

Abstract
Asymmetric total synthesis of bryostatin 16 was achieved in 26 steps in the longest linear sequence/39 total steps from aldehyde 10. A Pd-catalyzed alkyne-alkyne coupling was employed for the first-time as a macrocyclization method in a natural product synthesis. A route to convert bryostatin 16 to a new family of bryostatin analogues was developed. Toward the end, 20-epi-bryostatin 7, was synthesized from a bryostatin 16-like intermediate; and the key step involves a Re-catalyzed epoxidation/ring-opening reaction. Preliminary biological studies indicated that this new analogue exhibits nanomolar anti-cancer activity against several cancer cell lines.
Introduction
Bryostatins 1–20 were first isolated in 1968 by Pettit and co-workers from the marine bryozoan Bugula neritina (Figure 1).1,2 These structurally complex macrolides exhibit a remarkable range of biological activities, including antineoplastic activity,3 synergistic chemotheoreputic activity,4 cognition and memory enhancement,5 recovery of brain damage,6 etc. Stimulated by these appealing biological activities, total syntheses of bryostatins and their analogues have been an attractive goal. The structures of the bryostatins constitute significant synthetic challenges, which include a 26-membered lactone fused by three heavily substituted polyhydropyran (PHP) rings, two acid/base-sensitive exo-cyclic unsaturated esters, one congested C16–C17 trans-olefin, as well as numerous oxygen-containing functionalities and stereogenic centers. Previously, only three of the twenty bryostatins have been accessed by total synthesis. In 1990, Masamune and coworkers accomplished the synthesis of bryostatin 7.7 Their strategy involved use of a highly chemoselective macrolactonization to construct the macrocycle and use of Julia olefination8 to couple the northern fragment and southern fragment. In 1998, another family member, bryostatin 2, was synthesized by Evans et al.9 A key feature of Evans' synthesis is that they segregated the target into three polyhydropyran-containing subunits with similar complexity, and then assembled them via Julia olefination, sulfone alkylation and macrolactonization. Notably, Evans' synthesis also constitutes a formal synthesis of bryostatin 1.10 In 2000, bryostatin 3, a structurally unique family member, was synthesized by Ohmori, Nishiyama and Yamamura et al.11 Julia olefination was also employed to form the C16–C17 alkene, and a high yielding Yamaguchi esterification was used to furnish the 26-membered lactone. Recently, Hale and coworkers reported a concise synthesis of Masamune's southern fragment, which was recognized as a formal total synthesis of bryostatin 7.12 These elegant syntheses have illustrated the power of organic synthesis for the creation of molecules of extreme complexity; however, their lengths (>40 steps in the longest linear sequence and >70 total steps) have so far restrained them from serving as a practical supply source for this natural product.
Figure 1.

Bryostatins
An attractive goal in synthesis is to access multiple targets with a single route and multiple analogues via a common intermediate. This concept particularly bodes well for the bryostatin synthesis as this natural product contains twenty congeners and syntheses of their analogues are equally valuable.13 Among all the twenty bryostatins, we noticed that the structures of bryostatins 16 (1) and 1714 possess a unique feature (Figure 1): their C-ring contains a relatively reactive dihydropyran (DHP) moiety. By elaboration of this electron-rich and relatively reactive C19–C20 olefin, we envisioned that bryostatin 16 could act as a pivotal parent structure to allow access to almost all the other naturally occurring bryostatins (except bryostatin 3, 19 and 20).15 For example, oxidation of C19–C20 olefin could lead to bryostatins 1, 2, 4–9, 12, 14 and 15; a formal hydration of this C-C double bond should provide potential access to the C20-deoxy bryostatins (10, 11, 13, 18). Furthermore, bryostatin 16 also offers an ideal forum for us to employ a Pd-catalyzed tandem alkyne-alkyne coupling followed by 6-endo-dig cyclization methodology to access the C-ring DHP motif in an efficient and rapid fashion.16 In addition, simply by variations in this natural product's synthesis, we should be able to obtain new analogues that might not be easily available from other syntheses. In this full article, we provide a detailed account of our completion of the synthesis of bryostatin 16, and demonstrate a proof of principle in a concise synthesis of 20-epi-bryostatin 7 as a potent anticancer agent from a bryostatin 16-like intermediate.17
Results and Discussion
First Generation Strategy towards the Synthesis of Bryostatin 16
Given the difficulties of late-stage installation of the C16–C17 olefin18, we conceived of a strategy to introduce this sterically hindered alkene at an earlier stage. From a retrosynthetic viewpoint, the 26-membered lactone could be accessed via either macrolactonization of a seco acid or intramolecular transesterification of a seco methyl ester such as 2. The C-ring of bryostatin would be synthesized via the Pd-catalyzed diyne coupling between donor alkyne 3 and acceptor alkyne 4 followed by endo-cyclization of the secondary alcohol to form the DHP entity. The A-ring could be formed through acid-catalyzed tandem transesterification followed by methyl ketal formation from lactone 5. The 4-methylene-2,6-cis-tetrahydropyran (THP) moiety in 5 provides a perfect opportunity to examine another tandem transformation, the Ru-catalyzed alkene-alkyne coupling/Michael addition methodology (Scheme 1)19 between two rather complex fragments (6 and 7). Ideally, all of the three PHP rings of bryostatin 16 could be accessible via three tandem, catalytic, and atom-economical transformation.
Scheme 1.

First generation Strategy
Alkene 6, one of the key-coupling partners, has been previously synthesized in 16 overall steps from commercially available (R)-pantolactone (8) (Scheme 2).13f Although this synthesis is practical, to improve the efficiency our initial goal was to shorten the synthesis of fragment 6.
Scheme 2.

Previous Synthesis of Alkene 6
Starting from aldehyde 10, the same aldehyde used in the synthesis of fragment 7,20 aldehyde 9 was quickly afforded by asymmetric Brown allylation with (−)-β-allyldiisopinocampheylborane [Ipc2B(allyl)],21 followed by PMB protection with PMBBr/NaH, and oxidative cleavage of the terminal olefin (Scheme 3).22 As aldehyde 9 is a common intermediate with our previous route, with this modification alkene 6 is now available in 11 steps from aldehyde 10 (eq 1) which, in turn, is commercially available or derived in two steps from 2,2-dimethyl-1,3-propanediol.19
 |
(1) |
Scheme 3.

Improved Synthesis of Alkene 6
With both alkene 6 and alkyne 7 in hand, the Ru-catalyzed tandem alkene-alkyne coupling/Michael addition proceeded to generate cis-tetrahydropyran 5 (eq 2). The chemoselectivity was further demonstrated by the high compatibility of a β,γ-unsaturated ketone, a six-membered lactone, an unprotected allylic alcohol, a PMB ether and two different silyl ethers in this reaction. DCM was found to be the optimal solvent, while acetone or a DCM-DMF mixed solvent gave either lower conversion or more decomposition. Notably, only 1.2 equiv of alkene 6 was required in this coupling reaction. Though the yield is moderate, the yield based upon recovered starting material is high (80%, both of the starting materials can be recycled), presumably due to the fact that additional olefin functionality in the alkyne fragment could limit turnover of the Ru catalyst. This result has proved highly reproducible, and the reaction is scalable to several grams.
 |
(2) |
Advancement of lactone 5 is depicted in Scheme 4. Bromination of the exo-cyclic vinyl silane with NBS provided vinyl bromide 12 with retention of the olefin geometry in 98% yield. Subsequently, we attempted a one-pot acid-catalyzed transesterification–methyl ketalization–desilylation transformation. To our delight, treatment of lactone 12 with a catalytic amount of CSA in MeOH cleanly afforded the desired alcohol 13 containing both the A-ring and B-ring substructures in 93–96% yield (on a gram-scale). Use of the conditions (PPTS, HC(OMe)3, MeOH, reflux), previously reported for a similar transesterification/ketalization,13f gave a messy mixture. We envisaged that the vinyl bromide functionality would serve as a convenient handle in the future for the syntheses of bryostatin analogues via the use of metal-catalyzed coupling reactions. As the natural product contains a conjugated methyl ester at the C30 position, a carbonylation reaction of 13 was next examined. On a smaller scale (less than 100 mg), Pd(PPh3)4 acted as a good carbonylation catalyst, giving up to 78% yield (94% yield brsm); however, on a larger scale, these conditions suffered from poor conversion (less than 50% conversion). After surveying a number of Pd-catalysts, this problem was eventually solved by using PdCl2(CH3CN)2-dppf as the catalyst in which an 83% yield (90% yield brsm) of ester 14 was obtained on a 0.76 g scale.
Scheme 4.

