed at 2-fold higher levels (Supplementary figure 1). These results confirm that the Yaa-derived copy of TLR7 is expressed in an essentially normal fashion and maintains normal levels of TLR7 when expressed in association with a TLR7-null allele on the X chromosome. As shown in Figure 1, comparisons of B6.Sle1Yaa and B6.Sle1YaaTLR7− mice demonstrate that restoration of normal levels of expression of TLR7 eliminates the proteinuria and BUN (Figure 1a), severe GN (Figure 1b), splenomegaly and cellularity (Figure 1c and d) associated with the combination of Yaa with Sle1. These findings indicate that the gross pathological indications of transition to severe disease that are mediated by Yaa are eliminated by returning the expression of TLR7 to normal levels.
Figure 1. Comparative Pathology of B6Sle1YaaTLR7− and B6Sle1Yaa mice.
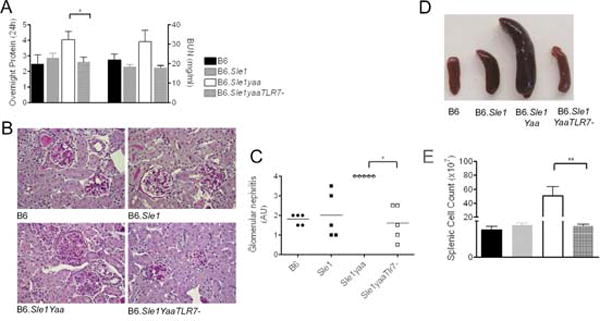
Proteinuria and BUN in 6month and 9 month cohorts of male mice (a) left and right panels respectively demonstrated that deletion of TLR7 eliminates the increase of B6Sle1Yaa. GN was assessed in 9 month mice using H&E staining (b) and graded as described in the methods (c). Splenic weights (d) and counts (e) of 9 month mice showed that the splenomegaly characteristic of B6Sle1Yaa mice was abolished with deletion of the additional copy of TLR7.
Splenomegaly, monocytosis and cellular activation are dependent on dysregulation of TLR7
Flow cytometric analysis of splenocytes revealed that the dysregulated activation and expansion of immune cell lineages that are characteristic of B6.Sle1Yaa return to levels seen in B6.Sle1 in B6.Sle1YaaTLR7− mice (Supplementary Table 1 and Supplementary Figure 1). Specifically, the characteristic monocytosis and subsequent cellular composition was restored to a splenic profile similar to B6.Sle1 (Figure 2a). Analysis of the myeloid lineage indicated that all myeloid populations, including PMNs, inflammatory Gr1+ monocytes and resident Gr1− monocytes, were increased in the B6.Sle1Yaa mice and deletion of TLR7 restored the levels to those observed in B6.Sle1 mice (Figure 2b). The Yaa-dependent increased activation of the lymphoid lineages, as exemplified by CD69 expression, was also abolished (Figure 2c and Supplementary Table 1). The increase in plasma cells observed in B6.Sle1Yaa mice was reduced on TLR7 deletion, and although the levels were higher than in B6.Sle1, this variation was not significant (Figure 2d and Supplementary figure 1). Furthermore, the follicular T cell phenotype, characteristic of late stage disease development in B6.Sle1Yaa, was eliminated on deletion of X-derived TLR7 (Figure 2e).
Figure 2. Splenic leukocyte lineage and activation in 9 month mice.
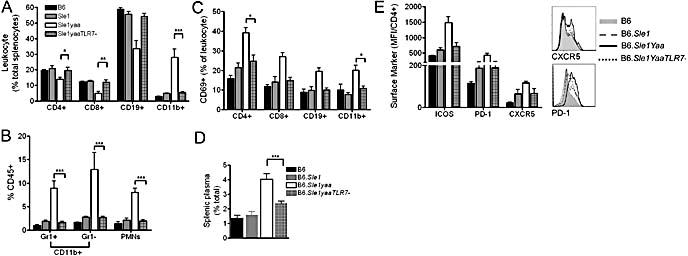
Lymphocyte (a) and myeloid (b) lineage and activation (c) were assessed in 9 month mice. The classic B6.Sle1Yaa monocytosis was not evident in B6.Sle1YaaTLR7- mice. The augmented B6Sle1Yaa plasma cell population was also reduced in B6Sle1YaaTLR7- mice (d). Increased expression of CXCR5, PD-1 and ICOS on CD4+ T cells, indicates a follicular phenotype in B6Sle1Yaa mice which is not present in B6Sle1YaaTLR7- counterparts(e).
