

Summary
Activation of precursor proteins by specific and limited proteolysis is a hallmark of the hemostatic process. The homologous coagulation factors (F)V and FVIII circulate in an inactive, quiescent state in blood. In this so-called procofactor state, these proteins have little, if any procoagulant activity and do not participate to any significant degree in their respective macromolecular enzymatic complexes. Thrombin is considered a key physiological activator, cleaving select peptide bonds in FV and FVIII which ultimately leads to appropriate structural changes that impart cofactor function. As the active cofactors (FVa and FVIIIa) have an enormous impact on thrombin and FXa generation, maintaining FV and FVIII as inactive procofactors undoubtedly plays an important regulatory role that has likely evolved to maintain normal hemostasis. Over the past three decades there has been widespread interest in studying the proteolytic events that lead to the activation of these proteins. While a great deal has been learned, mechanistic explanations as to how bond cleavage facilitates conversion to the active cofactor species remain incompletely understood. However, recent advances have been made detailing how thrombin recognizes FV and FVIII and also how the FV B-domain plays a dominant role in maintaining the procofactor state. Here we review our current understanding of the molecular process of procofactor activation with a particular emphasis on FV.
Keywords: factor V, factor VIII, FX activation, procofactor, proteolytic activation, prothrombin activation
Introduction
Blood coagulation factors (F)Va and FVIIIa are homologous cofactors for the prothrombinase (FXa, FVa, Ca2+ and anionic membranes) and intrinsic Xase complexes (FIXa, FVIIIa, Ca2+ and anionic membranes), respectively [1,2]. Prothrombinase catalyzes the conversion of prothrombin to thrombin, whereas the intrinsic Xase catalyzes the proteolytic conversion of FX to FXa, both pivotal steps in the coagulation cascade [3]. FXa and FIXa can both catalyze protein substrate cleavage in the absence of cofactor proteins. Yet, it is clear from biochemical studies that assembly of the cofactors into their respective macromolecular enzyme complexes results in dramatic rate enhancements [3]. It is for this reason that prothrombinase and intrinsic Xase are considered the physiologically relevant enzymes. The importance of these cofactors is further underscored by clinical findings which indicate that FV and FVIII deficiency states lead to parahemophilia and hemophilia A, respectively [4,5].
While the exact molecular mechanism by which FVa and FVIIIa accelerate protein substrate cleavage remains to be fully defined, considerable progress has been made over the past two decades identifying and elucidating how macromolecular binding sites on the active cofactor species contribute to their function (for recent reviews see [6–9]). There is mounting evidence that the cofactor not only provides protease binding sites, but also facilitates substrate docking thereby enforcing affinity and specificity.
For obvious reasons, maintaining enzymes and cofactors in an inactive state in the circulation is critical for the regulation of normal hemostasis. The zymogens and procofactors of coagulation lack key structural attributes required for enzyme complex formation and function. For FV and FVIII, it is well established that these functional sites, or cofactor exosites, are not readily available for productive interactions or are not poised to function [6,10–12]. After their discovery, it was recognized that FV and FVIII need to be proteolytically activated to fully participate in coagulation, with thrombin being identified as a key activator [13–16]. Because of difficulties in their isolation, it was not until many years later that meaningful correlations could be made between proteolysis of the procofactors and an increase in biological activity. Despite extensive investigation into procofactor activation mediated by thrombin or FXa over the past three decades [6,17,18], key mechanistic details regarding how the various proteolytic cleavage events facilitate the transition to the active cofactor species are lacking. Knowledge in this area has broad implications for better understanding cofactor function. For example, it may provide new insights into ways to engineer FVIII(a) and/or FV(a) derivatives with novel therapeutic properties or even provide new clues to develop therapeutically useful inhibitors targeting these important cofactors of coagulation. There are several excellent reviews on FV and FVIII which emphasize structure/function relationships, pathology, activated protein C (APC) resistance and the role of the cofactors in enzyme complex assembly/function [6,7,17–21]. This review will focus on FV and FVIII activation and the structural elements which assist in maintaining the procofactor state.
FVIII procofactor activation: transition to the active cofactor species
FVIII is synthesized as a large (Mr ≈ 300 000) single chain, multi-domain (A1-A2-B-A3-C1-C2) protein sharing significant homology with FV except in the B-domain region [2]. Prior to its secretion, FVIII is intracellularly processed to a series of metal ion-linked heterodimers produced by cleavage at the B-A3 junction as well as at additional sites in the B-domain [6]. These cleavages generate a variably sized heavy chain (A1-A2-B; 200–90 kDa) and a light chain (A3-C1-C2; 80 kDa) which are non-covalently associated (Fig. 1). Factor VIII also contains short segments (~30–40 amino acids) of negatively charged residues within the C-terminal regions of the A1 and A2 domains and the N-terminal portion of the A3 domain. These acidic regions are called a1 (337–372), a2 (711–740) and a3 (1649–1689), and are thought to function, in part, as binding sites for thrombin and other ligands (Fig. 1) [6]. The FVIII heterodimer is a procofactor and must be subjected to limited proteolysis to effect activation [16,22–28]. Even although FVIII has been shown to bind FIXa with high affinity, this complex does not efficiently activate FX and the binding interaction appears to be fundamentally different compared with the FVIIIa–FIXa complex [29]. The two principal activators of FVIII are thrombin and FXa which cleave at Arg372, Arg740 and Arg1689 generating FVIIIa, a heterotrimer composed of the A1 (50 kDa; 1–372), A2 (43 kDa; 373–740) and the light chain (A3-C1-C2; 73 kDa; 1689–2332) (Fig. 1) [28,30]. FXa also cleaves FVIII at Arg1721, Arg336 and Lys36, with proteolysis at the last two sites leading to a loss of cofactor activity [28,31]. Thrombin is thought to interact with FVIII via both exosites I and II [32,33]. The corresponding binding sites on FVIII are not well defined, but have been shown to involve acidic regions of the molecule [34,35]. Activation of FVIII results in a transient ~twenty- to fiftyfold increase in biological activity which decays over a short period of time. The rapid loss of activity is attributed to A2 domain dissociation from A1/A3-C1-C2, a mechanism which contributes to the regulation of FVIIIa cofactor activity [36–38].
Fig. 1.
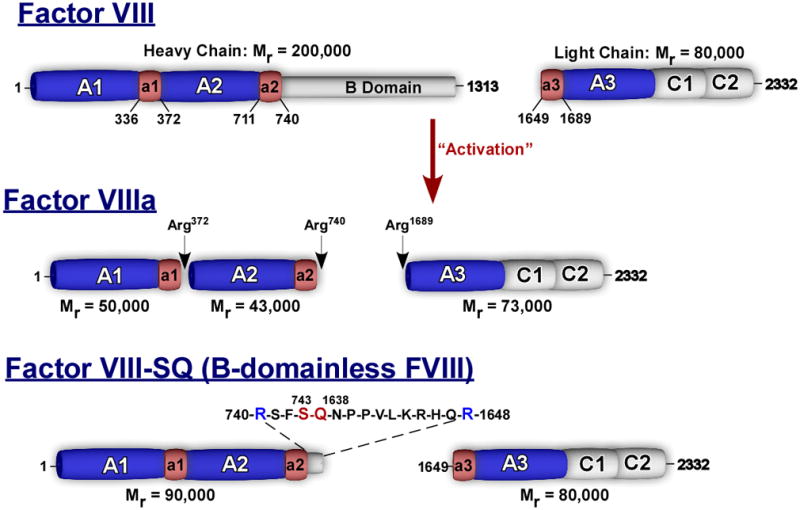
Schematic representation of factor (F)VIII, FVIIIa and FVIII-SQ. Boundaries of the acidic regions denoted by a1, a2, and a3 are indicated. ‘Activation’ represents thrombin-mediated proteolysis of FVIII and cleavage sites are indicated as well as the molecular weight of the various fragments. The ‘SQ-linker’ in rFVIII-SQ is given above the schematic.
