

Abstract
Structural fluctuations of a protein are essential for the function of native proteins and for protein folding. To understand how the main chain in the native state of a protein fluctuates on different time scales, we examined the rotational correlation functions (RCFs), C(t), of the backbone N-H bonds and of the dihedral angles γ built on four consecutive Cα atoms. Using molecular dynamics simulations of a model α/β protein (VA3) in its native state, we demonstrate that these RCFs decay as stretched exponentials, ln[C(t)] ≈ Dαtα with a constant Dα and an exponent α (0 < α < 0.35) varying with the free-energy profiles (FEPs) along the amino acid sequence. The probability distributions of the fluctuations of the main chain computed at short time scale (1 ps) were identical to those computed at large time scale (1 ns) if the time is rescaled by a factor depending on α < 1. This self-similar property and the nonexponential decays (α ≠ 1) of the RCFs are described by a rotational diffusion equation with a time-dependent diffusion coefficient D(t) = αDαtα-1. The present findings agree with observations of subdiffusion (α < 1) of fluorescent probes within a protein molecule. The subdiffusion of 15N-H bonds did not affect the value of the order parameter S2 extracted from the NMR relaxation data by assuming normal diffusion (α = 1) of 15N-H bonds on a nanosecond time scale. However, we found that the RCF does not converge to S2 on the nanosecond time scale for residues with multiple-minima FEPs.
Keywords: anomalous diffusion, model-free approach, power law, free-energy landscape
An important question for understanding protein dynamics, protein function, and protein folding is how the different segments of the backbone of a protein relax to a new conformation (1–3). On nanosecond and subnanosecond time scales, the dynamics of protein backbones can be probed in great detail experimentally by fluorescence (2) or by NMR relaxation studies (4–6), and theoretically by all-atom molecular dynamics (MD) simulations (7–12).
The orientation of the backbone of a protein around a residue n at any time t can be characterized by a unit vector un(t) representing the orientation of a local probe of the protein dynamics in a frame attached to the molecule. For example, the vector un(t) represents the orientation of a fluorophore linked to residue n in fluorescence anisotropy (2) or the orientation of a bond of the residue n in NMR (4–6). The local reorientation of a segment of the backbone of a protein moving from one conformation to another can be analyzed by recording the internal rotational correlation functions (RCFs) of un (2, 6, 13–15), which measure the average correlation of un with its initial orientation after a time t in the molecular frame. Different experimental techniques measure RCFs of different rank l, Cl(t), corresponding to averages of polynomials of un of different orders l. For example, dielectric spectroscopy measures the RCF C1(t) of a rotating dipole and, in fluorescence anisotropy and NMR, the RCF C2(t) for the fluctuations of a rotating dipole and of a bond, respectively, is obtained (2, 6). In experiments, the internal RCFs of un are extracted by assuming a model for the rotational diffusion of un. In the present work, we used all-atom MD simulations to establish the physical properties of the rotational diffusion of local probes un of the backbone dynamics. We address the following questions: How do the local probes un(t) diffuse on different time scales in a protein? How can a meaningful rotational diffusion constant of un be extracted from its RCF? How does the RCF of un reflect the free-energy profile (FEP) of the main chain of a protein along the sequence of the amino acids?
Two probes, un(t), of the backbone dynamics are considered here for each residue n (Fig. S1). First, un(t) represents the orientation of the main chain measured by a coarse-grained dihedral angle γn (16, 17) formed by the virtual bonds joining four consecutive Cα atoms (n - 1, n, n + 1, and n + 2) along the amino acid sequence; i.e., un(t) = { cos[γn(t)], sin[γn(t)]} with 2 < n < N - 2 and N being the number of residues. The dihedral angles γn (16) are coordinates used to describe (large) conformational changes of proteins (18) and are part of coarse-grained models of proteins (19, 20). The successive orientations of un(t) are represented by rotational diffusion on a circle (Fig. S1). Second, un(t) is a unit vector representing the orientation of the backbone amide (N-H)n bond in a frame attached to the molecule. The successive orientations of un(t) are represented by rotational diffusion on a sphere centered on the N atom (Fig. S1). The rotational diffusion of these local probes was analyzed here by using all-atom MD simulations of a 46-residue α/β model protein VA3 (PDB ID code 1ED0) in its native state (21) (see details in Materials and Methods and SI Text). Protein VA3 was chosen because it is highly homologous to the well-studied protein crambin, but it is soluble in water whereas crambin is not (7). Because of the presence of three disulfide bonds (namely 3-40, 4-32, and 16-26), VA3 remains folded in all MD trajectories, which enables one to apply with confidence the separation of internal motions of the protein from its overall motion as required to compute the internal RCFs of N-H bonds (11) (see SI Materials and Methods).