Incomplete resolution of Serum Autoimmune phenotypes with TLR7 deletion
Analysis of total serum IgG and IgM demonstrated that the high levels typical of the B6.Sle1Yaa strain were slightly reduced in the B6.Sle1YaaTLR7− mice, suggesting an incomplete resolution of the phenotype (Figures 3a and 3b). Examination of a cohort of 6 month old mice demonstrated that IgM autoantibodies remained high in B6.Sle1YaaTLR7- mice, when compared to Sle1 mice, although this difference was not significant. In a separate cohort of 9 month old mice, there was no difference in IgM ANAs, with all mice demonstrating detectable levels (Figure 3c). Furthermore, at 6 and 9 months all B6.Sle1YaaTLR7− mice demonstrated significantly higher levels of IgG ANA than in B6.Sle1, similar to their B6.Sle1Yaa counterparts (Figure 3d). Specific analysis of autoantigens using a protein array in the 9 month old mice showed that the major difference in IgM autoantibodies was that B6.Sle1Yaa had comparatively more specificity to subtypes of collagen, histones, sRNP and chromatin when compared to B6, B6.Sle1 and B6.Sle1YaaTLR7− sera (Figure 3e). Similar analysis of IgG ANAs demonstrated that B6.Sle1Yaa mice had high levels of IgG antibodies to ssRNA, H2B, ssDNA, dsDNA and glomerular basement membrane (Rat Glom) (Figure 3f). This analysis was repeated on a separate cohort of 9 month old mice, which produced similar results (Supplementary figure 2).
Figure 3. Serum Analysis of Ig in 6 and 9 month mice.
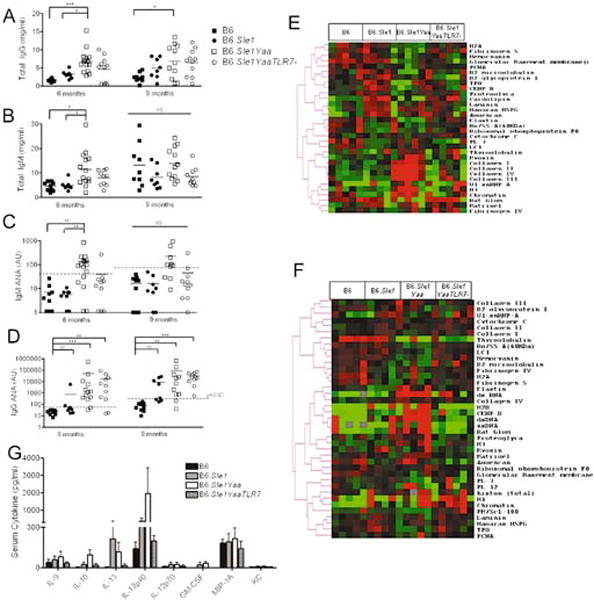
Total IgG (a) and IgM (b) were assessed in 9month old mice demonstrating increased levels in all B6Sle1Yaa mice. IgM ANAs were high in B6Sle1Yaa mice at 6 months compared to B6 and Sle1 control mice (c, left panel). There was no difference at 9 months (c, right panel). IgG ANAs were also determined in 6 and 9 month old mice (d, left and right panels respectively). NS: non-significant, *<p0.05, **p<0.01, ***p<0.001. The autoantigen specificity of these antibodies was assessed using a protein array, measuring IgM (e) and IgG (f) autoantigens. The data demonstrates different specificities of B6.Sle1Yaa mice compared to B6Sle1 and B6Sle1YaaTLR7−. Serum cytokines were determined using luminex which showed an increase in circulating IL-12p40 in B6Sle1Yaa mice (g).
Since TLR7 has been associated with U1_snRNP autoreactivity, we verified the autoantigen array data using an U1_snRNP Elisa (supplemental figure 3)[20–22]. We determined that between 30 and 50% of B6.Sle1Yaa mice were statistically positive at 6 and 9 months of age and thus it appears in this model, a high titre of anti-snRNP is not an overwhelming feature of disease in every mouse. Indeed earlier studies in MRL/lpr mice suggest that the penetrance of this antibody is less than 50% [21]. Moreover, approximately 30–40% of stimulatory immune complexes from SLE patients contain sn_RNP [23, 24] Taken together, these data suggest that snRNPs are not the only stimulatory factor in human disease or murine models. Elimination of TLR7 prevented this level of penetrance (Supplemental figure 3).