Synthesis of Aldehyde 15
The conditions for oxidizing primary alcohol 14 to aldehyde 15 were next optimized (Table 1). It is known that the methyl ketal of substrates like 14 is very sensitive to acidic conditions.23,24 Thus, Ley oxidation (TPAP-NMO)25, known as a non-acidic oxidation method, was initially employed. Using 0.1 equiv TPAP and 3 equiv NMO in CH3CN, the reaction gave full conversion but only 65% yield of aldehyde 15 (entry 1). By carefully tuning the reaction conditions, we found that lowering the catalyst loading (entry 3) or the oxidant loading (entry 4) or both (entry 5), gave cleaner reactions and higher yield (up to 77% yield). DCM was also discovered to be a more suitable solvent than CH3CN. Subsequently, a more effective oxidation was found; treatment of alcohol 14 with Dess-Martin periodinane (DMP) and excess NaHCO3, efficiently afforded aldehyde 15 in 88% yield on a 0.83-gram scale.
Table 1.
Oxidation of Alcohol 14
| Entry | Conditions | Yieldb |
|---|---|---|
| 1 | TPAP(0.1 equiv), NMO (3 equiv), 4A M.S., CH3CN | 65% |
| 2 | TPAP(0.1 equiv), NMO (2 equiv), 4A M.S., CH3CN | 67% |
| 2 | TPAP(0.1 equiv), NMO (2 equiv), 4A M.S., DCM | 56–70% |
| 3 | TPAP(0.08 equiv), NMO (2 equiv), 4A M.S., DCM | 74%a |
| 4 | TPAP(0.1 equiv), NMO (1.5 equiv), 4A M.S., DCM | 77%a |
| 5 | TPAP(0.05 equiv), NMO (1.5 equiv), 4A M.S., DCM | 77%a |
| 6 | DMP(3 equiv), NaHCO3 (20 equiv), DCM | 88% (0.83 g scale) |
The reaction did not give full conversion.
Isolated yield.
Advancement of aldehyde 15 to the key-coupling partner 3 was achieved in two steps (eq 3). Treatment of 15 with the Ohira-Bestmann reagent nicely provided the corresponding terminal alkyne in quantitative yield. As removal of the TBDPS protecting group at a late stage proved to be problematic,23 it was removed before the subsequent coupling with the other fragment. Due to its robust nature, the TBDPS deprotection required some experimentation. When 2.5 equiv TBAF (1.0 M in THF) was used, alcohol 3 was isolated in only 60% yield along with a significant amount of retro-aldol, β-hydroxyl elimination, and ester hydrolysis byproducts. Formation of those byproducts was likely caused by the strong basic nature of TBAF as well as the hydroxide present in commercial TBAF. To minimize those undesired reaction pathways, we envisioned that it would be helpful to add a buffer and to control the reaction pH and concentration. Indeed, treatment of the TBDPS ether with less TBAF (1.1 equiv) and buffered with 20 mol% HOAc at a lower concentration, led to the chemoselective cleavage of the TBDPS group and provided alcohol 3 in 90% yield (96% brsm).
 |
(3) |
With alkyne 3 in hand, we continued to examine the plan of uniting the two fragments together via diyne coupling and then closing the macrocycle via lactonization. Pd-catalyzed alkyne-alkyne coupling between fragments 3 and 4 proceeded very well, giving enyne 16 in 80% yield (Scheme 5). A gold-catalyzed 6-endo-dig cyclization was subsequently employed to build the C-ring subunit.26 A 4:1 mixture of DCM-CH3CN was found to be the optimal choice of solvent, and dihydropyran 17 was isolated as the major product in 65% yield.
Scheme 5.

End Game of the First Generation Strategy
The acetonide protecting group was efficiently removed with a catalytic amount of CSA in MeOH. However, under those conditions, the external C21–C34 olefin was isomerized, yielding a ~1:1 mixture of olefin geometric isomers (18). Transesterification of seco-ester 18 to form the macrolactone 19 was briefly explored. Treatment of 18 with 10 mol% Otera's catalyst27 in refluxing toluene or hexanes, however, did not provide any macrocycle, even when using 5Å molecular sieves to remove MeOH. Attempts to hydrolyze the C1 methyl ester of 18 followed by macrolactonization was not fruitful either. The major difficulties were due to the fragile nature of the external C21–C34 olefin leading to the existence of both the starting materials and the potential products as isomeric mixtures which complicated product identification and NMR spectral interpretation. Although this first generation plan still represents a valid route, given those late-stage difficulties, we resorted to a new strategy.
Second Generation Strategy towards the Synthesis of Bryostatin 16
Given the severe acid sensitivity of the C-ring, instead of carrying it through, we conceived of a strategy for constructing the C-ring of bryostatin 16 at the very end of the synthesis. The benefits of this strategy also include flexible late-stage variations for access to other bryostatins or analogues, as well as minimization of functional group transformations and protecting group usage. As early as in 1989, we have demonstrated the principle of using Pd-catalyzed α,ω-diyne cycloisomerization to form macrocycles.28 Use of this method in natural product synthesis, however, has not been previously established. While all the previous bryostatin total syntheses have relied on assembling the macrocycle by a demanding Julia olefination followed by a lactonization, we foresaw that we could employ the Pd-catalyzed alkyne-alkyne coupling as a novel macrocyclization method followed by a metal-catalyzed 6-endo-dig cyclization to construct both the macrolactone and the C-ring of byostatin 16 simultaneously (Scheme 6). Esterification between fragments 22 and 23 would provide the requisite diyne precursor. Acid 22 and alcohol 23 would be synthesized respectively from intermediates 3 and 4 from our first route.
Scheme 6.

Synthetic Plan of the Second Generation Strategy
Starting from the β-hydroxy methyl ester 3, the challenge was to hydrolyze the C1 methyl ester chemoselectively in the presence of the C31 methyl ester. We found that heating 3 with trimethyltin hydroxide in DCE29 provided the desired C1 acid (24) in 84% yield (Scheme 7). The desired chemoselectivity was attributed to two factors: first, the conjugated esters are generally less reactive towards hydrolysis than the non-conjugated ones due to the more delocalized π-systems; second, the Lewis acidity of trimethyltin hydroxide allows the adjacent β-hydroxy group to act as a directing group in this saponification reaction. Due to the acid-sensitivity of the methyl ketal functionality, acid 24 had to be handled with sufficient care.30 Subsequent TES protection of the secondary alcohol completed the synthesis of acid fragment 22.
Scheme 7.

Synthesis of Acid Fragment 22
Alcohol fragment 23 was synthesized in three straightforward steps from acetonide intermediate 4.31 Cu(OTf)2-catalyzed PMB protection of the secondary alcohol,32 acid-mediated hydrolysis of the acetonide moiety to provide the vicinal diol, and selective TBS-ether protection of the less hindered alcohol gave the alcohol fragment 23 (eq 4). A somewhat diminished yield was observed on a 0.4 gram scale in contrast to a 66% yield over three steps on a 3.7 mg scale, which is caused by incomplete conversion of the acetonide hydrolysis on a large scale. As a higher yield can be obtained, this step will be validated by future optimization studies.
 |
(4) |
Esterification between acid 22 and alcohol 23 was studied under several conditions (Scheme 8). Use of anhydride 27 as the coupling reagent has recently been developed by Shiina,33 which has been demonstrated as a mild and efficient esterification method. Indeed, use of Shiina's method provided the desired ester (26), albeit only in 15–19% yield. Under the reaction conditions, acid 22 was partially decomposed to its desilylated precursor 24. This reaction also suffered from reproducibility issues on larger scale. Switching to the more traditional Yamaguchi method,34 led to satisfactory and consistent results, and ester 26 was isolated in 70–92% yield.
Scheme 8.

Esterification between Acid 22 and Alcohol 23
Removal of both PMB protecting groups in one step from 26 proved to be non-trivial (Scheme 9). Treatment of ester 26 with excess DDQ under buffered conditions gave a mixture containing mono-deprotection product 28, diol 21 and lactol 29. One interesting observation was that cleavage of the C7 PMB ether was much faster than the one at the C23 position. However, this reaction did not afford complete conversion to diol 21 without generating byproduct 29. Addition of 2,2-dimethoxypropane has been attempted to prevent the undesirable ketal hydrolysis, but this did not help. After fine-tuning of reaction concentration and the DDQ stoichiometry, we settled on quenching the reaction at a point, which provided 52% yield of mono-deprotection product 28 and 46% yield of double-deprotection product 21 with minimum formation of byproduct 29. The mono-PMB ether 28 can be recycled and re-subjected to the DDQ-deprotection conditions to provide more diol 21 in an overall yield of around 72% from ester 26.
Scheme 9.