The internal RCFs at short time scale contain valuable information about the mechanism of rotational diffusion of the local probe. We demonstrated that the internal RCFs are related to the angular mean-square displacement (MSD) of the rotating vector un at short time scale, i.e., for which MSD(t) ≪ 1,
| [1] |
where K1 = 1/2, K2 = 2 for diffusion on a circle (dihedral angle γ) and K1 = 1/2, K2 = 3/2 for a diffusion on a sphere (N-H bond) (see Eqs. S4, S5, S9, and S10 of SI Text Sec. 3). An analogous relation (Eq. 1) is found at long times (t → ∞) if the motion of un is restricted to small angular displacements (4). Eq. 1 is valid for any model of rotational diffusion of the vector un. For example, assuming a free-rotational diffusion on a circle (dihedral angles γ) [on a sphere (N-H bonds)], the RCF is C2(t) = exp(-4Dt) [C2(t) = exp(-6Dt)] with a relaxation time defined by τf = 1/4D (τf = 1/6D) (22). At short times, i.e., t ≪ τf, the exponential can be replaced by its first-order expansion, i.e., C2(t) ≈ 1 - 4Dt [C2(t) ≈ 1 - 6Dt]. Comparing this expansion of the RCF at short times with Eq. 1, we deduce correctly that the MSD for free diffusion on a circle (on a sphere) is MSD(t) = 2Dt [MSD(t) = 4Dt] (22).
Using Eq. 1 on a short time scale (t < 100 ps), we demonstrate that the RCFs of the dihedral angles γ and of the N-H bonds computed from MD decay approximately as power laws of time, which do not correspond to free diffusion of un. Indeed, the MSD of the dihedral angles γ and of the backbone N-H bonds are power laws of time; MSD(t) = 2Dαtα (dihedral angles) (in agreement with ref. 17) and MSD(t) = 4Dαtα (N-H bonds), with a constant Dα (deg2 ps-α) and an exponent α < 0.4 varying along the amino acid sequence (Fig. S2). For each dihedral angle γn [or (N-H)n bond] along the amino acid sequence, the quantity 2Dα (4Dα) is by definition the variance of the distribution of the angular steps of a random walker on a circle (17) (on a sphere) moving each picosecond, whereas the exponent α is proportional to the speed of diffusion produced by these stochastic motions. An exponent α < 1 corresponds to a subdiffusive regime (23–26). The rotational subdiffusion found here agrees with translational subdiffusion found recently in proteins by fluorescence (27) and MD simulations (28). Subdiffusion was also found in the MD simulations of the dynamical structure factor of a protein (29).
We show that the RCFs computed from MD up to 1–2 ns are extremely well fitted by a stretched exponential (SE), ln[C(t)] ≈ Dαtα with exponents (α < 0.4) and constants Dα close to the ones computed from the MSDs (Fig. 1). The parameters [α,Dα] for each residue reflect the variation of the free-energy profiles of the dihedral angles and of the backbone N-H bonds along the sequence of the amino acids. Residues existing in multiple substates have exponents larger than 0.2. We show that the motions of the N-H bonds and of the dihedral angles γ can be described at short time by an (approximate) free diffusion equation with an effective time-dependent diffusion coefficient D(t) = αDαtα-1 and that the fluctuations of the orientation of the N-H bonds and of the main-chain dihedral angles are self-similar at least up to 1 ns (as shown for the N-H bonds in Fig. 2). Finally, we discuss the consequences of the anomalous diffusion (α ≠ 1) of the backbone N-H bonds for the interpretation of the RCFs extracted from NMR data, and we compare the present results to the predictions of the model-free (MF) approach (13–15) (Fig. 3 and SI Text Sec. 9).
Fig. 1.