Other autoantibodies, such as those specific to histone and chromatin in the IgG array, detectable in the ANA Elisa, were also detectable in B6.Sle1 and B6.Sle1YaaTLR7− mice. These results suggest that deletion of the X chromosome copy of TLR7 may not fully diminish the increased titers of humoral autoantibodies to chromatin seen in B6.Sle1Yaa, however, the transition to production of IgG antibodies specific for pathogenic autoantigens, such as dsDNA and glomerular autoantigens, is eliminated. Serum cytokine analysis using a 22-plex Luminex assay demonstrated detectable increases in the levels of IL-9, IL-10, IL-13, and IL-12p40 in B6.Sle1Yaa mice. These increases were eliminated on TLR7 deletion (Figure 3g).
Renal leukocyte infiltration and activation is restored on TLR7 deletion
A detailed flow cytometric analysis of the leukocyte infiltrate in the kidneys of our cohorts revealed that the kidneys of B6.Sle1Yaa mice are infiltrated by a characteristic profile of activated inflammatory cells and that these differences are dependent upon the dysregulation of TLR7 (Figure 4a and Supplementary Table 2). The major infiltrating cells into the kidney were CD4+ T cells and inflammatory Gr1high monocytes (Figure 4b and c respectively). The CD4+ T cells infiltrating the kidneys of B6.Sle1Yaa mice were activated, demonstrating an increase in surface expression of ICOS and PD-1, although there was no detectable expression of CXCR5 (Figure 4d and Supplementary Table 2). Analysis of the Gr1high monocytes demonstrated that they were activated, with increased expression of CD69 and CXCR4, a chemokine receptor whose upregulation is associated with renal damage (Figure 4e, [25, 26]). B cells were also activated, with increased expression of CD69 and CXCR4 (Figure 4f). The infiltrate and activation was absent in the B6.Sle1YaaTLR7− mice. Finally, analysis of cytokines from renal plasma demonstrated that IL-12p40 was upregulated within B6.Sle1Yaa mice, and that these levels were restored to levels observed in the mice without severe GN in B6.Sle1YaaTLR7− mice (Figure 4g).
Figure 4. Leukocyte infiltration of the Kidney.
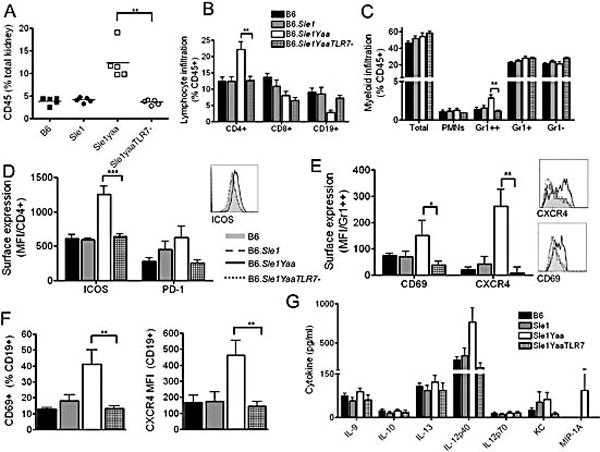
Kidneys were perfused and processed as described in the methods and flow cytometry was used to assess leukocyte infiltration and activation. B6.Sle1Yaa mice presented increased leukocyte infiltration, which was eliminated on deletion of the additional TLR7 (a). Lineage assessment of the kidney demonstrates that the primary infiltrating cells are CD4+ T cells (b) and Gr1high monocytes (c). Analysis of surface expression markers on the CD4 T cells demonstrated an increase in expression of follicular markers ICOS and PD-1 on B6Sle1Yaa mice, which was absent in B6Sle1YaaTLR7− mice (d). Analysis of the infiltrating Gr1high population in B6Sle1Yaa mice showed increased expression of CD69 and CXCR4 which was not present on deletion of the extra copy of TLR7 (e). B cells were activated in B6Sle1Yaa mice as demonstrated by expression of CD69 and CXCR4 and this too was eliminated in B6Sle1YaaTLR7 mice (f). Analysis of the supernatant in the kidney demonstrated that a primary cytokine in B6Sle1Yaa mice within the kidney was IL-12, with some mice producing high levels of MCP-1 (g). This was not evident in B6Sle1YaaTLR7− mice.