PMB Deprotection
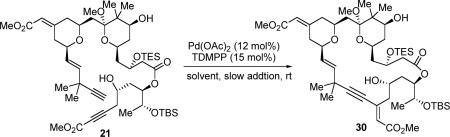
With diyne 21 in hand, the stage was now set for the macrocyclization (eq 5). To our delight, the desired macrocycle 30 was provided in 22% yield (44% brsm) when a solution of diyne 21 in benzene was slowly added via syringe pump to the catalyst solution that contains 12 mol % Pd(OAc)2 and 15 mol % tri(2,6-dimethoxyphenyl)-phosphine (TDMPP) (Table 2, entry 1). Note that a slightly higher ligand/Pd ratio (1.25:1) was used here, since a 1:1 ratio proved less efficient. Solvent effects were next examined. Use of toluene as the solvent proved to be most effective, providing up to 56% yield of macrocycle 30 (entry 3). In contrast, use of THF as the solvent gave a sluggish reaction and only 23% yield was obtained after 5 days (entry 2). Like other macrocyclizations, low concentration [0.002M] proved to be critical; otherwise, formation of the dimeric byproducts could be observed. This macrocyclization was also performed on a 45-mg scale, but the conversion was lower than the smaller scale reaction (entry 4). To the best of our knowledge, this represents the first example of using a Pd-catalyzed alkyne-alkyne coupling as a macrocyclization method for complex natural product synthesis, which illustrates a new avenue of using C-C bond formation to construct a macrocycle.
Table 2.
Macrocyclization via Alkyne-Alkyne Coupling
 |
(5) |
| Entry | Solvent | Yield |
|---|---|---|
| 1 | benzene | 22% (44% brsm) |
| 2 | THF | 23% (5 d) |
| 3a | toluene | 56% |
| 4b | toluene | 36% (57% brsm) |
on 3.2 mg scale
on 45 mg scale
Mechanistically,35 the Pd catalyst chemoselectively inserts into the C-H bond of the terminal alkyne and then eliminates one molecule of acetic acid. Subsequent coordination with the disubstituted alkyne then sets the stage for the chemo- and regioselective intramolecular migratory insertion. The resulting vinylpalladium motif is next protonated by the acetic acid to regenerate the Pd(OAc)2-TDMPP catalyst (Scheme 10).
Scheme 10.

Plausible Mechanism for the Pd-catalyzed Macrocyclization
The remaining challenge was to conduct a 6-endo-dig cyclization to form the C-ring of bryostatin. This cyclization has to proceed within the macrocycle, and the conformational bias of the macrocycle may interfere with the desired process. To our delight, use of the cationic gold catalyst gave satisfactory results (Scheme 11). When 20 mol% catalyst was employed, the THP product (34) was isolated in 56–73% yield. Notably, likely due to the Lewis acidity of the catalyst, the methyl ketal moiety was hydrolyzed under the cyclization conditions. Subsequent pivalation of the hindered secondary alcohol under rather forcing conditions (Piv2O 50 equiv, DMAP 80 equiv, 50 °C)36 did afford the pivalate ester (35) in 62% yield. Other acylation methods, such as use of pivaloyl chloride/Py/DMAP, proved to be less efficient.
Scheme 11.

Synthesis of THP 35
The following global deprotection turned out to be nontrivial. Treatment of pivalate ester 35 with HF-pyridine or aqueous HF provided a four-component mixture, which could be further separated into two mixtures: one containing bryostatin 16 (36) and one containing bryostatin 17 (37) (eq 6). The ratio between mixtures 36 and 37 was roughly 1.5 to 1, determined via 1H NMR spectroscopy. Bryostatin 17 was likely formed via the acid-catalyzed isomerization of the C21–C34 olefin; but what were the other two compounds isolated with bryostatin 16 and 17?
 |
(6) |
We hypothesized that these two mysterious compounds could be the C9 hemi-ketal diastereomers of the natural products. Although the hemiketal isomer of the THP A-ring drawn in the natural product could be the thermodynamically most stable one if secluded by itself, our previous bryostatin analogue synthesis13f indicated that this may not be true when the THP was fused in a macrocycle and there is a possibility to form both hemiketal isomers. In addition, when the A-ring conformation of the 36/37 mixtures was locked by forming the methyl ketals, the formed products contained only two compounds instead of four (eq 7), which serves as another evidence for our hypothesis, that the two unknown compounds are the C9 epimers of bryostatins 16 and 17.
 |
(7) |
Given the extreme acid-sensitivity of this natural product, we switched to basic desilylation conditions. Fortunately, treatment of 35 with 5 equiv of TBAF and direct purification by reverse phase HPLC37 successfully provided bryostatin 16 (eq 8), which was spectroscopically identical to the literature (Reported optical rotation [α]D: + 84, c 0.43, MeOH; Found [α]D: + 81, c 0.04, MeOH).14
 |
(8) |
Total Syntheses of 20-epi-Bryostatin 7
With completion of the total synthesis of bryostatin 16, we decided to address the question of how to convert brystatin 16 or a bryostatin 16-like compound to other structurally related bryostatins or their analogues (Figure 2).
Figure 2.
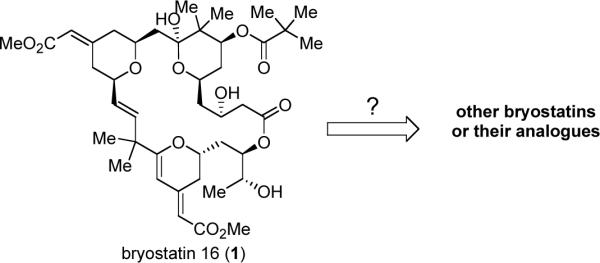
As illustrated in Scheme 12, during our bryostatin 16 synthesis, the yields for the last several steps were only moderate; and for certain steps, such as DDQ deprotection and macrocyclization, conversion was an issue. To permit enough materials to advance those intermediates, the initial goal was to improve or modify these challenging steps.
Scheme 12.

Some Challenging Steps in the Bryostatin 16 Synthesis
As we observed earlier, the PMB group on the C7 alcohol was cleaved faster than the one on the C23 alcohol. By carefully controlling the DDQ stoichiometry (2 equiv) and the reaction temperature (0 °C), the C7 PMB group was chemoselectively removed, providing alcohol 28 in 77% yield (Scheme 13). Subsequent acylation of the secondary C7 alcohol with acetic anhydride and pyridine afforded the corresponding ester in 91% yield. Due to the electron-withdrawing nature of the OAc group, the C9 ketal of the resultant ester became somewhat stabilized and thus less sensitive to acid than its precursor 28. Consequently, treatment with excess DDQ (10 equiv) at room temperature cleanly led to the second PMB-cleaved product (39) in 90% yield.
Scheme 13.