Typical results for RCFs of dihedral angles γn and of (N-H)n bonds of VA3 computed from MD (thick lines) up to t < 100 ps (A and B) and up to t < 1 ns (C and D). Results are presented in A and C for dihedral angles γ11, γ20, γ35, and γ39 having free-energy profiles (shown schematically in A, Inset) representative of all V(γ) calculated in MD (Fig. S3), and in B and D for corresponding (N-H)n amide bonds of residues n = 11, 20, 35, and 39 with typical free-energy profiles V(θ,φ) (shown schematically in B, Inset). For each dihedral angle γn (A) and each (N-H)n bond (B), the RCFs computed from MD are compared to RCFs computed from Eq. 1 (thin lines) using a power-law MSD (see text) and with a SE (dashed lines) with an exponent αn computed by fitting the RCF up to 50 ps (see text). The SE fit and the RCFs computed from MD are hardly distinguishable. For each dihedral angle γn (C) and each (N-H)n bond (D), the RCFs are compared with a SE with an exponent αn computed by fitting the RCF up to 50 ps (dashed lines) and by fitting the RCF up to 1 ns (dotted lines), respectively. The fit to a SE up to 1 ns is hardly distinguishable from the RCF computed from MD. In D, typical fits of the RCF up to 1 ns using the model-free approach (dot-dashed lines) for a monoexponential and biexponential function f2(t), respectively, are shown for the bonds (N-H)20 and (N-H)39.
Fig. 2.

Typical results for the time evolution of the probability distribution function (PDF) of Δξn computed from the fluctuations of the (N-H)n bonds extracted from MD trajectories of VA3. Results are presented for (N-H)11 and (N-H)35 with a typical harmonic FEP (Fig. 1B, Inset) and multiple-minima FEP, respectively. The superposed curves are the PDFs computed from MD and rescaled by using Eq. 4 at times t = 10, 20, 30, 40, 50, 60, 70, 80, 90, 100, 300, 500, 600, 800, and 1,000 ps choosing h = 1 at t = 100 ps (A) and h = 1 at t = 1 ns (B). The PDFs are rescaled by using an exponent α ≠ 1 calculated from a fit to a SE of 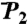 up to 50 ps (A) and up to 1 ns (B). The PDFs cannot be distinguished at the scale of the figure.
up to 50 ps (A) and up to 1 ns (B). The PDFs cannot be distinguished at the scale of the figure.
Fig. 3.

Convergence of RCF(t) to the generalized order parameter S2 of dihedral angles γn (A) and (N-H)n bonds (B) for each residue n along the amino acid sequence [values of 1 - RCF(t) and 1 - S2 are shown]. (A) The 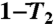 (γn) at t = 1 ns (empty squares) and extrapolated at t = 10 ns (empty diamonds), computed by using a SE with an exponent α and diffusion constant Dα extracted from a fit of
(γn) at t = 1 ns (empty squares) and extrapolated at t = 10 ns (empty diamonds), computed by using a SE with an exponent α and diffusion constant Dα extracted from a fit of 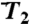 up to 1 ns, are compared to
up to 1 ns, are compared to 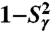 (open and filled circles) computed by using Eq. S54 of SI Text. The filled circles represent the angles γn with multiple-minima FEP. (B)
(open and filled circles) computed by using Eq. S54 of SI Text. The filled circles represent the angles γn with multiple-minima FEP. (B) 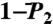 (N-H)n at t = 1 ns (empty squares) and extrapolated to t = 10 ns (empty diamonds), computed by using a SE with an exponent α and diffusion constant Dα extracted from a fit of
(N-H)n at t = 1 ns (empty squares) and extrapolated to t = 10 ns (empty diamonds), computed by using a SE with an exponent α and diffusion constant Dα extracted from a fit of 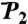 up to 1 ns, are compared to
up to 1 ns, are compared to 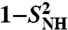 (open and filled circles) computed by using Eq. S67 of SI Text. The filled circles represent the (N-H)n amide bonds with multiple-minima 2D-free-energy landscapes. The values of
(open and filled circles) computed by using Eq. S67 of SI Text. The filled circles represent the (N-H)n amide bonds with multiple-minima 2D-free-energy landscapes. The values of 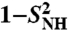 computed by using the MF approach (see text) and a monoexponential or a biexponential function f2(t) (empty triangles) are in excellent agreement with the values of
computed by using the MF approach (see text) and a monoexponential or a biexponential function f2(t) (empty triangles) are in excellent agreement with the values of 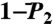 at 1 ns computed using a SE model. Thick lines β1, β2 and α1, α2 are the locations of the two β-sheets and the two α-helices, respectively, of VA3. Typical FEP of angles γn (A) and (N-H)n (B) bonds computed from MD are shown in Insets.
at 1 ns computed using a SE model. Thick lines β1, β2 and α1, α2 are the locations of the two β-sheets and the two α-helices, respectively, of VA3. Typical FEP of angles γn (A) and (N-H)n (B) bonds computed from MD are shown in Insets.
Results and Discussion
Computation of the RCFs for Diffusion on a Circle and on a Sphere.