Numerous studies have examined the role of the individual thrombin cleavage sites in the expression of FVIIIa cofactor activity and the results can be summarized as follows: first, mutagenesis studies indicate that cleavage at Arg740 appears to be of no consequence to the development of cofactor activity [39]. This is consistent with the observation that there are no known missense mutations at position 740 resulting in hemophilia A (http://europium.csc.mrc.ac.uk/WebPages/Main/main.htm) (Please note that it is the responsibility of the author(s) to ensure that all URLs given in this article are correct and useable.).. Second, cleavage of the light chain at Arg1689 results in the dissociation of von Willebrand factor (VWF) from the light chain and the exposure of a phospholipid binding site, steps which are required for the expression of cofactor activity [40,41]. Whether proteolysis at this site contributes to the potentiation of FVIIIa cofactor activity (e.g. apart from VWF removal) remains controversial. There is some evidence that cleavage at this site partially increases cofactor activity [42,43]; however, Pipe and Kaufman [44], using a single chain FVIII derivative (IR8), have shown that cofactor activity can be obtained even in the absence of the Arg1689 cleavage site. Third, cleavage at Arg740 and Arg1689 appear to facilitate cleavage within the heavy chain, as mutations at these sites slows subsequent cleavage at Arg372 [45,46]. Lastly, in addition to the results with IR8, mutagenesis and biochemical studies as well as descriptions of naturally occurring mutations clearly indicate that cleavage at Arg372 is essential for procofactor activation [39,47–50]. Biochemical data indicate that cleavage at this site exposes a functional FIXa binding site which promotes rapid FX activation by cofactor-bound FIXa [51].
The fundamental importance of the Arg372 cleavage site to the expression of cofactor activity suggests that sequences in and/or around acidic region 1 may be somehow involved in suppressing cofactor function. It is interesting to note that this region of FVIII is noticeably absent from FV (missing from exon 7), possibly suggesting a unique function [52,53]. Recent structural data on B-domain deleted FVIII indicate that this part of FVIII is highly flexible as no electron density was observed in this region [54,55]. It was suggested that acidic region 1 and possibly a portion of acidic region 3 (1649–1689), based on their location in the structure, could obscure functionally important surfaces on the molecule such as a FIXa binding site; results that are in line with functional studies [51]. Alternatively, cleavage at Arg372 could induce a change in conformation that is critical for the expression of FVIIIa cofactor function. Evidence for this comes from studies employing cross-linking agents and apolar probes as well as circular dichroism experiments. These studies support the idea that there are subtle, yet measureable changes in conformation in the vicinity of the A2 domain when FVIII is activated to FVIIIa [56–58]. Future biochemical and structural studies are needed to resolve the precise mechanism by which cleavage at Arg372 facilitates the FVIII procofactor to cofactor transition.
A somewhat surprising finding is that the FVIII B-domain does not appear to be involved in maintaining FVIII as a procofactor. While not sharing any sequence homology, the FVIII B-domain, like that of FV, is very large (908 residues), encoded by a single exon, heavily glycosylated, and is also removed after thrombin-mediated proteolysis. Yet, unlike FV (see below), several groups have established that removal of most of the B-domain (B-domainless FVIII; Fig. 1) yields a derivative, that like ful-length FVIII, has a low specific clotting activity and is predominantly synthesized as a heterodimer. Furthermore, in purified component assays, B-domainless FVIII has little, if any cofactor activity in FIXa catalyzed FX activation. Proteolytic processing of B-domain deleted FVIII by thrombin results in the expected increase in cofactor activity [44,59–62]. In some respects, this was unexpected considering the size of the FVIII B-domain and its potential for providing steric bulk which could obscure enzyme or substrate binding sites. As discussed below, these findings clearly distinguish the molecular mechanisms that regulate or prevent the potential cofactor activities of FV and FVIII.
FV procofactor activation: transition to the active cofactor species
Early work on the biochemistry of FV (Mr = 330 000; domain organization: A1-A2-B-A3-C1-C2) firmly established that it circulates as an inactive procofactor [63]. In whole blood, FV is distributed between two pools: approximately 80% is found in plasma, whereas the remaining 20% is found within the α-granules of platelets [64]. While megakaryocytes can synthesize FV [65–67], the vast majority of platelet FV is endocytozed from the plasma pool by megakaryocytes [68–70]. After endocytosis via a specific receptor-mediated process [71,72], FV is modified intracellularly such that it is functionally unique compared with its plasma-derived counterpart [70,73]. For example, platelet FV is stored in a partially proteolyzed state exhibiting significant procoagulant activity after its release by a variety of agonists and appears to be partially resistant to activated protein C [64,74–76]. Because of the inherent difficulties in preparing and working with homogeneous preparations of platelet-derived FV, most structure/function studies have focused on the plasma-derived material.
At physiological concentrations, purified plasma-derived single-chain FV is not known to bind FXa in a productive way and thus cannot assemble or function in prothrombinase [10,11,17,77,78]. As membrane-bound FXa is known to activate FV the two proteins must interact [11,79]; however, the data indicate that FV and FVa interact with membrane-bound FXa in a fundamentally different way, with active site interactions playing a dominant role in FV, but not FVa recognition [80]. Thrombin is considered the physiological activator of FV cleaving three peptide bonds within the B-domain at Arg709, Arg1018 and Arg1545 [77,81–83]. The resulting cofactor, FVa, is a heterodimer composed of an N-terminal heavy chain (Mr = 105 000) associated via Ca2+ ions to the C-terminal light chain (Mr = 74 000; Fig. 2) [77,81–84]. The large, heavily glycosylated central B-domain, spanning amino acids 710–1545, is not necessary for cofactor activity and is released during activation as two large fragments (Mr = 71 000 and Mr = 150 000) [77,82,83,85]. FVa has been very well characterized and is considered the ‘active’ cofactor species which participates in the rapid generation of thrombin under physiological conditions [3,18].
Fig. 2.

Schematic representation of factor (F)V, FVa and FV-810. ‘Activation’ represents thrombin-mediated proteolysis of FV and cleavage sites are indicated as well as the molecular weight of the various fragments. The sequence above FV-810 indicates which B-domain elements have been deleted.
Recent progress has been made delineating the interactions responsible for binding of FV to thrombin and the mechanism that regulates the specificity of the interaction. Using proteolytic derivatives of thrombin [85–87], thrombin mutants [32,88,89] and exosite I and II specific ligands [32,86,87,90–92], the findings indicate that both thrombin exosites contribute to FV activation to varying degrees. Bock and coworkers [91,93] made use of equilibrium binding studies to show that thrombin binds FV in an exosite I-dependent fashion through a site within the FV heavy chain region. The precise role of thrombin exosite II seems less clear, but recent studies suggest it plays an important role in cleavage at Arg1545 [92]. While the thrombin binding site on FV remains to be defined, there is some data to suggest that it lies within the acidic C-terminal region of the FVa heavy chain [87,91,93–96].
Major advances in our understanding of FV activation followed from the work of Nesheim and Mann [83,97] as well as Esmon [82] who provided definitive evidence for the proteolytic activation of FV. After these studies, most approaches aimed at understanding how FV is activated were principally based on correlating bond cleavage within the B-domain with the development of procoagulant activity. These studies have largely relied on the kinetic appearance of proteolytic fragments during activation [77,79,82,83,98,99], reconstituted FV activation products [82,85], FV(a) derivatives generated by a variety of proteases [77,99–111] and recombinant FV derivatives with specific modifications to thrombin cleavage sites to establish this correlation [94,112–115]. While somewhat conflicting results have emerged, most data support the idea that variable amounts of cofactor activity will be observed depending on which region of the B-domain is cleaved and which assay is employed to evaluate activity. For example, cleavage at Arg709 and Arg1018 yields a FV derivative with significant, but partial cofactor activity [82,85,113,115]. However, individual cleavage at these sites does not lead to any substantial increase in cofactor activity [113–115]. Maximal activity was observed to correlate with cleavage at Arg1545, as mutagenesis studies have shown that isolated cleavage at this site is sufficient for complete activation [113–115]. This is also consistent with the observation that a protease from Russell’s viper venom (RVV-V), which cleaves FV at Arg1545, results in full activation [77,99,111,116]. These studies suggest that single cleavage at Arg1545 is sufficient for activation of FV and that release of the B-domain from the heavy chain is not a necessary requirement for the expression of cofactor activity.
Careful evaluation of these studies indicates that the role of proteolysis and B-domain removal in driving FV activation is complex and far from understood. This, in part, stems from numerous factors including: the difficulty of evaluating three cleavage sites, failure to remove B-domain fragment(s) from cleaved FV preparations, associated problems with activity measurements (e.g. preventing feedback activation) and the inherent difficulty in preparing well-defined products using proteolysis. These problems clearly impose limitations in correlating proteolysis within the B-domain with the development of cofactor activity.
An alternative way of looking at this problem is to evaluate how FV is preserved as an inactive procofactor. One possibility is that binding sites on the heavy and/or light chain which are important for cofactor function are in a conformational state that precludes FXa/prothrombin binding. Proteolysis could then drive cofactor activation by facilitating essential conformational changes in a manner analogous to the activation strategy used by the chymotrypsin-like serine proteases [117]. A second possibility is that B-domain sequences serve an inhibitory function by rendering binding sites on the heavy and/or light chain inaccessible to FXa or prothrombin. Proteolysis would then promote dissociation of inhibitory B-domain sequences effecting activation in a so-called release for inhibition mechanism. Aspartic and cysteine proteases use this approach to control the inactivity of the zymogen [117]. Studies over the past several years into the function of the FV B-domain have provided key insights into discriminating between these possible mechanisms.