Synthesis of Key Intermediate 41
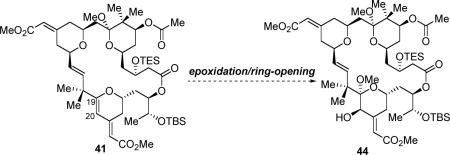
Macrocyclization with diyne precursor 39 under the conditions developed earlier was effective; however, the yield for macrocycle 40 was only moderate: 41% (71% brsm), and the reaction was slow (3 days). Having surveyed several factors for this Pd-catalyzed alkyne-alkyne coupling, we eventually found that addition of a mild proton source, like an alcohol, was beneficial. As precursor 39 has a methyl acetal moiety, methanol was used as the additive. With this variation, the yield for this macrocyclization was increased to 65% (72% brsm), and the reaction rate was also enhanced (40 h). The exact reason why an alcohol additive improved both the reaction yield and rate is unclear. We conjecture that a proton source might facilitate protonation of the vinyl palladium intermediate generated in the catalytic cycle (see Scheme 10), which eventually increases the turnover efficiency of the catalyst. The conditions for the 6-endo-dig cyclization were next optimized. When macrocycle 40 was subjected to the conditions developed earlier [AuCl(PPh3) (20 mol%), AgSbF6 (20 mol%), NaHCO3, DCM/CH3CN, 0 °C to rt], in contrast to the reaction of 30, a complex mixture, containing the desired product (41), the product with methyl-ketal hydrolyzed, the starting material (40) and the starting material with methyl-ketal hydrolyzed was observed. Switching to a platinum catalyst38 [(PtCl2(CH2=CH2))2 (20 mol%), NaHCO3, DCM or ether, rt] did save the methyl ketal, however, the C21–C34 olefin was isomerized under those reaction conditions. Finally, the problem was solved by adding 2,2-dimethoxy propane to the Au-catalyzed cyclization reaction. To our delight, under the modified conditions, dihydropyran 41 was obtained in 80–83% yield with the methyl ketal remaining intact. During this reaction, 2,2-dimethoxy propane was believed to act as both an acid scavenger and a methyl ketal repairer.
With a scalable route to access the bryostatin 16 intermediate (41), efforts were next taken to explore the conditions for the chemoselective and diastereoselective oxidation of the C19–C20 olefin in the presence of three other olefins. An asymmetric dihydroxylation reaction seemed to be the most straightforward method to install two oxygen functionalities stereoselectively on both of the C19 and C20 carbons. Under the Sharpless' asymmetric dihydroxylation (SAD) conditions,39 to our surprise, the oxidation occurred almost exclusively at the C13–C30 olefin, the olefin that was supposed to be least reactive towards oxidation due to its electron-deficient nature (eq 9). As a result, diol 43 was isolated as the major product in 70% yield; while the more electron-rich C19–20 alkene remains intact. The structure of diol 43 has been carefully confirmed by 1H NMR, IR, gCOSY, gHSQC, gHMBC, ROSEY and HRMS. The stereochemistry of the formed diols was tentatively assigned according to the Sharpless model.39 This unusual chemoselectivity could be explained plausibly by the fact that SAD reaction is more sensitive to steric factors than electronic ones due to the bulkiness of the chiral Os-catalyst.
 |
(9) |
Epoxidation is known to be more sensitive to electronic effects compared to steric effects, thereby a strategy of epoxidation followed by ring-opening with an alcohol was next pursued (eq 10). Although trifluoroperacetic acid (TFPAA) was used to oxidize a less complicated substrate containing a similar C-ring motif,13f reaction of 41 under the same conditions only resulted in decomposition of the starting material (Table 3, entry 1). Payne oxidation,40 with use of either CH3CN or CCl3CN as the oxidant presursor, failed to provide any identifiable products (entries 2 and 3). On the other hand, a Re-catalyzed epoxidation, using methyl rhenium trioxide (MTO) as the catalyst and urea-hydrogen peroxide (UHP) as the oxidant was promising.41 Under these conditions, the epoxidation/ring-opening product (44) was isolated in around 10% yield, along with some TES-cleavage products (entry 4). Although the yield was low, we were encouraged about the chemoselectivity.
Table 3.
An Epoxidation/Ring-Opening Strategy
 |
(10) |
| Entry | Conditions | Results |
|---|---|---|
| 1 | TFPAA, CH3CN, Na2HPO4, DCM/MeOH 0 °C to rt | decomposition of 41 |
| 2 | CCI3CN, UHP, Na2HPO4, DCM/MeOH 0 °C to rt | complete decomposition |
| 3 | CH3CN, UHP, KHCO3, MeOH rt | complex mixture |
| 4 | MTO (10 mol%), UHP (2 equiv), MeOH, 0 °C | 44 (ca 10%)+TES-cleavage products |
We assumed that TES-cleavage was caused by the acid generated during the reaction, thus buffering of the reaction with a base should minimize the undesirable desilylation. Moreover, it is also known that Lewis base can act as a ligand to accelerate the Re-catalyzed epoxidation reaction.42 Indeed, when N-methylimidazole (50 mol%) was added, the reaction proceed with full conversion in two hours at 0 °C. Surprisingly, epoxide 45 turned out to be very stable under those reaction conditions, and it could be isolated via aqueous workup (Scheme 14).43 Epoxide 45 was subsequently ring-opened with MeOH, either by addition of ZnCl2 solution to the reaction mixture in a one-pot fashion or treatment of the crude epoxide with dilute HOAc in MeOH, providing C20 alcohol 44 in 48% or 64% yield respectively. It is not totally unexpected that the stereochemistry of the C20 alcohol in 44 was opposite to the one in the natural bryostastin,44 as the Re catalyst likely attacks at the less hindered face of the C-ring at the epoxidation stage. However, it represented the feasibility to function the C19–C20 olefin chemoselectively in a bryostatin-16 like compound!
Scheme 14.

Synthesis of Alcohol 44
Synthesis of 20-epi-Bryostatin 7
We envisaged that the unnatural C20-epimer of bryostatins, could serve as an attractive new family of bryostatin analogues for two reasons: first, they might retain the biological activities that are comparable or complementary to those of the natural products due to their closely related structures; second, study of these analogues could contribute to our understanding of the structure-activity relationships (SAR) of bryostatins, especially about the role of the C-ring unit generally and the C20 stereocenter specifically.
Consequently, we explored the synthesis of the analogue 20-epi-bryostatin 7. Acylation of the C20 alcohol with acetic anhydride, followed by global deprotection with aqueous HF, afforded 20-epi-bryostatin 7 (47) in 63% yield over two steps (Scheme 15). The structure of 47 was confirmed by 1H, gCOSY, gHSQC, gHMBC, IR and HRMS.45 Note that we are able to install the C7 and C20 ester groups in 47 at different stages, thus this route also represents a valid access to other bryostatin analogues.
Scheme 15.