The persistence of the orientation of un(t) is measured by computing the RCF Cl,n(t) of rank l = 1 and l = 2. For circular geometry, they are defined by
| [2] |
where Tl is a Tchebychev polynomial, namely T1(x) = x and T2(x) = 2x2 - 1 (22). In Eq. 2, xn ≡ un(t′)•un(t′ + t) is the cosine of the angular displacement Δγn of un between t′ and t′ + t (Fig. S1), and Tl is averaged over all possible initial orientations (at all times t′).
For spherical geometry, the RCFs of rank l = 1 and l = 2 are computed (22) by using
| [3] |
where Pl is a Legendre polynomial, namely P1(x) = x and P2(x) = 3x2/2 - 1/2 (22). In Eq. 3, xn ≡ un(t′)•un(t′ + t) is the cosine of the angular displacement Δξn of un between t′ and t′ + t (Fig. S1), and Pl is averaged over all possible initial orientations (at all times t′).
The RCFs 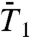 and
and 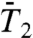 of all 43 γn angles and
of all 43 γn angles and 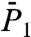 and
and 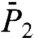 of the 39 amide (N-H)n bonds (excluding the nonexisting N-H bonds of PRO5, PRO22, PRO24, and PRO41) (a total of 820 RCFs) were computed by applying Eqs. 2 and 3, respectively, to five 80-ns MD trajectories of VA3 at 300 K in explicit solvent (see Materials and Methods). The 80-ns MD trajectories provide enough statistics to compute the RCFs accurately up to about 1–2 ns (30) (at large times, spurious oscillations in the RCFs of some residues are observed. See SI Text Sec. 1 and Fig. S4).
of the 39 amide (N-H)n bonds (excluding the nonexisting N-H bonds of PRO5, PRO22, PRO24, and PRO41) (a total of 820 RCFs) were computed by applying Eqs. 2 and 3, respectively, to five 80-ns MD trajectories of VA3 at 300 K in explicit solvent (see Materials and Methods). The 80-ns MD trajectories provide enough statistics to compute the RCFs accurately up to about 1–2 ns (30) (at large times, spurious oscillations in the RCFs of some residues are observed. See SI Text Sec. 1 and Fig. S4).
Relation Between RCF and MSD at Short Time (up to 100 ps).
The MSDs of the dihedral angles γ and of the N-H bonds were computed from the five MD trajectories by using Eqs. S6 and S11, respectively. The MSD of γ was shown previously (17) to be 2Dαtα (with α < 1) by using all-atom MD. Therefore, the MSDs were fitted up to 50 ps by using MSD(t) = 2Dαtα for the dihedral angles γ (diffusion on a circle) and MSD(t) = 4Dαtα for the N-H bonds (diffusion on a sphere). All MSDs (a total of 410) were represented perfectly well by these power laws with α < 1 up to at least 100 ps (Fig. S2). We conclude that the diffusion of γ dihedral angles (17) and the diffusion of the N-H bonds are anomalous. Consequently, the RCFs should decay approximately as power laws at short time. The RCFs computed from MD were compared up to 100 ps to those computed by using the power-law MSD in Eq. 1; i.e., 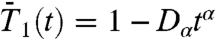 (Fig. S5a),
(Fig. S5a), 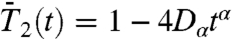 (Fig. 1A),
(Fig. 1A), 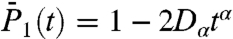 (Fig. S6a), and
(Fig. S6a), and 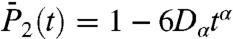 (Fig. 1B). The RCFs computed from the MSD by using Eq. 1 are good approximations for most of the residues at least up to 100 ps with a maximum deviation of about 15% (as shown for
(Fig. 1B). The RCFs computed from the MSD by using Eq. 1 are good approximations for most of the residues at least up to 100 ps with a maximum deviation of about 15% (as shown for 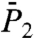 of N-H39 in Fig. 1B).
of N-H39 in Fig. 1B).
RCFs Decay as Stretched Exponentials.
The RCFs computed from MD were fitted to stretched exponentials, namely 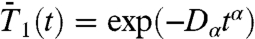 ,
, 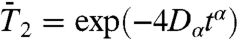 ,
, 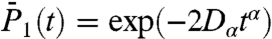 , and
, and 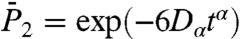 , on two time scales: up to 50 ps [Fig. S5a (
, on two time scales: up to 50 ps [Fig. S5a (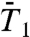 ), Fig. 1A (
), Fig. 1A (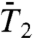 ), Fig. S6a (
), Fig. S6a (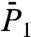 ), Fig. 1B (
), Fig. 1B (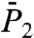 )], and up to 1 ns [Fig. 1C (
)], and up to 1 ns [Fig. 1C (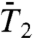 ) and Fig. 1D (
) and Fig. 1D (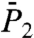 )]. These SEs are hardly discernible from the RCFs computed from MD to which they were fitted.