Role of the FV B-domain in maintaining the procofactor state
After the isolation of bovine [82,83,97] and human FV [77,98,99,118], it was apparent that a large part of the molecule was not necessary for activity. Elucidation of the primary structure of FV revealed that this region, termed the B-domain or connecting region, links the heavy and light chain regions of the molecule [81,119,120]. The human FV B-domain is 836 amino acids long, makes up ~50% of the mass of the protein, and has no homology to any other known protein, including the FVIII B-domain (Fig. 3) [81,120]. It is heavily glycosylated and has unusual regions of tandem repeats, the function of which is still unknown. Because of these unusual properties and lack of importance in FVa procoagulant activity, less attention has been given to studying its functional significance. It has been suggested, however, that the B-domain may play a role in the anticoagulant function of FV by stimulating the activated protein C-mediated inactivation of FVIIIa (for review see [20]).
Fig. 3.

Schematic representation of the factor (F)V B-domain. The B-domain is defined by residues 710–1545 which are liberated after thrombin-mediated proteolysis. An expanded view of the B-domain is indicated along with the sequence of the basic region (963–1008) implicated in preserving the FV procofactor state. Yellow circles, potential N-linked glycosylation sites; green box, 31X-9 amino acid tandem repeat region; red box, 2X-17 amino acid repeat region; blue box residues 963–1008.
The FV B-domain does not appear to contribute in a substantial way to thrombin binding [91,93]; however, it does appear to influence the rate and possibly order of bond cleavage. For example, the thrombin-mediated activation pathway of human FV follows a kinetically preferred order in which cleavage at Arg709 is followed by cleavage at Arg1018, and generation of the light chain is accompanied by cleavage at Arg1545 [17,18]. It is possible that this preference results from steric and/or conformational restrictions imposed by the B-domain; coordinate removal of these constraints exposes subsequent cleavage sites. Evidence for this comes from studies showing that mutating Arg1018 significantly delays cleavage at Arg1545, suggesting that proteolysis at Arg1018 causes a conformational change at or near position 1545 which then makes it more susceptible to thrombin [94,113–115].
As FV has little, if any activity and thrombin-mediated proteolysis of FV unmasks binding sites for FXa and prothrombin [77,78], an obvious role for the B-domain is to somehow prevent activity in the procofactor. Based on its size, it is easy to imagine how it could physically separate the heavy and light chains. Initial electron microscope (EM) images of FV and FVa suggested that the physical separation model was plausible, thus providing an adequate explanation for the inactivity of FV [121,122]. However, these observations were not consistent with several other EM images [123–126]. These last studies suggested that the B-domain appears as an appendage stemming from a globular core, presumed to be the heavy/light chain, which remains closely associated and essentially unchanged after the conversion from FV to FVa [126]; results that were consistent with physical studies of FV and FVa [97,127].
If the heavy and light chain regions of FV are conformationally similar to FVa, a role for the B-domain would then be to sterically block or conceal functional binding sites important to cofactor function. Initial evidence for this came from the Kane laboratory, who used a FV derivative in which a large segment of the B-domain was deleted (FVdes811–1491 or FV-810; Fig. 2) [113,128]. This recombinant single chain FV derivative was found to have constitutive, but partial activity of 30–38% compared with FVa when transiently expressed in COS-7 cells. The molecular basis for these findings was not investigated, yet full activity was achieved after proteolysis at Arg1545 by RVV-V or thrombin. These studies suggested that a clear function of the B-domain is to somehow prevent expression of procoagulant activity prior to proteolytic processing [113]. More recently, our laboratory has further investigated the molecular properties of this B-domain-deleted FV variant [80]. Using purified preparations derived from baby hamster kidney cells, we found that FV-810, as well as a thrombin-resistant derivative, yielded complementary but somewhat different results. Direct binding measurements and functional assays revealed that FV-810 interacts with membrane-bound FXa with high affinity and was functionally equivalent to FVa in the absence of intentional proteolysis [80]. These data suggested that proteolysis within the B-domain, while necessary, is incidental to the mechanism by which cofactor function is actually realized. Instead, proteolytic activation of FV simply eliminates steric and/or conformational constraints contributed by the B-domain that otherwise interfere with discrete binding interactions which govern the eventual function of FVa. Removal of these inhibitory constraints through recombinant truncation bypasses the requirement for proteolysis to activate the molecule.
These studies suggested that there must be regions of the B-domain that directly or indirectly contribute to keeping FV inactive. Using a panel of progressively finer B-domain truncated variants, we were able to identify a discrete region of the B-domain that appears to play a critical role in stabilizing the procofactor state [129]. This region of the B-domain (residues 963–1008; Fig. 3) is unusually basic with 18 out of 46 residues being Arg or Lys and is well conserved across the vertebrate lineage (see below). As expected, disruption of these sequences by mutagenesis or through deletion yielded derivatives with cofactor-like properties in the absence of intentional proteolysis. While still unclear, it is likely that other, as yet to be indentified components of the B-domain, also play a role. Thus, discrete sequences in the FV B-domain serve to stabilize the inactive procofactor state and suggest that the length of the B-domain per se is not a primary factor in preserving the procofactor state. The role of proteolysis in FV activation is therefore to facilitate removal of these inhibitory B-domain sequences in a release from the inhibition mechanism.
Sequence analysis and evolution of the FV B-domain
The finding that a discrete region of the B-domain appears to play such a fundamental role in regulating the procofactor to cofactor transition is surprising at first glance, as there is generally weak homology between the B-domains of mammalian species (<50%) [81,120,130–132]. However, careful inspection of these and other sequences from lower vertebrates has revealed some very interesting findings (Table 1). In mammalian species, certain features of the FV B-domain such as the number of tandem repeats, the overall length (~850 a.a.) and glycosylation content are generally conserved. However, this does not appear to be the case in lower vertebrates as both B-domain length and sequences can vary dramatically between species (Table 1). Major differences generally lie at the C-terminal half of the domain (e.g. sequences C-terminal to the equivalent Arg1018 thrombin cleavage site), which contains a variable number (9–36X) of 9-amino-acid tandem repeats; there are also several examples in which these repeats appear to be absent [81,133–136]. While there is weak homology throughout most of the B-domain when you compare various FV sequences, several short motifs are strongly conserved. Two of these regions lie near the Arg709 and Arg1545 cleavage sites. A third region was identified as the highly basic region detailed above which is remarkably well conserved from fish to mammals (Fig. 4) [135,136]. While it is unknown whether this sequence motif functions the same way in other species, the observation that this part of the B-domain is one of only a few portions that is highly conserved across the vertebrate lineage points to its functional significance.
Table 1.
Factor V properties from different species
| Species | Common Name | 1 Sequence Length | 2 B-domain Length | 3 Tandem Repeats | 41018 Site | 5 Length of N-term. B-domain | 6 Length of C-term. B-domain |
|---|---|---|---|---|---|---|---|
| Homo sapiens | Man | 2196 | 836 | 31X | LSPRT | 309 | 527 |
| Sus scrofa | Pig | 2230 | 874 | 37X | LSPRS | 292 | 582 |
| Bos taurus | Cow | 2183 | 823 | 29X | LSPRS | 293 | 530 |
| Mus musculus | Mouse | 2155 | 797 | 26X | LSPRG | 293 | 504 |
| Ornithorhynchus anatinus | Platypus | 1971 | 608 | 9X | MSPRG | 316 | 292 |
| Gallus gallus | Chicken | 1880 | 528 | None | LNPRS | 407 | 118 |
| Anolis carolinensis | Lizard | 1991 | 620 | None | LTPRT | 536 | 84 |
| Xenopus tropicalis | Frog | 1983 | 638 | None | FSPRG | 578 | 60 |
| Danio rerio | Zebrafish | 2095 | 709 | None | FSPRG | 621 | 88 |
| Takifugu rubripes | Pufferfish | 1816 | 471 | None | LSPRG | 375 | 96 |
| Pseudonaja textills | Brown Snake | 1430 | 46 | None | None | N/A | N/A |
The sequence length represents the mature sequence without the signal sequence.
Length of the B-domain is defined as sequences between the first (e.g. Arg709) and last thrombin-cleavage site (e.g. Arg1545).
Tandem repeats represent 9-amino-acid repeats with the human consensus sequence QT(T/N)LSPDLS in the C-terminal portion of the B domain.
The 1018 site refers to the equivalent Arg1018 cleavage site in human FV.
Length of N-terminal B-domain is defined as sequences between the first and second thrombin cleavage sites.