Discussion
In the present study, we sought to determine whether TLR7 was responsible for the autoimmune phenotypes conferred by Yaa in the B6.Sle1Yaa strain. We and others have previously shown that the genetic lesion underlying Yaa is an X to Y translocation involving a cluster of tightly linked genes adjacent to the psuedoautosomal region of the X chromosome [8, 27]. Several of the genes in the translocated segment are expressed at a 2-fold higher level in Yaa-bearing males, many of these in B cells. Thus, although the presence of TLR7 within the translocated cluster made it a strong candidate for the autoimmune phenotypes associated with Yaa, the potential involvement of other translocated genes with autoimmunity could not be excluded.
The results presented in this study indicate that the dysregulated expression of TLR7 is responsible for the bulk of the autoimmune phenotypes elicited by Yaa in combination with Sle1. Our analyses indicate that the inclusion of a TLR7 deletion in B6.Sle1Yaa mice eliminates virtually all of the severe disease phenotypes associated with Yaa in this model system. This basic finding is in accordance with a recent publication which eliminated TLR7 in FcγRII−/−Yaa mice [28]. Taken together, these findings firmly establish that a very modest increase in the level of expression of TLR7 can mediate a rapid and highly penetrant transition to severe pathogenesis in mice that are prone to the development of spontaneous humoral autoimmunity.
Our serum analysis of autoantibodies suggests that levels of IgM and IgG ANAs at 6 months remained high in B6.Sle1YaaTLR7− mice and were not fully restored to levels demonstrated by their B6.Sle1 counterparts. However, although the titer of autoantibodies was still somewhat elevated, the transition to pathogenic autoantibodies, such as IgG antibodies to dsRNA, dsDNA and glomerular basement membrane did not occur in B6.Sle1YaaTLR7− (Figure 3F). This could reflect the contribution of one of the other translocated genes in the Yaa locus, or reflect slight aberrations in the expression of TLR7 when it is dictated solely by the translocated copy in the Yaa locus. In this regard, we did not observe gross abnormalities in the expression of TLR7 in B6.Sle1YaaTLR7−mice (Supplementary Fig. 1), although we cannot exclude possible variations in expression following specific cell activation events. Indeed, recent investigations have demonstrated that administration of IFNα to purified B cells increases TLR7 mRNA expression and function [20, 29]. Thus, further work will be needed to identify the causes of these minor phenotypic variations.
The results we report provide important new insights into the impact of TLR7 upregulation on end organ targeting in systemic autoimmunity. In the B6.Sle1Yaa model, the impact of upregulating TLR7 is most clearly apparent in CD4+ T cell and myeloid lineages. Thus, the development of CD4+ TFH populations in the spleen is a prominent feature in B6.Sle1Yaa mice and these cells are not expanded in B6.Sle1YaaTLR7− mice. TFH cells are also a prominent feature of systemic autoimmunity in the san roquin mutant mouse, [30] suggesting that the activation of this T cell lineage may be an important element in the development of severe autoimmunity. Interestingly, a significant increase in infiltrating activating CD4+ T cells into the kidney is a dominant feature of B6.Sle1Yaa disease that is completely eliminated in B6.Sle1YaaTLR7− mice. This suggests that the development of these highly activated infiltrating CD4+ T cells is dependent upon the dysregulation of the innate immune system by TLR7 and that their generation is essential for severe end organ disease. An earlier report demonstrated that TLR7 activation predominantly impacted the development of kidney pathology, suggesting that activation of the innate immune system strongly influenced end organ disease [12]. Taken together, these findings suggest that an upregulation of TLR7 drives end organ disease in our model system, predominantly via the activation of the T cell lineage.
In summary, our analysis of the impact of TLR7 upregulation on disease development in B6.Sle1Yaa mice clearly indicates that subtle variations in signaling in the innate immune system are sufficient to drive severe disease pathogenesis in genetically predisposed mice. This firmly establishes that variations in signaling within the innate immune system can profoundly influence lupus development. Although mutations similar to Yaa do not appear to be a common feature of human SLE [31], allelic variants of IRF5 are strongly associated with SLE susceptibility [32]. IRF5 is a transcription factor that is induced in response to TLR signaling, most notably via TLR7, and the disease associated allele mediates increased expression of IRF5. Whether this IRF5 polymorphism in humans causes a dysregulation of TLR7 signaling analogous to that of Yaa, remains to be determined.