End Game of the Synthesis of 20-epi-Bryostatin 7 (47)
20-epi-Bryostatin 7 was next tested against several cancer cell lines in a biological assay. Initial biological studies showed that this bryostatin analogue possessed nanomolar potency against DOHH2—a Lymphoma cancer cell line, Granta 519—a Lymphoma cancer cell line, and Jurkat— a T-lymphocyte cancer cell line (Table 4). These biological studies indicate that this new bryostatin analogue could be a potential drug lead for anti-cancer chemotherapy.
Table 4.
Anticancer Activity of 20-epi-Bryostatin 7 (47)a
| Cell lines | DOHH2 | Granta 519 | Jurkat |
|---|---|---|---|
| IC50 (nM) | 22.5 | 17.6 | 44.7 |
DOHH2: a Lymphoma cancer cell line; Granta 519: a Lymphoma cancer cell line; Jurkat: a T-lymphocyte cancer cell line
Conclusion
In summary, we have developed a unique and highly concise strategy (26 steps in the longest linear sequence, 39 total steps from aldehyde 10, eq 11) for the asymmetric total synthesis of bryostatin 16. A Pd-catalyzed alkyne-alkyne coupling was employed for the first-time as a macrocyclization method in natural product synthesis. The efficiency of our synthesis can also be attributed to a tandem Ru-catalyzed alkene-alkyne coupling/Michael addition to form the B-ring, an acid-catalyzed one-pot cascade to form the A-ring, a directed chemoselective ester-hydrolysis, and a palladium/gold-catalyzed cascade to form the C-ring of bryostatin 16. These atom-economical and/or chemoselective approaches not only are useful in bryostatin syntheses, but should also be indicative for the synthesis of numerous other polyacetate-polypropionate derived natural products.
 |
(11) |
In addition, we demonstrated the feasibility to functionalize the C-ring of bryostatin 16 by a highly chemo- and stereoselective Re-catalyzed epoxidation/ring-opening reaction. By accomplishing a concise synthesis of a potent anti-cancer agent 20-epi-bryostatin 7, we also proved the principle for the first time that novel bryostatin analogues can be derived from a bryostatin 16-like intermediate (eq 12). Extension of this strategy in the synthesis of various other natural bryostatins and their analogues, and performance of more systematic biological studies are currently being undertaken.
 |
(12) |
Experimental
Compound 5
CpRu(CH3CN)3PF6 (4.0 mg, 0.0092 mmol) was added to a solution of compound 6 (50 mg, 0.080 mmol) and compound 7 (24.5 mg, 0.069 mmol) in DCM (0.4 ml) at 0 °C. The resulting yellow solution was stirred at rt for 12 h. Compound 5 was purified directly via silica gel flash column chromatography (10%, then 15% ethyl acetate/petroleum ether) to give a colorless foam (23.1 mg, 34%; 80% brsm, 45.6 mg 6 + 7 can be recovered).
At a larger scale, compound 6 (969 mg, 1.59 mmol) and compound 7 (471 mg, 1.33 mmol) with CpRu(CH3CN)3PF6 (86 mg, 0.199 mmol) in DCM (3 ml), according to the same procedure, gave compound 5 (0.45 g, 35%, 0.19 g, 7 was recovered). RF: 0.3 (ethyl acetate:petroleum ether, 1:9 v/v); [α]D20(deg cm3 g−1 dm−1): −21.1 (c 1.27 g cm−3 in DCM); 1H NMR (CDCl3, 500 MHz) δ 7.63–7.59 (m, 4H), 7.47–7.44 (m, 2H), 7.43–7.37 (m, 4H), 7.13 (d, J = 8.5 Hz, 2H), 6.76 (d, J = 8.5 Hz, 2H), 5.63 (dd, J = 0.5, 16 Hz, 1H), 5.40 (dd, J = 7.0, 16 Hz, 1H), 5.26 (s, 1H), 4.55 (d, J = 11 Hz, 1H), 4.34 (d, J = 11 Hz, 1H), 4.03–3.98 (m, 2H), 3.88–3.80 (m, 2H), 3.73 (m, 1H), 3.73 (s, 3H), 3.25 (s, 2H), 3.00 (dd, J = 5.5, 17 Hz, 1H), 2.53 (dd, J = 7.0, 18 Hz, 1H), 2.47–2.38 (m, 3H), 2.23 (br d, J = 13 Hz, 1H), 1.97 (br dd, J = 12, 24 Hz, 2H), 1.90 (ddd, J = 2.5, 5.5, 14 Hz, 1H), 1.58–1.55 (m, 2H), 1.48 (dd, J = 1.5, 10.5, Hz, 1H), 1.18 (s, 3H), 1.08 (s, 3H), 1.03 (s, 9H), 0.93 (s, 3H), 0.92 (s, 3H), 0.88 (s, 9H), 0.09 (s, 9H), 0.00 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 212.3, 170.4, 159.2, 152.9, 139.2, 135.73, 135.69, 133.4, 133.2, 130.7, 130.12, 130.06, 129.6, 128.0, 127.9, 127.7, 123.7, 113.8, 79.4, 79.3, 75.1, 74.6, 73.2, 71.8, 65.2, 55.3, 52.6, 45.3, 45.2, 40.6, 39.7, 38.9, 38.1, 37.9, 30.4, 26.9, 26.0, 24.0, 23.7, 20.9, 20.8, 19.1, 18.3, 0.4, −5.4; IR (film): 2956, 2858, 1744, 1702, 1612, 1514, 1249, 1094, 838 cm−1; HRMS (C57H86O8Si3): Calc'd. 1005.5528 (M+Na+), Found 1005.5520.
Compound 13
CSA (18 mg, 0.080 mmol) was added to a solution of compound 12 (0.88 g, 0.89 mmol) in MeOH (dry, 18 ml) at 0 °C. The resulting solution was stirred at rt for 12 h, before it was poured into saturated aqueous NaHCO3. The mixture was extracted with ethyl acetate three times and the combined organic fractions were dried over Na2SO4. Compound 13 was purified via silica gel flash column chromatography (15%, then 30% ethyl acetate/petroleum ether) to give a colorless oil (0.76 g, 93%). RF: 0.2 (ethyl acetate:petroleum ether, 1:4 v/v); [α]D20(deg cm3 g−1 dm−1): +27.5 (c 0.85 g cm−3 in DCM); 1H NMR (C6D6, 400 MHz) δ 7.84–7.80 (m, 4H), 7.26–7.21 (m, 6H), 6.80 (d, J = 8.8 Hz, 2H), 5.98 (s, 1H), 5.71 (dd, J = 1.2, 16 Hz, 1H), 5.52 (dd, J = 5.2, 16 Hz, 1H), 4.58 (m, 1H), 4.38 (d, J = 11.2, 1H), 4.18 (d, J = 11.2 Hz, 1H), 3.71–3.64 (m, 2H), 3.57 (m, 1H), 3.45 (m, 1H), 3.38 (s, 3H), 3.31 (s, 3H), 3.13 (s, 2H), 2.97 (m, 1H), 2.90 (s, 3H), 2.75–2.65 (m, 2H), 2.26 (brd, J = 12 Hz, 1H), 2.18 (dd, J = 4.8, 16 Hz, 1H), 1.89 (m, 1H), 1.81–1.65 (m, 4H), 1.51 (m, 1H), 1.20 (s, 3H), 1.17 (s, 9H), 1.14 (s, 3H), 0.91 (s, 3H), 0.90 (s, 3H); 13C NMR (C6D6, 100 MHz) δ 171.6, 159.6, 140.8, 138.2, 136.30, 136.26, 134.4, 134.2, 131.8, 130.1, 129.3, 129.2, 128.0, 127.9, 114.0, 104.3, 100.5, 78.1, 77.2, 74.6, 71.4, 71.2, 69.7, 66.5, 54.7, 51.2, 48.0, 44.7, 43.6, 43.5, 42.4, 39.0, 38.2, 37.6, 33.2, 27.1, 23.9, 23.7, 21.1, 19.5, 16.8; IR (film): 3444, 2933, 1738, 1614, 1514, 1248 cm−1; HRMS (C50H69O9BrSi): Calc'd. 943.3792 (M+Na+), Found 943.3801.
Compound 26
To a solution of hydroxyacid 24 (74 mg, 0.12 mmol) in DCM (2.5 ml) was added freshly distilled 2,6-lutidine (62 mg, 0.58 mmol) at −10 °C, followed by dropwise addition of freshly distilled TESOTf (67 mg, 0.25 mmol). The resulting solution was stirred at the same temperature for 20 min, before poured into pH 7.0 buffer. The mixture was extracted with ethyl acetate five times and the combined organic fractions were dried over Na2SO4. The TES ether-acid 22 was purified via quick silica gel flash column chromatography (10%, 20% then 30% ethyl acetate/petroleum ether) to give a colorless foam (73 mg, 79%). (Significant decomposition has been observed when slower chromatography was applied.)