Length of C-terminal B-domain is defined as sequences between the second and last thrombin cleavage sites.
Fig. 4.

Alignment of factor (F)V B-domain sequences from the highly basic region. The conserved basic region (residues 963–1008) derived from the human FV sequence was aligned using a modified CLustal W algorithm (AlignX Module; Invitrogen Corporation town, state (if applicable), and country.) to corresponding regions from several select vertebrates (see Table 1 for common names).
An exception to these findings has been found in an unusual form of FV derived from the venom of three different Australian elapids [137–139]. These snakes (Pseudonaja textilis, Oxyuranus scutellatus scutellatus and Oxyuranus microlepidotus), which are the most venomous in Australia, are known to have a powerful prothrombin-activating component in their venom. These venom proteins are multi-subunit complexes consisting of a FXa-like and FV-like component and are highly procoagulant [140–142]. Venom-derived FV from these snakes share ~44% homology with mammalian FV and have a similar domain structure (A1-A2-B-A3-C1-C2) [137–139]. Surprisingly, their B-domains are remarkably short (46 residues), and more importantly they lack the conserved basic region. These findings raise the possibility that these snake species have shed these regulatory components to synthesize a constitutively active form of FV. To examine this, we recently expressed and characterized recombinant venom FV derived from P. textilis (pt-FV). We were able to show that pt-FV is synthesized in an active state and unlike human FV does not require proteolytic removal of the B-domain to express procoagulant activity [143]. Thus, pt-FV represents a biological correlate to structure/function studies with human FV and is a naturally occurring example of a protein that has acquired a new functional state through loss of inhibitory sequences. Remarkably, this protein can also function in the absence of anionic membranes and is completely resistant to activated protein C, despite being cleaved within the heavy chain at the equivalent Arg506 and Arg709 sites [143]. We speculate that this functional resistance to activated protein C is likely because of non-covalent interactions which contribute to the stabilization of activity. Additionally, it is also possible that a unique disulfide bond linking the A2 and the A3 domains may play some role in stabilizing pt-FV thereby preventing dissociation of the C-terminal heavy chain region from the rest of the molecule after activated protein C cleavage. Thus, pt-FV represents an exceptional example of a protein that has adapted into a potent biological weapon for host defense and envenomation of prey.
Concluding remarks
FV and FVIII both circulate in blood as inactive procofactors and only express activity after limited proteolysis. Once activated, they serve as two important cofactors in coagulation by dramatically enhancing the catalytic rate of their respective macromolecular enzyme complexes. This similarity in function is not surprising, considering that FV and FVIII are thought to descend from a common ancestral A1-A2-A3-containing protein through a gene-duplication event [133,134]. After the acquisition of C-type domains as well as the B-domain, a second gene-duplication ultimately separated ancestral genes for FV and FVIII. Despite this common origin and functional equivalence, the mechanisms by which these proteins are kept in an inactive procofactor state are fundamentally different. For FVIII, the B-domain does not appear to be involved in regulating cofactor activity. Rather, cleavage between the A1 and A2 domains at position Arg372 is critical for the generation of cofactor activity. Furthermore, FVIII association with VWF also plays an important role, not only in stabilizing FVIII, but also in obscuring functional binding sites important to cofactor function. In contrast, the FV B-domain plays a fundamentally important role as discrete conserved B-domain sequences are involved in the mechanism by which FV persists as an inactive procofactor. Elimination of these sequences bypasses the requirement for FV proteolysis to activate the molecule; a clear example of this as observed in nature is venom FV derived from the common brown snake, P. textilis. This implies that the B-domain serves an inhibitory function which, under normal physiological conditions, is efficiently removed upon proteolytic processing.
The molecular process of maintaining FV and FVIII as inactive procofactors plays a critical regulatory role which has evolved to limit the expression of cofactor activity. Despite its significance, clear mechanistic insight by which the various proteolytic events lead to expression of FVa and FVIIIa procoagulant activity has proven difficult to pinpoint. However, novel approaches coupled with structural information have provided some new clues. While several questions remain, these recent studies lay the groundwork for uncovering the precise molecular mechanism by which FV and FVIII transition from the procofactor to cofactor state.
Acknowledgments
We are grateful to Dr Hans Vos from the Leiden University Medical Center for providing us with the FV sequences of Gallus gallus, Anolis carolinensis and Xenopus tropicalis. We would also like to thank our colleague, Dr Sriram Krishnaswmay for critical review of the manuscript. We would like to acknowledge funding from the National Institutes of Health (HL-88010 and HL-74124, Project 2; to R.M.C.) and the Judith Graham Pool Postdoctoral Research Fellowship (to M.H.A.B.).
Footnotes
Disclosure of Conflicts of Interest
R.M.C. receives research support and royalties from Wyeth Pharmaceuticals for technology related to FXa. M.H.A.B. declares no conflicts of interest.
References
- 1.Mann KG, Jenny RJ, Krishnaswamy S. Cofactor proteins in the assembly and expression of blood clotting enzyme complexes. Annual Rev Biochem. 1988;57:915–56. doi: 10.1146/annurev.bi.57.070188.004411. [DOI] [PubMed] [Google Scholar]
- 2.Kane WH, Davie EW. Blood Coagulation factors V and VIII: Structural and functional similarities and their relationship to hemorrhagic and thrombotic disorders. Blood. 1988;71:539–55. [PubMed] [Google Scholar]
- 3.Mann KG, Nesheim ME, Church WR, Haley PE, Krishnaswamy S. Surface dependent reactions of the vitamin K-dependent enzyme complexes. Blood. 1990;76:1–16. [PubMed] [Google Scholar]
- 4.Jacquemin M, De Maeyer M, D’Oiron R, Lavern’Homme R, Peerlinck K, Saint-Remy J-M. Molecular mechanisms of mild and moderate hemophilia A. J Thromb Haemost. 2003;1:456–63. doi: 10.1046/j.1538-7836.2003.00088.x. [DOI] [PubMed] [Google Scholar]
- 5.Mannucci PM, Duga S, Peyvandi F. Recessively inherited coagulation disorders. Blood. 2004;104:1243–52. doi: 10.1182/blood-2004-02-0595. [DOI] [PubMed] [Google Scholar]
- 6.Fay PJ. Activation of factor VIII and mechanisms of cofactor action. Blood Reviews. 2004;18:1–15. doi: 10.1016/s0268-960x(03)00025-0. [DOI] [PubMed] [Google Scholar]
- 7.Lollar P. Structure and function of Factor VIII. Adv Exp Med Biol. 1995;386:3–17. doi: 10.1007/978-1-4613-0331-2_1. [DOI] [PubMed] [Google Scholar]
- 8.Krishnaswamy S. Exosite-driven substrate specificity and function in coagulation. J Thromb Haemost. 2005;3:54–67. doi: 10.1111/j.1538-7836.2004.01021.x. [DOI] [PubMed] [Google Scholar]
- 9.Bock PE, Panizzi P, Verhamme IM. Exosites in the substrate specificity of blood coagulation reactions. J Thromb Haemost. 2007;5 (Suppl 1):81–94. doi: 10.1111/j.1538-7836.2007.02496.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nesheim ME, Taswell JB, Mann KG. The contribution of bovine factor V and factor Va to the activity of prothrombinase. J Biol Chem. 1979;254:10952–62. [PubMed] [Google Scholar]