Materials and Methods
Reagents and mice
All mice were bred in the University of Texas Southwestern Medical Center’s specific pathogenic free (SPF) facility. The care and use of laboratory animals conformed to the National Institutes of Health guidelines and all experimental procedures conformed to an IACUC approved animal protocol. Breeding pairs for C57BL/6J (B6) mice were originally obtained from the Jackson Laboratory. The derivation of B6.Sle1Yaa has been described previously [8, 33]. Gross pathological changes in tissues such as the liver, lung and skin were not observed in the B6.Sle1Yaa mice, therefore further examination of these tissues was not carried out. Breeding pairs for TLR7−/− mice were obtained from Dr. I Rifkin at Boston University, with Dr. S Akira’s permission [34]. Resultant mice were at least 7 generations backcrossed to B6. They were tested for contaminating 129-derived Sles1 alleles which were confirmed to be of B6 origin.
Assessment of Renal Disease
Mice were caged in metabolic cages and urine was collected over a 24 hr period. Protein was measured using the Coommassie® Plus Protein Assay kit (Pierce Biotechnology Inc. Rockford, IL) as per the manufacturer’s instructions. BSA (Pierce) was used as a standard. Blood Urea Nitrogen (BUN) was assessed using the QuantiChrom Urea Assay Kit (BioAssay Systems, CA). Following euthanasia, kidneys were analyzed by an independent pathologist (JZ), as previously described [35] The severity of GN was graded using the following guidelines set by the World Health Organization: 0, normal; 1, mild increase in mesangial cellularity and matrix; 2, moderate increase in mesangial cellularity and matrix, with thickening of the GBM; 3, focal endocapillary hypercellularity with obliteration of capillary lumina and substantial increase in the thickness and irregularity of the GBM; 4, diffuse endocapillary hypercellularity, with segmental necrosis, crescents, and hyalinized end-stage glomeruli). Tubular interstitial nephritis (TIN) was graded on a 0–4 scale (Supplemental figure 4).
Serology
Autoantigen arrays on serum samples were completed at the UT Southwestern Microarray Core facility [36]. Serum ANAs were analyzed using an anti-histone-DNA antibody Elisa as described previously [37].
Cytokine Multiplex Analysis: Following an initial 22-plex screening of B6.Sle1Yaa male sera, 9 cytokines were analyzed in sera and supernatants from kidney using the Bio-Plex cytokine assay kit (Bio-Rad) as per manufacturer’s protocol at BIIR Luminex Core, Dallas.
Cell Preparation and Flow Cytometry
Peripheral Blood was taken retro-orbitally, or by cardiac puncture. Kidneys were prepared as described previously [25]. Briefly they were minced and resuspended into 0.75mls of PBS. Cells were spun down and the supernatant was kept at −20°C for cytokine analysis. Cells were resuspended in digestion buffer, consisting of Collagenase (1mg/ml) and DNase (1ug/ml) in RPMI Complete Media and incubated at 37°C for 30mins. Cells were centrifuged and filtered through a 70uM mesh and then mixed 1:1 with 40% Percoll solution. This was centrifuged 3000rpm/20mins/RT/brake off. The loose pellet was washed, counted and resuspended in staining buffer. Analysis of myeloid subtypes was based upon analyses described by Geismann and colleagues [38], with the gating strategy shown in Supplementary Figure 5. Acquisition and analysis was completed using a BD LSR II with Diva software, and Flowjo 7.2 for Windows, Treestar.
Statistical Analyses
Results are expressed as the arithmetic mean ± standard error of the mean (SE). Normality was tested using Kolmogorov and Smirnov, followed by 1-way parametric ANOVA & Tukey post hoc comparison. If normality test failed, 1-way ANOVA and Dunn’s multiple comparison test was used to test multiple comparisons. Analyses were completed using InSTAT version 3.0 and Prism 4.0 for Windows (GraphPad Software, San Diego, CA, USA).
Supplementary Material
Acknowledgments
This work was supported by funding from the National Institutes of Health and the Alliance for Lupus Research. JC is supported by NIH U19 AI057234. We thank Jane Zheng for genotyping, and Elizabeth Curry and Angela Mobley from the UT Southwestern Flow Cytometry Core.