To a solution of TES ether-acid 22 (17.0 mg, 0.0225 mmol) in dry toluene (0.5 ml) was added Et3N (4.8 mg, 0.047 mmol) at rt under N2, followed by dropwise addition of freshly distilled 2,4,6-trichlorobenzoyl chloride (5.6 mg, 0.024 mmol) at rt. The resulting solution was stirred at rt for 1 h, before a solution of alcohol 23 (10.1 mg, 0.0225 mmol) and DMAP (6.9 mg, 0.056 mmol) in toluene (0.75 ml) was added. The resulting mixture was stirred at rt for another 1h, before poured into pH 7.0 buffer. The mixture was extracted with ethyl acetate four times and the combined organic fractions were dried over Na2SO4. The ester 26 was purified via silica gel flash column chromatography (10%, then 20% ethyl acetate/petroleum ether) to give a colorless foam (25 mg, 92%). RF: 0.35 (ethyl acetate:petroleum ether, 1:4 v/v); [α]D20(deg cm3 g−1 dm−1): +62.6 (c 0.11 g cm−3 in DCM); 1H NMR (C6D6, 500 MHz) δ 7.41 (d, J =8.5 Hz, 2H), 7.24 (d, J = 8.5 Hz, 2H), 6.85 (d, J = 8.5 Hz, 2H), 6.79 (d, J = 8.5 Hz, 2H), 6.07 (dd, J = 5.0, 15.5 Hz, 1H), 5.93 (s, 1H), 5.79 (dd, J = 1.5, 15.5 Hz, 1H), 5.48 (ddd, J = 2.0, 4.0, 10.5 Hz, 1H), 4.60 (m, 1H), 4.53 (d, J = 10.5, 1 H), 4.47 (d, J = 11.5 Hz, 1H), 4.42 (d, J = 10.5 Hz, 1H), 4.32 (brd, J = 15.5 Hz, 1H), 4.27 (d, J = 11.5 Hz, 1H), 3.97 (m, 1H), 3.88–3.83 (2H), 3.79 (m, 1H), 3.67 (m ,1H), 3.45 (s, 3H), 3.33 (s, 3H), 3.30 (s, 3H), 3.28 (s, 3H), 3.27 (s, 3H), 2.70 (br s, 1H), 2.69 (d, J = 2.0 Hz, 1H), 2.38–2.25 (4H), 2.07–1.89 (6H), 2.01 (s, 1H), 1.84 (dd, J = 5.0, 16 Hz, 1H), 1.80 (m, 1H), 1.65 (m, 1H), 1.57 (dd, J = 9.5, 19.5 Hz, 2H), 1.29 (s, 3H), 1.22 (s, 3H), 1.21 (s, 6H), 1.18 (d, J = 6.0 Hz, 3H), 1.07 (t, J = 6.5 Hz, 9H), 1.00 (3, 9H), 0.72 (m, 6H), 0.17 (s, 3H), 0.12 (s, 3H); 13C NMR (C6D6, 125 MHz) δ 170.9, 166.7, 159.8, 159.6, 157.7, 153.9, 136.8, 131.7, 130.5, 130.1, 129.3, 128.7, 128.3, 128.1, 127.9, 114.9, 114.1, 114.03, 113.96, 104.5, 89.4, 85.8, 78.5, 77.5, 75.6, 75.1, 73.6, 73.4, 72.1, 71.5, 70.5, 68.7, 67.8, 66.2, 54.74, 54.70, 52.0, 50.6, 48.3, 44.9, 44.3, 43.7, 43.6, 39.5, 36.4, 34.4, 33.8, 33.5, 29.8, 26.01, 25.96, 24.5, 21.2, 18.6, 18.2, 17.0, 7.3, 5.7, −4.6, −4.7; IR (film): 2927, 2240, 1717, 1651, 1614, 1514, 1463, 1377, 1250, 1075 cm−1; HRMS (C66H100O15Si2): Calc'd. 1211.6499 (M+Na+), Found 1211.6484.
Compound 30
To a mixture of Pd(OAc)2 (4.4 mg, 0.02 mmol) and TDMPP [tris(2,6-dimethoxyphenyl)phosphine] (11.2 mg, 0.025 mmol) was added freshly distilled toluene (1 ml). The mixture was stirred at rt for 30 min, and the resulting red solution (0.02 ml, ca. 0.0004 mmol) was slowly added a solution of diyne 21 (3.2 mg, 0.0034 mmol) in freshly distilled toluene (1.6 ml) under N2. The reaction was stirred at rt for 3 days, before it was filtered through a short plug of silica gel. The solvent was removed under vacuum and the macrocycle 30 was purified via silica gel flash column chromatography (20%, 30% then 40% ethyl acetate/petroleum ether) to give a white paste (1.8 mg, 56%).
The same reaction was also carried out with Pd(OAc)2 (1.1 mg, 0.0048 mmol), TDMPP (1.6 mg, 0.0075 mmol), diyne 21 (45 mg, 0.048 mmol) in toluene (20 ml) to give macrocycle 30 (16.0 mg, 36% yield; 16.7 mg 21 was recover, 57% yield brsm). RF: 0.35 (ethyl acetate:petroleum ether, 3:7 v/v); [α]D20(deg cm3 g−1 dm−1): −43.8 (c 0.21 g cm−3 in DCM); 1H NMR (C6D6, 500 MHz) δ 6.32 (s, 1H), 6.12 (dd, J = 3.5, 15.5 Hz, 1H), 5.89 (d, J = 15 Hz, 1H), 5.73 (s, 1H), 5.34 (d, J = 10.5 Hz, 1H), 4.67 (m, 1H), 4.36 (d, J = 13 Hz, 1H), 4.31 (m, 1H), 4.12 (d, J = 11 Hz, 1H), 4.03–4.00 (2H), 3.91 (m ,1H), 3.86 (m ,1H), 3.40 (s, 3H), 3.38 (t, J = 7.5 Hz, 1H), 3.26 (s, 3H), 3.21 (s, 3H), 3.16 (dd, J = 5.0, 14.5 Hz, 1H), 2.68 (dd, J = 3.0, 16 Hz, 1H), 2.62 (dd, J = 9.0, 16 Hz, 1H), 2.21 (dd, J = 8.0, 16 Hz, 1H), 2.00 (m 1H), 1.92–1.84 (4H), 1.65–1.53 (3H), 1.46 (m, 1H), 1.36–1.29 (2H), 1.24 (s, 3H), 1.22 (s, 3H), 1.19 (d, J = 6.5 Hz, 3H), 1.10 (s, 3H), 1.09 (s, 3H), 1.07 (t, J = 8.0 Hz, 9H), 0.98 (s, 9H), 0.66 (m, 6H), 0.16 (s, 3H), 0.11 (s, 3H); 13C NMR (C6D6, 125 MHz) δ 172.3, 166.5, 157.7, 140.2, 134.9, 129.1, 128.2, 127.9, 125.1, 114.8, 103.5, 102.0, 83.9, 76.4, 75.1, 74.3, 70.0, 68.7, 67.4, 66.9, 65.9, 50.9, 50.6, 49.1, 46.5, 44.6, 43.2, 42.8, 40.6, 40.2, 37.2, 36.8, 36.4, 34.6, 30.2, 29.8, 29.6, 26.0, 20.6, 18.5, 18.3, 16.6, 7.35, 6.0, −4.68, −4.72; IR (film): 3442 (br), 2924, 2853, 1717, 1650, 1614, 1435, 1376, 1256, 1151, 1107 cm−1; HRMS (C50H84O13Si2): Calc'd. 971.5348 (M+Na+), Found 971.5341.
Bryostatin 16
To a solution of pivalate ester 35 (1.0 mg, 0.001 mmol) in THF (0.05 ml) was added TBAF (0.005 ml, 0.005 mmol, 1M) at 0 °C. The resulting solution was allowed to slowly warm to rt and stirred for 4 h. The reaction mixture was diluted with ethyl acetate and pH 7.0 buffer was added. The mixture was extracted with ethyl acetate five times and the combined organic fractions were dried over Na2SO4. The residue was purified by reverse phase HPLC (RP C-18 column, CH3CN in H2O from 65% to 95%) to give 1 as a white paste (0.4 mg, ca 52%). RF: 0.35 (ethyl acetate:petroleum ether, 4:1 v/v); [α]D20(deg cm3 g−1 dm−1): +81 (c 0.04 g cm−3 in MeOH); For NMR data, see the supporting information; IR (film): 3359 (br), 2958, 2917, 2849, 1722, 1702, 1605, 1614, 1433, 1375, 1259, 1154, 1099 cm−1; HRMS (C42H62O14): Calc'd. 913.4037 (M+Na+), Found 913.4038.
Compound 41
To a mixture of Au(PPh3)3Cl (10.2 mg, 0.020 mmol) and AgSbF6 (7.0 mg, 0.020 mmol) was added dry DCM (0.5 ml) at rt under N2. The resulting mixture was stirred in the dark for 15 min, and a purple precipitate was formed. The supernatant solution (0.015 ml, ca 0.0006 mmol) was transferred to a mixture of compound 40 (2.9 mg, 0.0030 mmol) and NaHCO3 (2.4 mg, 0.03 mmol) in DCM/CH3CN/2,2-dimethoxypropane, (10:1:2, 0.4 ml) at 0 °C under N2. The resulting reaction mixture was stirred vigorously overnight, before it was poured into a mixture of saturated aqueous NaHCO3 and saturated aqueous Na2S2O3 (ca 1:1), and then the mixture was extracted with ethyl acetate four times and the combined organic fractions were dried over Na2SO4. Dihydropyran 41 was purified via quick silica gel flash column chromatography (10%, then 20% ethyl acetate/petroleum ether) to give a colorless foam (2.4 mg, 83%): Rf: 0.3 (10% ethyl acetace in petroleum ether); [α]D: 42.5 (c 0.17, DCM); 1H NMR (C6D6, 500 MHz): δ 6.13 (d, J = 15.5 Hz, 1H), 5.74 (s, 1H), 5.73 (dd, J = 4.5, 15.5 Hz, 1H), 5.67 (dd, J = 5.0, 12 Hz, 1H), 5.61 (s, 1H), 5.46 (dd, J = 4.5, 11 Hz, 1H), 5.37 (s, 1H), 4.63 (m, 1H), 4.37 (d, J = 13 Hz, 1H), 4.02–3.91 (m, 4H), 3.77 (t, J = 12 Hz, 1H), 3.43 (s, 3H), 3.41 (s, 3H), 3.11 (s, 3H), 2.68 (dd, J = 5.0, 15 Hz, 1H), 2.42–2.34 (m, 2H), 2.18 (dd, J = 8.5, 16 Hz, 1H), 2.05–1.84 (m, 5H), 1.78 (m, 1H), 1.75 (s, 3H), 1.65–1.45 (5H) 1.30 (s, 3H), 1.24 (s, 3H), 1.12 (s, 3H), 1.09 (s, 3H), 1.05 (d, J = 7.0 Hz, 3H), 1.03 (s, 9H), 0.97 (t, J = 8.0 Hz, 9H), 0.57 (m, 6H), 0.23 (s, 3H), 0.13 (s, 3H); 13C-NMR(C6D6, 125 MHz) δ 170.0, 169.8, 169.3, 167.3, 158.2, 150.5, 136.2, 129.1, 128.5, 127.8, 125.0, 114.7, 108.9, 103.2, 100.9, 77.0, 73.8, 73.7, 73.4, 72.4, 68.1, 66.8, 64.7, 50.5, 50.4, 48.2, 44.8, 44.0, 42.7, 42.2, 41.0, 39.7, 37.0, 33.8, 33.7, 32.4, 30.2, 26.0, 25.0, 24.9, 20.72, 20.70, 18.3, 18.0, 17.9, 7.2, 6.0, −4.7; IR (film) 2953, 2929, 1734, 1608, 1614, 1435, 1375, 1245, 1150, 1102 cm−1; HRMS (C52H86O14Si2): Calc'd. 1013.5454 ([M+Na]+), Found 1013.5453.