- 11.Foster WB, Nesheim ME, Mann KG. The factor Xa-catalyzed activation of factor V. J Biol Chem. 1983;258:13970–7. [PubMed] [Google Scholar]
- 12.Lollar P, Knutson G, Fass D. Activation of porcine FVIII:C by thrombin and FXa. Biochemistry. 1985;24:8056–64. doi: 10.1021/bi00348a033. [DOI] [PubMed] [Google Scholar]
- 13.Ware AG, Guest M, Seegers WH. Plasma accelerator factor and purified prothrombin activation. Science. 1947;106:41–2. doi: 10.1126/science.106.2741.41-a. [DOI] [PubMed] [Google Scholar]
- 14.Ware AG, Murphy R, Seegers WH. The function of Ac-globulin in blood clotting. Science. 1947;106:618–9. doi: 10.1126/science.106.2764.618-a. [DOI] [PubMed] [Google Scholar]
- 15.Owren PA. Parahaemophila: haemorrhagic diathesis due to absence of a previously unknown clotting factor. Lancet. 1947;249:446–8. [PubMed] [Google Scholar]
- 16.Rapaport SI, Schiffman S, Patch MJ, Ames SB. The importance of activation of anti-haemophilic globulin and proaccelerin by traces of thrombin in the generation of intrinsic prothrombinase activity. Blood. 1963;21:221–36. [PubMed] [Google Scholar]
- 17.Mann KG, Kalafatis M. Factor V: A combination of Dr. Jekyll and Mr. Hyde. Blood. 2002;101:20–30. doi: 10.1182/blood-2002-01-0290. [DOI] [PubMed] [Google Scholar]
- 18.Kane WH, Factor V. Hemostasis and Thrombosis. 5. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 177–92. Xxxx XX, Xxxxxx XX, eds. [Google Scholar]
- 19.Fay PJ, Jenkins PV. Mutating factor VIII: lessons from structure to function. Blood Rev. 2005;19:15–27. doi: 10.1016/j.blre.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 20.Segers K, Dahlback B, Nicolaes GA. Coagulation factor V and thrombophilia: background and mechanisms. Thromb Haemost. 2007;98:530–42. [PubMed] [Google Scholar]
- 21.Lenting PJ, Van Mourik JA, Mertens K. The life cycle of coagulation factor VIII in view of its structure and function. Blood. 1998;92:3983–96. [PubMed] [Google Scholar]
- 22.Vehar GA, Davie EW. Preparation and properties of bovine factor VIII (antihemophilic factor) Biochemistry. 1980;19:401–10. doi: 10.1021/bi00544a001. [DOI] [PubMed] [Google Scholar]
- 23.Fass DN, Knutson GJ, Katzmann JA. Monoclonal antibodies to porcine factor VIII coagulant and their use in the isolation of active coagulant protein. Blood. 1982;59:594–600. [PubMed] [Google Scholar]
- 24.Fulcher CA, Roberts J, Zimmerman TS. Thrombin proteolysis of purified FVIII procoagulant protein: Correlation of activation with generation of specific polypeptides. Blood. 1983;60:807. [PubMed] [Google Scholar]
- 25.Lollar P, Knutson G, Fass D. Stabilization of thrombin activated porcine Factor VIII:C by Factor IXa and phospholipid. Blood. 1984;63:1303–8. [PubMed] [Google Scholar]
- 26.Toole JJ, Knopf JL, Wozney JM, Sultman LA, Buecker JL, Pittman DD, Kaufman RJ, Brown E, Shoemaker C, Orr EC, Amphlett GW, Foster WB, Coe ML, Knutson GJ, Fass DN, Hewick RM. Molecular cloning of a cDNA encoding human antihaemophilic factor. Nature. 1984;312:342–7. doi: 10.1038/312342a0. [DOI] [PubMed] [Google Scholar]
- 27.Wood WI, Capon DJ, Simonsen CC, Eaton DL, Gitschier J, Keyt B, Seeburg PH, Smith DH, Hollingshead P, Wion KL, Delwart E, Tuddenham EGD, Vehar GA, Lawn RM. Expression of active human factor VIII from recombinant DNA clones. Nature. 1984;312:330–6. doi: 10.1038/312330a0. [DOI] [PubMed] [Google Scholar]
- 28.Eaton D, Rodriguez H, Vehar G. Proteolytic processing of human FVIII. Correlation of specific cleavages by thrombin FXa and activated protein C with activation and inactivation of Factor VIII coagulant activity. Biochemistry. 1986;25:505–12. doi: 10.1021/bi00350a035. [DOI] [PubMed] [Google Scholar]
- 29.Duffy EJ, Parker ET, Mutucumarana VP, Johnson AE, Lollar P. Binding of factor VIIIa and factor VIII to factor IXa on phospholipid vesicles. J Biol Chem. 1992;267:17006–11. [PubMed] [Google Scholar]
- 30.Vehar G, Keyt B, Eaton D, Rodriguez H, O’Brien DP, Rotblat F, Oppermann H, keck R, Wood W, Harkins R, Tuddenham EGD, Lawn R, Capon D. Structure of human factor VIII. Nature. 1984;312:337–42. doi: 10.1038/312337a0. [DOI] [PubMed] [Google Scholar]
- 31.Nogami K, Wakabayashi H, Schmidt K, Fay PJ. Altered interactions between the A1 and A2 subunits of factor VIIIa following cleavage of A1 subunit by factor Xa. J Biol Chem. 2003;278:1634–41. doi: 10.1074/jbc.M209811200. [DOI] [PubMed] [Google Scholar]
- 32.Esmon CT, Lollar P. Involvement of thrombin anion-binding exosites 1 and 2 in the activation of factor V and factor VIII. J Biol Chem. 1996;271:13882–7. doi: 10.1074/jbc.271.23.13882. [DOI] [PubMed] [Google Scholar]
- 33.Myles T, Yun TH, Leung LL. Structural requirements for the activation of human factor VIII by thrombin. Blood. 2002;100:2820–6. doi: 10.1182/blood-2002-03-0843. [DOI] [PubMed] [Google Scholar]
- 34.Newell JL, Fay PJ. Acidic residues C-terminal to the A2 domain facilitate thrombin-catalyzed activation of factor VIII. Biochemistry. 2008;47:8786–95. doi: 10.1021/bi8007824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nogami K, Zhou Q, Myles T, Leung LL, Wakabayashi H, Fay PJ. Exosite-interactive regions in the A1 and A2 domains of factor VIII facilitate thrombin-catalyzed cleavage of heavy chain. J Biol Chem. 2005;280:18476–87. doi: 10.1074/jbc.M412778200. [DOI] [PubMed] [Google Scholar]
- 36.Lollar P, Parker C. XxxxpH-dependent denaturation of thrombin-activated porcine factor VIII. J Biol Chem. 1990;265:1688–92. [PubMed] [Google Scholar]
- 37.Fay PJ, Haidari PJ, Smudzin TM. Human factor VIIIa subunit structure. Reconstitution of factor VIIIa from the isolated A1/A3-C1-C2 dimer and A2 subunit. J Biol Chem. 1991;266:8957–62. [PubMed] [Google Scholar]
- 38.Lollar P, Parker ET. Structural basis for the decreased procoagulant activity of human factor VIII compared to the porcine homolog. J Biol Chem. 1991;266:12481–6. [PubMed] [Google Scholar]
- 39.Pittman DD, Kaufman RJ. Proteolytic requirements for thrombin activation of anti-hemophilic factor (factor VIII) Proc Natl Acad Sci USA. 1988;85:2429–33. doi: 10.1073/pnas.85.8.2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hamer RJ, Koedam JA, Beeser-Visser NH, Bertina RM, Van Mourik JA, Sixma JJ. Factor VIII binds to von Willebrand factor via its Mr-80,000 light chain. Eur J Biochem. 1987;166:37–43. doi: 10.1111/j.1432-1033.1987.tb13480.x. [DOI] [PubMed] [Google Scholar]
- 41.Hill-Eubanks DC, Parker CG, Lollar P. Differential proteolytic activation of factor VIII-von Willebrand factor complex by thrombin. Proc Natl Acad Sci USA. 1989;86:6508–12. doi: 10.1073/pnas.86.17.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Regan LM, Fay PJ. Cleavage of factor VIII light chain is required for maximal generation of factor VIIIa activity. J Biol Chem. 1995;270:8546–52. doi: 10.1074/jbc.270.15.8546. [DOI] [PubMed] [Google Scholar]
- 43.Donath M-JSH, Lenting PJ, Van Mourik JA, Mertens K. The role of cleavage of the light chain at positions Arg1689 or Arg1721 in subunit interactions and activation of human blood coagulation factor VIII. J Biol Chem. 1995;270:3648–55. doi: 10.1074/jbc.270.8.3648. [DOI] [PubMed] [Google Scholar]