Abbreviations
- SLE
Systemic Lupus Erythematosus
- ANA
anti-nuclear antibodies
- IC
Immune complex
- GN
glomerulonephritis
- DC
Dendritic Cell
Footnotes
Conflict of Interest
The authors declare that they have no conflict of interest.
References
- 1.Fairhurst AM, Wandstrat AE, Wakeland EK. Systemic lupus erythematosus: multiple immunological phenotypes in a complex genetic disease. Adv Immunol. 2006;92:1–69. doi: 10.1016/S0065-2776(06)92001-X. [DOI] [PubMed] [Google Scholar]
- 2.Wakeland EK, Liu K, Graham RR, Behrens TW. Delineating the genetic basis of systemic lupus erythematosus. Immunity. 2001;15:397–408. doi: 10.1016/s1074-7613(01)00201-1. [DOI] [PubMed] [Google Scholar]
- 3.Morel L, Rudofsky UH, Longmate JA, Schiffenbauer J, Wakeland EK. Polygenic control of susceptibility to murine systemic lupus erythematosus. Immunity. 1994;1:219–229. doi: 10.1016/1074-7613(94)90100-7. [DOI] [PubMed] [Google Scholar]
- 4.Rudofsky UH, Evans BD, Balaban SL, Mottironi VD, Gabrielsen AE. Differences in expression of lupus nephritis in New Zealand mixed H-2z homozygous inbred strains of mice derived from New Zealand black and New Zealand white mice. Origins and initial characterization. Lab Invest. 1993;68:419–426. [PubMed] [Google Scholar]
- 5.Wandstrat AE, Nguyen C, Limaye N, Chan AY, Subramanian S, Tian XH, Yim YS, Pertsemlidis A, Garner HR, Jr, Morel L, Wakeland EK. Association of extensive polymorphisms in the SLAM/CD2 gene cluster with murine lupus. Immunity. 2004;21:769–780. doi: 10.1016/j.immuni.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 6.Morel L, Mohan C, Yu Y, Croker BP, Tian N, Deng A, Wakeland EK. Functional dissection of systemic lupus erythematosus using congenic mouse strains. J Immunol. 1997;158:6019–6028. [PubMed] [Google Scholar]
- 7.Mohan C, Alas E, Morel L, Yang P, Wakeland EK. Genetic dissection of SLE pathogenesis. Sle1 on murine chromosome 1 leads to a selective loss of tolerance to H2A/H2B/DNA subnucleosomes. J Clin Invest. 1998;101:1362–1372. doi: 10.1172/JCI728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Subramanian S, Tus K, Li QZ, Wang A, Tian XH, Zhou J, Liang C, Bartov G, McDaniel LD, Zhou XJ, Schultz RA, Wakeland EK. A Tlr7 translocation accelerates systemic autoimmunity in murine lupus. Proc Natl Acad Sci U S A. 2006;103:9970–9975. doi: 10.1073/pnas.0603912103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morel L, Blenman KR, Croker BP, Wakeland EK. The major murine systemic lupus erythematosus susceptibility locus, Sle1, is a cluster of functionally related genes. Proc Natl Acad Sci U S A. 2001;98:1787–1792. doi: 10.1073/pnas.031336098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murphy ED, Roths JB. A Y chromosome associated factor in strain BXSB producing accelerated autoimmunity and lymphoproliferation. Arthritis Rheum. 1979;22:1188–1194. doi: 10.1002/art.1780221105. [DOI] [PubMed] [Google Scholar]
- 11.Subramanian S, Yim YS, Liu K, Tus K, Zhou XJ, Wakeland EK. Epistatic suppression of systemic lupus erythematosus: fine mapping of Sles1 to less than 1 mb. J Immunol. 2005;175:1062–1072. doi: 10.4049/jimmunol.175.2.1062. [DOI] [PubMed] [Google Scholar]
- 12.Pawar RD, Patole PS, Zecher D, Segerer S, Kretzler M, Schlondorff D, Anders HJ. Toll-like receptor-7 modulates immune complex glomerulonephritis. J Am Soc Nephrol. 2006;17:141–149. doi: 10.1681/ASN.2005070714. [DOI] [PubMed] [Google Scholar]