Compound 44
UHP (37.6 mg, 0.4 mmol) was added to a solution of MTO (5.0 mg, 0.02 mmol), N-methylimidazole (8.2 mg, 0.1 mmol) in freshly distilled MeOH (2 ml) at rt. The resulting solution was stirred at rt for 5 min, during which the color of the reaction turned to yellow. A portion of the above solution (0.026 ml) was added to a solution of 41 (2.5 mg, 0.0026 mmol) in MeOH (0.2 ml) at 0 °C. The resulting reaction mixture was stirred at °C for 6h (monitored by TLC) before it was quenched with saturate aqueous NaHCO3 and Na2S2O3. The mixture was extracted with ethyl acetate four times and the combined organic fractions were dried over Na2SO4. The crude epoxide product was obtained after the solvent was removed under vacuum. To the above crude epoxide was added a solution of HOAc in MeOH (0.2 ml, obtained from 1 drop of HOAc in 1 ml MeOH) at 0 °C. The resulting solution was stirred at 0 °C for 3 h (monitored by TLC), before it was quenched with saturate aqueous NaHCO3. The mixture was extracted with ethyl acetate four times and the combined organic fractions were dried over Na2SO4. Compound 44 was purified via silica gel flash column chromatography (10%, then 20% ethyl acetate/petroleum ether) to give a colorless foam (1.7 mg, 64%).
A one-pot protocol
UHP (19 mg, 0.2 mmol) was added to a solution of MTO (5.0 mg, 0.02 mmol), N-methylimidazole (8.2 mg, 0.1 mmol) in freshly distilled MeOH (1 ml) at rt. The resulting solution was stirred at rt for 5 min, during which the color of the reaction turned to yellow. A portion of the above solution (0.021 ml, 10 mol% MTO) was added to a solution of 41 (4.3 mg, 0.0044 mmol) in MeOH (0.3 ml) and DCM (0.2 ml) at 0 °C for 30 min, before another portion of the oxidant (0.022 ml) was added. The resulting reaction mixture was stirred at °C for 3h (monitored by TLC) before it was cooled to −78 °C and ZnCl2 (0.020 ml, 1.0 M in ether) was added. The resulting solution was stirred at 4 °C for 4 h (monitored by TLC), before it was quenched with saturate aqueous NaHCO3 and Na2S2O3. The mixture was extracted with ethyl acetate four times and the combined organic fractions were dried over Na2SO4. Compound 44 was purified via silica gel flash column chromatography (10%, then 20% ethyl acetate/petroleum ether) to give a colorless foam (2.2 mg, 48%): Rf: 0.33 (15% ethyl acetace in petroleum ether); [α]D: 57.7 (c 0.17, DCM); For NMR data, see the supporting information; IR (film): 3400 (br), 2954, 2928, 2856, 1722, 1651, 1378, 1247, 1098 cm−1; HRMS (C53H90O16Si2): Calc'd. 1061.5665 ([M+Na]+), Found 1061.5640.
Compound 47
To a solution of alcohol 44 (1.0 mg, 0.001 mmol) in pyridine (0.15 ml) was added acetic anhydride (0.1 ml) at 0 °C, followed by DMAP (1.0 mg, 0.008 mmol). The resulting solution was stirred at rt for 4 h, before it was quenched with MeOH (0.1 ml), followed by pH 7.0 Buffer at 0 °C. The mixture was extracted with ethyl acetate four times and the combined organic fractions were dried over Na2SO4. The residue was purified by silica gel column chromatography (10%, then 20% ethyl acetate/petroleum ether) to give compound 46 (ca 1.0 mg) as a colorless thick oil.
To a solution of the above acetate (46, ca 1.0 mg) in CH3CN (0.2 ml) was added aqueous HF (2 drops, conc. 48–53%) at 0 °C. The resulting solution was stirred at a warming ice-bath for 2.5 h, before solid K2HPO4 and saturated aqueous NaHCO3 were added. The mixture was extracted with ethyl acetate four times and the combined organic fractions were dried over Na2SO4. The residue was purified by preparative TLC (60% ethyl acetace in petroleum ether) to give 20-epi-bryostatin 7 (47) as a white paste (0.5 mg, ca 63% over two steps): Rf: 0.30 (60% ethyl acetace in petroleum ether); [α]D: 27.6 (c 0.07, DCM); For NMR data, see the supporting information; IR (film): 3455, 3300(br), 2924, 2853, 1722, 1652, 1374, 1243, 1153, 1077 cm−1; HRMS (C41H60O17): Calc'd. 847.3728 ([M+Na]+), Found 847.3741.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge the National Institutes of Health (GM 13598) for their support of our programs. G.D. was a Stanford Graduate Fellow. We thank Dr. H. Yang for discussions and for help characterizing compound 35, and we additionally thank him and Dr. C. S. Brindle for providing synthetic intermediates. Palladium and ruthenium salts were supplied by Johnson Matthey. We are grateful to Dr. J. Flygare and Genentech for performing the preliminary biological test of 20-epi-bryostatin 7. We also acknowledge Dr. S. R. Lynch for his help with two-dimensional nuclear magnetic resonance analysis. We also thank the Wender group's assistance with reverse-phase high-performance liquid chromatography.
Footnotes
Supporting Information Available. Experimental procedures and characterization data for all new compounds (PDF). This material is available free of charge via the internet at http://pubs.acs.org.
REFERENCES
- 1.Pettit GR, Day JF, Hartwell JL, Wood HB. Nature. 1970;227:962. doi: 10.1038/227962a0. [DOI] [PubMed] [Google Scholar]
- 2.Pettit GR, Herald CL, Doubek DL, Herald DL, Arnold E, Clardy J. J. Am. Chem. Soc. 1982;104:6846. [Google Scholar]
- 3.For some recent reviews, see:Hale KJ, Hummersone MG, Manaviazar S, Frigerio M. Nat. Prod. Rep. 2002;19:413. doi: 10.1039/b009211h. Wender PA, Baryza JL, Hilinski MK, Horan JC, Kan C, Verma VA. Beyond Natural Products: Synthetic Analogues of Bryostatin 1. In: Huang Z, editor. Drug Discovery Research: New Frontiers in the Post-Genomic Era. Wiley-VCH; Hoboken, NJ: 2007. pp. 127–162.
- 4.For a review, see: Newman DJ, Cragg GM. J. Nat. Prod. 2004;67:1216. doi: 10.1021/np040031y. For ongoing studies, see http://clinicaltrials.gov/ Dowlati A, Lazarus HM, Hartman P, Jacobberger JW, Whitacre C, Gerson SL, Ksenich P, Cooper BW, Frisa PS, Gottlieb M, Murgo AJ, Remick SC. Clin. Cancer Res. 2003;9:5929.