- 44.Pipe SW, Kaufman RJ. Characterization of genetically engineered inactivation-resistant coagulation factor VIIIa. Proc Natl Acad Sci USA. 1997;94:11851–6. doi: 10.1073/pnas.94.22.11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Newell JL, Fay PJ. Cleavage at Arg-1689 influences heavy chain cleavages during thrombin-catalyzed activation of factor VIII. J Biol Chem. 2009;284:11080–9. doi: 10.1074/jbc.M900234200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newell JL, Fay PJ. Proteolysis at Arg740 facilitates subsequent bond cleavages during thrombin-catalyzed activation of factor VIII. J Biol Chem. 2007;282:25367–75. doi: 10.1074/jbc.M703433200. [DOI] [PubMed] [Google Scholar]
- 47.Shima M, Ware J, Yoshioka A, Fukui H, Fulcher CA. An arginine to cysteine amino acid substitution at a critical thrombin cleavage site in dysfunctional factor VIII molecule. Blood. 1989;74:1612–7. [PubMed] [Google Scholar]
- 48.Arai M, Inaba H, Higuchi M, Antonarakis SE, Kazazian HH, Fujimaki M, Hoyer LW. Direct characterization of factor VIII in plasma: Detection of a mutation altering a thrombin cleavage site (arginine-372 to histidine) Proc Natl Acad Sci USA. 1989;86:4277–81. doi: 10.1073/pnas.86.11.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.O’Brien DP, Pattinson JK, Tuddenham EGD. Purification and characterization of factor VIII 372-Cys: A hypofunctional cofactor from a patient with moderately severe haemophilia A. Blood. 1990;75:1664–72. [PubMed] [Google Scholar]
- 50.Nogami K, Zhou Q, Wakabayashi H, Fay PJ. Thrombin-catalyzed activation of factor VIII with His substituted for Arg372 at the P1 site. Blood. 2005;105:4362–8. doi: 10.1182/blood-2004-10-3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fay PJ, Mastri M, Koszelak ME, Wakabayashi H. Cleavage of factor VIII heavy chain is required for the functional interaction of a2 subunit with factor IXa. J Biol Chem. 2001;276:12434–9. doi: 10.1074/jbc.M009539200. [DOI] [PubMed] [Google Scholar]
- 52.Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, Eaton DH, Vehar GA, Capon DJ, Lawn RM. Characterization of the human factor VIII gene. Nature. 1984;312:326–30. doi: 10.1038/312326a0. [DOI] [PubMed] [Google Scholar]
- 53.Cripe LD, Moore D, Kane WH. Structure of the gene for human coagulation factor V. Biochemistry. 1992;31:3777–85. doi: 10.1021/bi00130a007. [DOI] [PubMed] [Google Scholar]
- 54.Shen BW, Spiegel PC, Chang CH, Huh JW, Lee JS, Kim J, Kim YH, Stoddard BL. The tertiary structure and domain organization of coagulation factor VIII. Blood. 2008;111:1240–7. doi: 10.1182/blood-2007-08-109918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ngo JC, Huang M, Roth DA, Furie BC, Furie B. Crystal structure of human factor VIII: implications for the formation of the factor IXa-factor VIIIa complex. Structure. 2008;16:597–606. doi: 10.1016/j.str.2008.03.001. [DOI] [PubMed] [Google Scholar]
- 56.Curtis JE, Helgerson SL, Parker ET, Lollar P. Isolation and characterization of thrombin-activated human factor VIII. J Biol Chem. 1994;269:6246–51. [PubMed] [Google Scholar]
- 57.O’Brien LM, Huggins CF, Fay PJ. Interacting regions in the A1 and A2 subunits of factor VIIIa identified by zero-length cross-linking. Blood. 1997;90:3943–50. [PubMed] [Google Scholar]
- 58.Sudhakar K, Fay PJ. Exposed hydrophobic sites in factor VIII and isolated subunits. J Biol Chem. 1996;271:23015–21. doi: 10.1074/jbc.271.38.23015. [DOI] [PubMed] [Google Scholar]
- 59.Toole JJ, Pittman DD, Orr EC, Murtha P, Wasley LC, Kaufman RJ. A large region (~95 kDa) of human factor VIII is dispensable for in vitro procoagulant activity. Proc Natl Acad Sci USA. 1986;83:5939–42. doi: 10.1073/pnas.83.16.5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eaton DL, Wood WI, Eaton D, Hass PE, Hollingshead P, Wion KL, Mather J, Lawn RM, Vehar GA, Gorman C. Construction and characterization of an active factor VIII variant lacking the central one-third of the molecule. Biochemistry. 1986;25:8343–7. doi: 10.1021/bi00374a001. [DOI] [PubMed] [Google Scholar]
- 61.Pittman DD, Alderman EM, Tomkinson KN, Wang JH, Giles AR, Kaufman RJ. Biochemical, immunological, and in vivo functional characterization of B-domain-deleted FVIII. Blood. 1993;81:2925–35. [PubMed] [Google Scholar]
- 62.Lind P, Larsson K, Spira J, Sydow-Backman M, Almstedt A, Gray E, Sandberg H. Novel forms of B-domain deleted recombinant factor VIII molecules. Construction and biochemical characterization. Eur J Biochem. 1995;232:19–27. doi: 10.1111/j.1432-1033.1995.tb20776.x. [DOI] [PubMed] [Google Scholar]
- 63.Jenny RJ, Mann KG. Factor V: a prototype pro-cofactor for vitamin K-dependent enzyme complexes in blood clotting. Bailliere’s Clin Haem. 1989;2:919–44. doi: 10.1016/s0950-3536(89)80052-6. [DOI] [PubMed] [Google Scholar]
- 64.Tracy PB, Eide LL, Bowie EJW, Mann KG. Radioimmunoassay of factor V in human plasma and platelets. Blood. 1982;60:59–63. [PubMed] [Google Scholar]
- 65.Gewirtz AM, Keefer M, Doshi K, Anyanayaki A, Hiu C, Colman RW. Biology of human megakaryocyte Factor V. Blood. 1986;67:1639–42. [PubMed] [Google Scholar]
- 66.Chiu HC, Schick P, Colman RW. Biosynthesis of Factor V in isolated guinea pig megakaryocytes. J Clin Invest. 1985;75:339–46. doi: 10.1172/JCI111706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giampaolo A, Vulcano F, Macioce G, Mattia G, Barca A, Milazzo L, Ciccarelli C, Hassan HJ. Factor-V expression in platelets from human megakaryocytic culture. Br J Haematol. 2005;128:108–11. doi: 10.1111/j.1365-2141.2004.05279.x. [DOI] [PubMed] [Google Scholar]
- 68.Camire RM, Pollak ES, Kaushansky K, Tracy PB. Secretable human platelet-derived factor V originates from the plasma pool. Blood. 1998;92:3035–41. [PubMed] [Google Scholar]
- 69.Christella M, Thomassen LG, Castoldi E, Tans G, Magdeleyns EJ, Delaunoit C, Debusscher L, Van Assche KJ, Rosing J. Endogenous factor V synthesis in megakaryocytes contributes negligibly to the platelet factor V pool. Haematologica. 2003;88:1150–6. [PubMed] [Google Scholar]
- 70.Gould WR, Simioni P, Silveira JR, Tormene D, Kalafatis M, Tracy PB. Megakaryocytes endocytose and subsequently modify human factor V in vivo to form the entire pool of a unique platelet-derived cofactor. J Thromb Haemost. 2005;3:450–6. doi: 10.1111/j.1538-7836.2005.01157.x. [DOI] [PubMed] [Google Scholar]
- 71.Bouchard BA, Williams JL, Meisler NT, Long MW, Tracy PB. Endocytosis of plasma-derived factor V by megakaryocytes occurs via a clathrin-dependent, specific membrane binding event. J Thromb Haemost. 2005;3:541–51. doi: 10.1111/j.1538-7836.2005.01190.x. [DOI] [PubMed] [Google Scholar]
- 72.Bouchard BA, Meisler NT, Nesheim ME, Liu CX, Strickland DK, Tracy PB. A unique function for LRP-1: a component of a two-receptor system mediating specific endocytosis of plasma-derived factor V by megakaryocytes. J Thromb Haemost. 2008;6:638–44. doi: 10.1111/j.1538-7836.2008.02894.x. [DOI] [PubMed] [Google Scholar]
- 73.Gould WR, Silveira JR, Tracy PB. Unique in vivo modifications of coagulation factor V produce a physically and functionally distinct platelet-derived cofactor: characterization of purified platelet-derived factor V/Va. J Biol Chem. 2004;279:2383–93. doi: 10.1074/jbc.M308600200. [DOI] [PubMed] [Google Scholar]
- 74.Monkovic DD, Tracy PB. Functional characterization of human platelet-released factor V and its activation by factor Xa and thrombin. J Biol Chem. 1990;265:17132–40. [PubMed] [Google Scholar]
- 75.Camire RM, Kalafatis M, Cushman M, Tracy RP, Mann KG, Tracy PB. The mechanism of inactivation of human platelet factor Va from normal and activated protein C-resistant individuals. J Biol Chem. 1995;270:20794–800. doi: 10.1074/jbc.270.35.20794. [DOI] [PubMed] [Google Scholar]
- 76.Camire RM, Kalafatis M, Simioni P, Girolami A, Tracy PB. Platelet-derived factor Va/VaLeiden cofactor activities are sustained on the surface of activated platelets despite the presence of activated protein C. Blood. 1998;91:2818–29. [PubMed] [Google Scholar]