- 13.Anders HJ, Banas B, Linde Y, Weller L, Cohen CD, Kretzler M, Martin S, Vielhauer V, Schlondorff D, Grone HJ. Bacterial CpG-DNA aggravates immune complex glomerulonephritis: role of TLR9-mediated expression of chemokines and chemokine receptors. J Am Soc Nephrol. 2003;14:317–326. doi: 10.1097/01.asn.0000042169.23931.73. [DOI] [PubMed] [Google Scholar]
- 14.Anders HJ. A Toll for lupus. Lupus. 2005;14:417–422. doi: 10.1191/0961203305lu2102rr. [DOI] [PubMed] [Google Scholar]
- 15.Patole PS, Grone HJ, Segerer S, Ciubar R, Belemezova E, Henger A, Kretzler M, Schlondorff D, Anders HJ. Viral double-stranded RNA aggravates lupus nephritis through Toll-like receptor 3 on glomerular mesangial cells and antigen-presenting cells. J Am Soc Nephrol. 2005;16:1326–1338. doi: 10.1681/ASN.2004100820. [DOI] [PubMed] [Google Scholar]
- 16.Anders HJ, Vielhauer V, Eis V, Linde Y, Kretzler M, Perez de Lema G, Strutz F, Bauer S, Rutz M, Wagner H, Grone HJ, Schlondorff D. Activation of toll-like receptor-9 induces progression of renal disease in MRL-Fas(lpr) mice. Faseb J. 2004;18:534–536. doi: 10.1096/fj.03-0646fje. [DOI] [PubMed] [Google Scholar]
- 17.Jorgensen TN, Thurman J, Izui S, Falta MT, Metzger TE, Flannery SA, Kappler J, Marrack P, Kotzin BL. Genetic susceptibility to polyI:C-induced IFNalpha/beta-dependent accelerated disease in lupus-prone mice. Genes Immun. 2006;7:555–567. doi: 10.1038/sj.gene.6364329. [DOI] [PubMed] [Google Scholar]
- 18.Pawar RD, Ramanjaneyulu A, Kulkarni OP, Lech M, Segerer S, Anders HJ. Inhibition of Toll-like receptor-7 (TLR-7) or TLR-7 plus TLR-9 attenuates glomerulonephritis and lung injury in experimental lupus. J Am Soc Nephrol. 2007;18:1721–1731. doi: 10.1681/ASN.2006101162. [DOI] [PubMed] [Google Scholar]
- 19.Barrat FJ, Meeker T, Chan JH, Guiducci C, Coffman RL. Treatment of lupus-prone mice with a dual inhibitor of TLR7 and TLR9 leads to reduction of autoantibody production and amelioration of disease symptoms. Eur J Immunol. 2007;37:3582–3586. doi: 10.1002/eji.200737815. [DOI] [PubMed] [Google Scholar]
- 20.Thibault DL, Chu AD, Graham KL, Balboni I, Lee LY, Kohlmoos C, Landrigan A, Higgins JP, Tibshirani R, Utz PJ. IRF9 and STAT1 are required for IgG autoantibody production and B cell expression of TLR7 in mice. J Clin Invest. 2008 doi: 10.1172/JCI30065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 22.Vollmer J, Tluk S, Schmitz C, Hamm S, Jurk M, Forsbach A, Akira S, Kelly KM, Reeves WH, Bauer S, Krieg AM. Immune stimulation mediated by autoantigen binding sites within small nuclear RNAs involves Toll-like receptors 7 and 8. J Exp Med. 2005;202:1575–1585. doi: 10.1084/jem.20051696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Savarese E, Chae OW, Trowitzsch S, Weber G, Kastner B, Akira S, Wagner H, Schmid RM, Bauer S, Krug A. U1 small nuclear ribonucleoprotein immune complexes induce type I interferon in plasmacytoid dendritic cells through TLR7. Blood. 2006;107:3229–3234. doi: 10.1182/blood-2005-07-2650. [DOI] [PubMed] [Google Scholar]
- 24.Migliorini P, Baldini C, Rocchi V, Bombardieri S. Anti-Sm and anti-RNP antibodies. Autoimmunity. 2005;38:47–54. doi: 10.1080/08916930400022715. [DOI] [PubMed] [Google Scholar]
- 25.Wang A, Fairhurst A-M, Tus K, Subramanian S, Liu Y, Lin F, Igarashi P, Zhou XJ, Batteux F, Wong D, Wakeland EK, Mohan C. CXCR4/CXCL12 dysregulation plays a pivotal role in murine lupus nephritis. Submitted. 2008 doi: 10.4049/jimmunol.0801920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Togel F, Isaac J, Hu Z, Weiss K, Westenfelder C. Renal SDF-1 signals mobilization and homing of CXCR4-positive cells to the kidney after ischemic injury. Kidney Int. 2005;67:1772–1784. doi: 10.1111/j.1523-1755.2005.00275.x. [DOI] [PubMed] [Google Scholar]