- 5.(a) Etcheberrigaray R, Tan M, Dewachter I, Kuiperi C, Van der Auwera I, Wera S, Qiao L, Bank B, Nelson TJ, Kozikowski AP, Van Leuven F, Alkon DL. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11141. doi: 10.1073/pnas.0403921101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Alkon DL, Sun M-K, Nelson TJ. Trends Pharmacol. Sci. 2007;28:51. doi: 10.1016/j.tips.2006.12.002. [DOI] [PubMed] [Google Scholar]; (c) Alkon DL, Epstein H, Kuzirian A, Bennett MC, Nelson TJ. Proc. Natl. Acad. Sci. U.S.A. 2005;102:16432. doi: 10.1073/pnas.0508001102. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Sun M-K, Alkon DL. Eur. J. Pharmacol. 2005;512:43. doi: 10.1016/j.ejphar.2005.02.028. [DOI] [PubMed] [Google Scholar]; (e) Hongpaisan J, Alkon DL. Proc. Natl. Acad. Sci. U.S.A. 2007;104:19571. doi: 10.1073/pnas.0709311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun M-K, Hongpaisan J, Nelson TJ, Alkon DL. Proc. Natl. Acad. Sci. U.S.A. 2008;105:13620. doi: 10.1073/pnas.0805952105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kageyama M, Tamura T, Nantz MH, Roberts JC, Somfai P, Whritenour DC, Masamune S. J. Am. Chem. Soc. 1990;112:7407. [Google Scholar]
- 8.(a) Julia M, Paris JM. Tetrahedron Lett. 1973:4833. [Google Scholar]; (b) Kocienski PJ, Lythgoe B, Ruston S. J. Chem. Soc., Perkin Trans. 1. 1978:829. [PubMed] [Google Scholar]; (c) Kocienski P. Phosphorus Sulfur. 1985;24:97. [Google Scholar]
- 9.(a) Evans DA, Carter PH, Carreira EM, Prunet JA, Charette AB, Lautens M. Angew. Chem., Int. Ed. 1998;37:2354. doi: 10.1002/(SICI)1521-3773(19980918)37:17<2354::AID-ANIE2354>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]; (b) Evans DA, Carter PH, Carreira EM, Charrette AB, Prunet JA, Lautens M. J. Am. Chem. Soc. 1999;121:7540. [Google Scholar]
- 10.Pettit GR, Sengupta D, Herald CL, Sharkey NA, Blumberg PM. Can. J. Chem. 1991;69:856. [Google Scholar]
- 11.Ohmori K, Ogawa Y, Obitsu T, Ishikawa Y, Nishiyama S, Yamamura S. Angew. Chem., Int. Ed. 2000;39:2290. [PubMed] [Google Scholar]
- 12.Manaviazar S, Frigerio M, Bhatia GS, Hummersone MG, Aliev AE, Hale KJ. Org. Lett. 2006;8:4477. doi: 10.1021/ol061626i. [DOI] [PubMed] [Google Scholar]
- 13.For recent examples of bryostatin analogues, see: Keck, Gary E, Li Wei, Kraft,Matthew B, Kedei Noemi, Lewin, Nancy E, Blumberg, Peter M. Org. Lett. 2009;11:2277. doi: 10.1021/ol900585t. Keck GE, Poudel YB, Welch DS, Kraft MB, Truong AP, Stephens JC, Kedei N, Lewin NE, Blumberg PM. Org. Lett. 2009;11:593. doi: 10.1021/ol8027253. Wender PA, Horan JC, Verma VA. Org. Lett. 2008;10:3331. doi: 10.1021/ol801235h. Wender PA, DeChristopher BA, Schrier AJ. J. Am. Chem. Soc. 2008;130:6658. doi: 10.1021/ja8015632. Keck GE, Kraft MB, Truong AP, Li W, Sanchez CC, Kedei N, Lewin NE, Blumberg PM. J. Am. Chem. Soc. 2008;130:6660. doi: 10.1021/ja8022169. Trost BM, Yang H, Thiel OR, Frontier AJ, Brindle CS. J. Am. Chem. Soc. 2007;129:2206. doi: 10.1021/ja067305j.
- 14.Pettit GR, Gao F, Blumberg PM, Herald CL, Coll JC, Kamano Y, Lewin NE, Shmidt JM, Chapuis J-C. J. Nat. Prod. 1996;59:286. doi: 10.1021/np960100b. [DOI] [PubMed] [Google Scholar]
- 15.For biosynthetic approaches, see: Sudek S, Lopanik NB, Waggoner LE, Hildebrand M, Anderson C, Liu H, Patel A, Sherman DH, Haygood MG. J. Nat. Prod. 2007;70:67. doi: 10.1021/np060361d. Kerr RG, Lawry J, Gush KA. Tetrahedron Lett. 1996;37:8305.
- 16.Trost BM, Frontier AJ. J. Am. Chem. Soc. 2000;122:11727. [Google Scholar]
- 17.For a preliminary report of a portion of this work, see: Trost BM, Dong G. Nature. 2008;456:485. doi: 10.1038/nature07543.
- 18.For an unsuccessful macrocyclic RCM or a RRCM strategy, see: Ball M, Bradshaw BJ, Dumeunier R, Gregson TJ, MacCormick S, Omori H, Thomas EJ. Tetrahedron. Lett. 2006;47:2223. b) Refs 7, 9, 11 and 13f.
- 19.Trost BM, Yang H, Wuitschik G. Org. Lett. 2005;7:4761. doi: 10.1021/ol0520065. [DOI] [PubMed] [Google Scholar]
- 20.For a synthesis of compound 7, see ref 17.
- 21.Brown HC, Jadhav PK. J. Am. Chem. Soc. 1983;105:2092. [Google Scholar]
- 22.Yu W, Mei Y, Kang Y, Hua Z, Jin Z. Org. Lett. 2004;6:3217. doi: 10.1021/ol0400342. [DOI] [PubMed] [Google Scholar]
- 23.Yang H. Ph.D. thesis. Stanford University; 2006. [Google Scholar]
- 24.Similar observation has also been reported by Evans, see ref 9b.
- 25.Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994:639. [Google Scholar]
- 26.For a related model study, see: Dong G. Ph.D. thesis. Stanford University; 2009.
- 27.Otera J, Danoh N, Nozaki H. J. Org. Chem. 1991;56:5307. [Google Scholar]
- 28.Trost BM, Matsubara S, Caringi JJ. J. Am. Chem. Soc. 1989;111:8745. [Google Scholar]
- 29.Nicolaou KC, Estrada AA, Zak M, Lee SH, Safina BS. Angew. Chem., Int. Ed. 2005;44:1378. doi: 10.1002/anie.200462207. [DOI] [PubMed] [Google Scholar]
- 30.For example, either extended exposure of 24 to silica gel or storage in neat form led to the loss of the methyl ketal. It was also found that the most desirable way to store acid 24 was to freeze it as a benzene solution.
- 31.For the synthesis of compound 4, see ref 13f.
- 32.Rai AN, Basu A. Tetrahedron Lett. 2003;44:2267. [Google Scholar]
- 33.(a) Shiina I, Kubota M, Oshiumi H, Hashizume M. J. Org. Chem. 2004;69:1822. doi: 10.1021/jo030367x. [DOI] [PubMed] [Google Scholar]; (b) Shiina I, Kubota M, Ibuka R. Tetrahedron Lett. 2002;43:7535. [Google Scholar]; (c) Shiina I, Ibuka R, Kubota M. Chem. Lett. 2002:286. [Google Scholar]
- 34.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull. Chem. Soc. Jpn. 1979;52:1989. [Google Scholar]
- 35.For a mechanistic discussion and a full paper, see: Trost BM, Sorum MT, Chan C, Ruehter G. J. Am. Chem. Soc. 1997;119:698.
- 36.For a similar pivalation, see ref. 10.
- 37.Isomerization occurred even when trying to purify bryostatin 16 via prep-TLC.
- 38.Liu B, De Brabander JK. Org. Lett. 2006;8:4907. doi: 10.1021/ol0619819. [DOI] [PubMed] [Google Scholar]
- 39.For a review, see: Kolb HC, Van Nieuwenhze MS, Sharpless KB. Chem. Rev. 1994;94:2483.
- 40.a) Payne GB, Deming PH, Williams PH. J. Org. Chem. 1961;26:659. [Google Scholar]; b) Arias LA, Adkins S, Nagel CJ, Bach RD. J. Org. Chem. 1983;48:888. [Google Scholar]
- 41.For a review, see: Kuhn FE, Santos AM, Herrmann WA. Dalton Trans. 2005:2483. doi: 10.1039/b504523a. Also, see: Herrmann WA, Fischer RW, Marz DW. Angew. Chem. Int. Ed. 1991;30:1638. Boehlow TR, Spilling CD. Tetrahedron Lett. 1996;37:2717. Soldaini G, Cardona F, Goti A. Tetrahedron Lett. 2003;44:5589.
- 42.Rudolph J, Reddy KL, Chiang JP, Sharpless KB. J. Am. Chem. Soc. 1997;119:6189. [Google Scholar]
- 43.Epoxide 45 decomposed on silica gel.
- 44.A very small ratio of the desired diastereomer was observed, and the dr was about 7:1 favoring the undesired one.
- 45.The stereochemistry of the C19 hemi-ketal was tentatively assigned as the same one in the natural product.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.