- 77.Suzuki K, Dahlback B, Stenflo J. Thrombin-catalyzed activation of human coagulation factor V. J Biol Chem. 1982;257:6556–64. [PubMed] [Google Scholar]
- 78.Esmon CT, Owen WG, Duiguid D, Jackson CM. The action of thrombin on blood clotting factor V: Conversion of factor V to a prothrombin-binding protein. Biochim Biophys Acta. 1973;310:289–94. doi: 10.1016/0005-2795(73)90034-2. [DOI] [PubMed] [Google Scholar]
- 79.Monkovic DD, Tracy PB. Activation of human factor V by factor Xa and thrombin. Biochemistry. 1990;29:1118–28. doi: 10.1021/bi00457a004. [DOI] [PubMed] [Google Scholar]
- 80.Toso R, Camire RM. Removal of B-domain sequences from factor V rather than specific proteolysis underlies the mechanism by which cofactor function is realized. J Biol Chem. 2004;279:21643–50. doi: 10.1074/jbc.M402107200. [DOI] [PubMed] [Google Scholar]
- 81.Jenny RJ, Pittman DD, Toole JJ, Kriz RW, Aldape RA, Hewick RM, Kaufman RJ, Mann KG. Complete cDNA and derived amino acid sequence of human factor V. Proc Natl Acad Sci USA. 1987;84:4846–50. doi: 10.1073/pnas.84.14.4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Esmon CT. The subunit structure of thrombin-activated factor V. Isolation of activated factor V, separation of subunits and reconstitution of biological activity. J Biol Chem. 1979;254:964–73. [PubMed] [Google Scholar]
- 83.Nesheim ME, Mann KG. Thrombin-catalyzed activation of single chain bovine factor V. J Biol Chem. 1979;254:1326–34. [PubMed] [Google Scholar]
- 84.Krishnaswamy S, Russell GD, Mann KG. The reassociation of factor Va from its isolated subunits. J Biol Chem. 1989;264:3160–8. [PubMed] [Google Scholar]
- 85.Nesheim ME, Foster WB, Hewick R, Mann KG. Characterization of factor V activation intermediates. J Biol Chem. 1984;259:3187–96. [PubMed] [Google Scholar]
- 86.Arocas V, Lemaire C, Bouton MC, Bezeaud A, Bon C, Guillin MC, Perrus MJ. Inhibition of thrombin-catalyzed factor V interaction by bothrojaracin. Thromb Haemost. 1998;79:1157–61. [PubMed] [Google Scholar]
- 87.Bukys MA, Orban T, Kim PY, Beck DO, Nesheim ME, Kalafatis M. The structural integrity of anion binding exosite I of thrombin is required and sufficient for timely cleavage and activation of factor V and factor VIII. J Biol Chem. 2006;281:18569–80. doi: 10.1074/jbc.M600752200. [DOI] [PubMed] [Google Scholar]
- 88.Henriksen RA, Owen WG, Nesheim ME, Mann KG. Identification of a congenital dysthrombin, thrombin Quick. J Clin Invest. 1980;66:934–40. doi: 10.1172/JCI109961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Myles T, Yun TH, Hall SW, Leung LL. An extensive interaction interface between thrombin and factor V is required for factor V activation. J Biol Chem. 2001;276:25143–9. doi: 10.1074/jbc.M011324200. [DOI] [PubMed] [Google Scholar]
- 90.Esmon CT, Esmon NL, Harris KW. Complex formation between thrombin and thrombomodulin inhibits both thrombin-catalyzed fibrin formation and factor V activation. J Biol Chem. 1982;257:7944. [PubMed] [Google Scholar]
- 91.Dharmawardana KR, Bock PE. Demonstration of exosite I-dependent interactions of thrombin with human factor V and factor Va involving the factor Va heavy chain: Analysis by affinity chromatography employing a novel method for active-site selective immobilization of serine proteinases. Biochemistry. 1998;37:13143–52. doi: 10.1021/bi9812165. [DOI] [PubMed] [Google Scholar]
- 92.Segers K, Dahlback B, Bock PE, Tans G, Rosing J, Nicolaes GA. The role of thrombin exosites I and II in the activation of human coagulation factor V. J Biol Chem. 2007;282:33915–24. doi: 10.1074/jbc.M701123200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Dharmawardana KR, Olson ST, Bock PE. Role of regulatory exosite I in binding of thrombin to human factor V, factor Va, factor Va subunits, and activation fragments. J Biol Chem. 1999;274:18635–43. doi: 10.1074/jbc.274.26.18635. [DOI] [PubMed] [Google Scholar]
- 94.Marquette KA, Pittman DD, Kaufman RJ. The factor V B-domain provides two functions to facilitate thrombin cleavage and release of the light chain. Blood. 1995;86:3026–34. [PubMed] [Google Scholar]
- 95.Pittman DD, Tomkinson KN, Michnick D, Seligsohn U, Kaufman RJ. Posttranslational sulfation of factor V is required for efficient thrombin cleavage and activation and for full procoagulant activity. Biochemistry. 1994;33:6952–9. doi: 10.1021/bi00188a026. [DOI] [PubMed] [Google Scholar]
- 96.Beck DO, Bukys MA, Singh LS, Szabo K, Kalafatis M. The contribution of amino acid region Asp695-Asp698 of factor V to procofactor activation and factor Va function. J Biol Chem. 2004;279:3084–95. doi: 10.1074/jbc.M306850200. [DOI] [PubMed] [Google Scholar]
- 97.Nesheim ME, Myrmel KH, Hibbard L, Mann KG. Isolation and characterization of single chain bovine factor V. J Biol Chem. 1979;254:508–17. [PubMed] [Google Scholar]
- 98.Dahlback B. Human coagulation factor V purification and thrombin-catalyzed activation. J Clin Invest. 1980;66:583–91. doi: 10.1172/JCI109890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kane WH, Majerus PW. Purification and characterization of human coagulation factor V. J Biol Chem. 1981;256:1002–7. [PubMed] [Google Scholar]
- 100.Oates AM, Salem HH. The regulation of human factor V by a neutrophil protease. Blood. 1987;70:846–51. [PubMed] [Google Scholar]
- 101.Bradford HN, Annamalai A, Doshi K, Colman RW. Factor V is activated and cleaved by platelet calpain: Comparison with thrombin proteolysis. Blood. 1988;71:388–94. [PubMed] [Google Scholar]
- 102.Lee CD, Mann KG. Activation/Inactivation of human factor V by plasmin. Blood. 1989;73:185–90. [PubMed] [Google Scholar]
- 103.Yukelson LY, Tans G, Thomassen MCLGD, Hemker HC, Rosing J. Procoagulant activities in venoms from central Asian snakes. Toxicon. 1991;29:491–502. doi: 10.1016/0041-0101(91)90023-k. [DOI] [PubMed] [Google Scholar]
- 104.Gerads I, Tans G, Yukelson LY, Zwaal RFA, Rosing J. Activation of bovine factor V by an activator purified from the venom of Naja Naja Oxiana. Toxicon. 1992;30:1065–79. doi: 10.1016/0041-0101(92)90052-7. [DOI] [PubMed] [Google Scholar]
- 105.Bakker HM, Tans G, Thomassen MCLGD, Yukelson LY, Ebberink R, Hemker HC, Rosing J. Functional properties of human factor Va lacking the Asp683-Arg709 domain of the heavy chain. J Biol Chem. 1994;269:20662–7. [PubMed] [Google Scholar]
- 106.Tans G, Nicolaes GAF, Christella M, Thomassen LGD, Hemker HC, van Zonneveld A-J, Pannekoek H, Rosing J. Activation of human factor V by meizothrombin. J Biol Chem. 1994;269:15969–72. [PubMed] [Google Scholar]
- 107.Allen DH, Tracy PB. Human coagulation factor V is activated to the functional cofactor by elastase and cathepsin G expressed at the monocyte surface. J Biol Chem. 1995;270:1408–15. doi: 10.1074/jbc.270.3.1408. [DOI] [PubMed] [Google Scholar]
- 108.Samis JA, Garrett M, Manuel RP, Nesheim ME, Giles AR. Human neutrophil elastase activates human factor V but inactivates thrombin-activated human factor V. Blood. 1997;90:1065–74. [PubMed] [Google Scholar]
- 109.Camire RM, Kalafatis M, Tracy PB. Proteolysis of factor V by cathepsin G and elastase indicates that cleavage at Arg1545 optimizes cofactor function by facilitating factor Xa binding. Biochemistry. 1998;37:11896–906. doi: 10.1021/bi980520v. [DOI] [PubMed] [Google Scholar]
- 110.Rosing J, Govers-Riemslag JWP, Yukelson LY, Tans G. Factor V activation and inactivation by venom proteases. Haemostasis. 2001;31:241–6. doi: 10.1159/000048069. [DOI] [PubMed] [Google Scholar]