- 27.Pisitkun P, Deane JA, Difilippantonio MJ, Tarasenko T, Satterthwaite AB, Bolland S. Autoreactive B cell responses to RNA-related antigens due to TLR7 gene duplication. Science. 2006;312:1669–1672. doi: 10.1126/science.1124978. [DOI] [PubMed] [Google Scholar]
- 28.Deane JA, Pisitkun P, Barrett RS, Feigenbaum L, Town T, Ward JM, Flavell RA, Bolland S. Control of toll-like receptor 7 expression is essential to restrict autoimmunity and dendritic cell proliferation. Immunity. 2007;27:801–810. doi: 10.1016/j.immuni.2007.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bekeredjian-Ding IB, Wagner M, Hornung V, Giese T, Schnurr M, Endres S, Hartmann G. Plasmacytoid dendritic cells control TLR7 sensitivity of naive B cells via type I IFN. J Immunol. 2005;174:4043–4050. doi: 10.4049/jimmunol.174.7.4043. [DOI] [PubMed] [Google Scholar]
- 30.Vinuesa CG, Cook MC, Angelucci C, Athanasopoulos V, Rui L, Hill KM, Yu D, Domaschenz H, Whittle B, Lambe T, Roberts IS, Copley RR, Bell JI, Cornall RJ, Goodnow CC. A RING-type ubiquitin ligase family member required to repress follicular helper T cells and autoimmunity. Nature. 2005;435:452–458. doi: 10.1038/nature03555. [DOI] [PubMed] [Google Scholar]
- 31.Kelley J, Johnson MR, Alarcon GS, Kimberly RP, Edberg JC. Variation in the relative copy number of the TLR7 gene in patients with systemic lupus erythematosus and healthy control subjects. Arthritis Rheum. 2007;56:3375–3378. doi: 10.1002/art.22916. [DOI] [PubMed] [Google Scholar]
- 32.Graham RR, Kozyrev SV, Baechler EC, Reddy MV, Plenge RM, Bauer JW, Ortmann WA, Koeuth T, Gonzalez Escribano MF, Pons-Estel B, Petri M, Daly M, Gregersen PK, Martin J, Altshuler D, Behrens TW, Alarcon-Riquelme ME. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet. 2006;38:550–555. doi: 10.1038/ng1782. [DOI] [PubMed] [Google Scholar]
- 33.Morel L, Croker BP, Blenman KR, Mohan C, Huang G, Gilkeson G, Wakeland EK. Genetic reconstitution of systemic lupus erythematosus immunopathology with polycongenic murine strains. Proc Natl Acad Sci U S A. 2000;97:6670–6675. doi: 10.1073/pnas.97.12.6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hemmi H, Kaisho T, Takeuchi O, Sato S, Sanjo H, Hoshino K, Horiuchi T, Tomizawa H, Takeda K, Akira S. Small anti-viral compounds activate immune cells via the TLR7 MyD88-dependent signaling pathway. Nat Immunol. 2002;3:196–200. doi: 10.1038/ni758. [DOI] [PubMed] [Google Scholar]
- 35.Xie C, Zhou XJ, Liu X, Mohan C. Enhanced susceptibility to end-organ disease in the lupus-facilitating NZW mouse strain. Arthritis Rheum. 2003;48:1080–1092. doi: 10.1002/art.10887. [DOI] [PubMed] [Google Scholar]
- 36.Li QZ, Xie C, Wu T, Mackay M, Aranow C, Putterman C, Mohan C. Identification of autoantibody clusters that best predict lupus disease activity using glomerular proteome arrays. J Clin Invest. 2005;115:3428–3439. doi: 10.1172/JCI23587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mohan C, Morel L, Yang P, Watanabe H, Croker B, Gilkeson G, Wakeland EK. Genetic dissection of lupus pathogenesis: a recipe for nephrophilic autoantibodies. J Clin Invest. 1999;103:1685–1695. doi: 10.1172/JCI5827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.