- 111.Kalafatis M, Beck DO, Mann KG. Structural requirements for expression of factor Va activity. J Biol Chem. 2004;278:33550–61. doi: 10.1074/jbc.M303153200. [DOI] [PubMed] [Google Scholar]
- 112.Pittman DD, Marquette KA, Kaufman RJ. Role of the B domain for factor VIII and factor V expression and function. Blood. 1994;84:4214–25. [PubMed] [Google Scholar]
- 113.Keller FG, Ortel TL, Quinn-Allen MA, Kane WH. Thrombin-catalyzed activation of recombinant human factor V. Biochemistry. 1995;34:4118–24. doi: 10.1021/bi00012a030. [DOI] [PubMed] [Google Scholar]
- 114.Thorelli E, Kaufman RJ, Dahlback B. Cleavage requirements for activation of factor V by factor Xa. Eur J Biochem. 1997;247:12–20. doi: 10.1111/j.1432-1033.1997.00012.x. [DOI] [PubMed] [Google Scholar]
- 115.Steen M, Dahlback B. Thrombin-mediated proteolysis of factor V resulting in gradual B-domain release and exposure of the factor Xa-binding site. J Biol Chem. 2002;277:38424–30. doi: 10.1074/jbc.M204972200. [DOI] [PubMed] [Google Scholar]
- 116.Smith CM, Hanahan DJ. The activation of factor V by factor Xa or α-chymotrypsin and comparison with thrombin and RVV-V action. An improved factor V isolation procedure. Biochemistry. 1976;15:1830–7. doi: 10.1021/bi00654a007. [DOI] [PubMed] [Google Scholar]
- 117.Khan AM, James MNG. Molecular mechanisms for the conversion of zymogens to active proteolytic enzymes. Prot Sci. 1998;7:815–36. doi: 10.1002/pro.5560070401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Katzmann JA, Nesheim ME, Hibbard LS, Mann KG. Isolation of functional human coagulation Factor V by using a hybridoma antibody. Proc Natl Acad Sci USA. 1981;78:162–6. doi: 10.1073/pnas.78.1.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kane WH, Davie EW. Cloning of a cDNA coding for human factor V, a blood coagulation factor homologous to factor VIII and ceruloplasmin. Proc Natl Acad Sci USA. 1986;83:6800–4. doi: 10.1073/pnas.83.18.6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kane WH, Ichinose A, Hagen FS, Davie EW. Cloning of cDNAs coding for the heavy chain region and connecting region of human Factor V, a blood coagulation factor with four types of internal repeats. Biochemistry. 1987;26:6508. doi: 10.1021/bi00394a033. [DOI] [PubMed] [Google Scholar]
- 121.Dahlback B. Ultrastructure of human coagulation factor V. J Biol Chem. 1985;260:1347–9. [PubMed] [Google Scholar]
- 122.Dahlback B. Bovine coagulation factor V visualized with electron microscopy. Ultrastructure of the isolated activated forms and of the activtion fragments. J Biol Chem. 1986;261:9495–501. [PubMed] [Google Scholar]
- 123.Lampe PD, Pusey ML, Wei J, Nelsestuen GL. Electron microscopy and hydrodynamic properties of blood clotting factor V and activation fragments of factor V with phospholipid vesicles. J Biol Chem. 1984;259:9959–64. [PubMed] [Google Scholar]
- 124.Mosesson MW, Nesheim ME, DiOrio JP, Hainfeld JF, Wall JS, Mann KG. Studies on the structure of bovine factor V by scanning transmission electron microscopy. Blood. 1985;65:1158–62. [PubMed] [Google Scholar]
- 125.Fowler WE, Fay PJ, Arvan DS, Marder VJ. Electro microscopy of human factor V and factor VIII: Correlation of morphology with domain structure and localization of factor V activation fragments. Proc Natl Acad Sci USA. 1990;87:7648–52. doi: 10.1073/pnas.87.19.7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mosesson MW, Church WR, DiOrio JP, Krishnaswamy S, Mann KG, Hainfeld JF, Wall JS. Structural model of factors V and Va based on scanning transmission electron microscope images and mass analysis. J Biol Chem. 1990;265:8863–8. [PubMed] [Google Scholar]
- 127.Laue TM, Johnson AE, Esmon CT, Yphantis DA. Structure of bovine blood coagulation factor Va. Determination of the subunit associations, molecular weights, and asymmetries by analytical ultracentrifugation. Biochemistry. 1984;23:1339–48. doi: 10.1021/bi00302a001. [DOI] [PubMed] [Google Scholar]
- 128.Kane WH, Devore-Carter D, Ortel TL. Expression and characterization of recombinant human factor V and a mutant lacking a major portion of the connecting region. Biochemistry. 1990;29:6762–8. doi: 10.1021/bi00481a003. [DOI] [PubMed] [Google Scholar]
- 129.Zhu H, Toso R, Camire RM. Inhibitory sequences within the B-domain stabilize circulating factor V in an inactive state. J Biol Chem. 2007;282:15033–9. doi: 10.1074/jbc.M701315200. [DOI] [PubMed] [Google Scholar]
- 130.Guinto ER, Esmon CT, Mann KG, MacGillivray RTA. The complete cDNA sequence of bovine coagulation factor V. J Biol Chem. 1992;267:2971–8. [PubMed] [Google Scholar]
- 131.Yang TL, Cui J, Rehumtulla A, Yang A, Moussalli M, Kaufman RJ, Ginsburg D. The structure and function of murine factor V and its inactivation by protein C. Blood. 1998;91:4593–9. [PubMed] [Google Scholar]
- 132.Grimm DR, Colter MB, Braunschweig M, Alexander LJ, Neame PJ, Kim HK. Porcine factor V: cDNA cloning, gene mapping, three-dimensional protein modeling of membrane binding sites and comparative anatomy of domains. Cell Mol Life Sci. 2001;58:148–59. doi: 10.1007/PL00000775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jiang Y, Doolittle RF. The evolution of vertebrate blood coagulation as viewed from a comparison of puffer fish and sea squirt genomes. Proc Natl Acad Sci U S A. 2003;100:7527–32. doi: 10.1073/pnas.0932632100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Davidson CJ, Tuddenham EG, McVey JH. Xx450 million years of hemostasis. J Thromb Haemost. 2003;1:1487–94. doi: 10.1046/j.1538-7836.2003.00334.x. [DOI] [PubMed] [Google Scholar]
- 135.Vos HL. Alignment of factor V sequences in twelve mammalian species shows several strongly conserved motifs in the B-domain. J Thromb Haemost. 2005;3:P0041. [Google Scholar]
- 136.Vos HL, van Wijngaarden A. Variation and conservation of the B-domain of factor V. J Thromb Haemost. 2009;7:PP-MO-151. [Google Scholar]
- 137.Rao VS, Swarup S, Kini RM. The nonenzymatic subunit of pseutarin C, a prothrombin activator from eastern brown snake (Pseudonaja textilis) venom, shows structural similarity to mammalian coagulation factor V. Blood. 2003;102:1347–54. doi: 10.1182/blood-2002-12-3839. [DOI] [PubMed] [Google Scholar]
- 138.Welton RE, Burnell JN. Full length nucleotide sequence of a factor V-like subunit of oscutarin from Oxyuranus scutellatus scutellatus (coastal Taipan) Toxicon. 2005;46:328–36. doi: 10.1016/j.toxicon.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 139.St Pierre L, Masci PP, Filippovich I, Sorokina N, Marsh N, Miller DJ, Lavin MF. Comparative analysis of prothrombin activators from the venom of Australian elapids. Mol Biol Evol. 2005;22:1853–64. doi: 10.1093/molbev/msi181. [DOI] [PubMed] [Google Scholar]
- 140.Walker FJ, Owen WG, Esmon CT. Characterization of the prothrombin activator from the venom of Oxyuranus scutellatus scutellatus (taipan venom) Biochemistry. 1980;19:1020–3. doi: 10.1021/bi00546a029. [DOI] [PubMed] [Google Scholar]
- 141.Speijer H, Govers-Riemslag JW, Zwaal RF, Rosing J. Prothrombin activation by an activator from the venom of Oxyuranus scutellatus (Taipan snake) J Biol Chem. 1986;261:13258–67. [PubMed] [Google Scholar]
- 142.Rao VS, Kini RM. Pseutarin C, a prothrombin activator from Pseudonaja textilis venom: its structural and functional similarity to mammalian coagulation factor Xa-Va complex. Thromb Haemost. 2002;88:611–9. [PubMed] [Google Scholar]
- 143.Bos MH, Boltz M, St PL, Masci PP, de JJ, Lavin MF, Camire RM. Venom factor V from the common brown snake escapes hemostatic regulation through procoagulant adaptations. Blood. 2009;114:686–92. doi: 10.1182/blood-2009-02-202663. [DOI] [PMC free article] [PubMed] [Google Scholar]