)]. These SEs are hardly discernible from the RCFs computed from MD to which they were fitted.
The parameters Dα and α computed along the sequence of the amino acids from the SEs fitted to the RCFs up to 50 ps are very close to those extracted independently from the power laws fitted to the MSD up to 100 ps except for the exponents α of a few residues in loops (Figs. S5 b and c and S6 b and c). Therefore, we could, in principle, extract the constant Dα and the exponent α of the MSD of each N-H bond by fitting its RCF measured at short time (up to 50 ps) by a stretched exponential, keeping in mind that the exponent extracted from the RCF of an N-H bond within a loop might be slightly lower than the exponent of the MSD of the bond (Fig. S6).
The SEs fitted to the RCF up to 50 ps (Fig. 1 A and B) nicely reproduce the typical behavior of the RCFs up to 1 ns as shown in Fig. 1 C and D, but not their exact values (similar results are found for 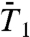 and
and 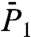 ). For most of the residues, the values of Dα computed from the RCFs up to 50 ps and up to 1 ns are nearly identical, and the exponents α computed from the RCFs up to 50 ps are larger than those calculated up to 1 ns (as illustrated in Fig. 1). The profiles of the variations of α and Dα along the sequence of the amino acids computed from the RCFs up to 50 ps and up to 1 ns are similar (see SI Text Sec. 2 and Figs. S5 and S6).
). For most of the residues, the values of Dα computed from the RCFs up to 50 ps and up to 1 ns are nearly identical, and the exponents α computed from the RCFs up to 50 ps are larger than those calculated up to 1 ns (as illustrated in Fig. 1). The profiles of the variations of α and Dα along the sequence of the amino acids computed from the RCFs up to 50 ps and up to 1 ns are similar (see SI Text Sec. 2 and Figs. S5 and S6).
Variation of α and Dα Along the Amino Acid Sequence, FEP, and RCF.
The values of [α,Dα] of the RCF of a dihedral angle γ are governed by the shape of its effective 1D FEP, V(γ), as shown previously (17). The FEP was computed by using V(γ) = -kT ln[P(γ)], where k and T = 300 K are the Boltzmann constant and temperature, respectively, and where P(γ) is the PDF of each dihedral angle γ computed over the whole MD trajectory (17). The FEP, averaged over five trajectories, is represented along the primary sequence in Fig. S3. The FEPs are similar between the different trajectories, as shown by the comparison between the average FEP and the one computed from run 1 in Fig. S3. Typical FEPs, representative of all V(γ), are reproduced schematically in Fig. 1A, Inset: V(γ11) is a stiff harmonic FEP, V(γ20) is a single-minimum soft FEP, V(γ35) is a triple-well FEP, and V(γ39) is a double-well FEP.
The constant Dα has a tendency toward smaller values in stiffer (harmonic) FEPs [as V(γ11)] (Dα ≈ 10 deg2 ps-α) and large values (Dα ≈ 50–110 deg2 ps-α) in wide FEPs [as V(γ20)] and in multiple-minima FEPs [as V(γ35) and V(γ39)] (Figs. S3 and S5b). For each residue, the value of 2Dα is very close to the variance of the distribution of the displacements Δγ computed at 1 ps. All residues moving on multiple-minima FEPs [as V(γ35) and V(γ39) in Fig. 1A] have large exponents α (fast diffusion) (α≥0.2): They are generally found in loops but some (γ17, γ26, and γ34) (see Fig. S3) are in secondary structures (Fig. S5c). Visual inspection of the structures produced by MD shows that the two minima of V(γ26) are two possible orientations of the disulfide bond 16-26 corresponding to the formation or not of a hydrogen bond between the PRO24 and LYS28 both in the shortest helix of VA3. Rupture of the hydrogen bond implies a motion of ARG17 and of the two other disulfide bonds 4-32 and 3-40 and explains the multiple minima observed. This might be an artifact of the force field or an actual effect. Missing or nonnative hydrogen bonds in current force fields may be responsible for disagreements between the long-time limit of the RCFs computed by MD and the relaxation NMR data as illustrated in ref. 11. However, MD using current force fields (including the one used here) reproduce reasonably well most of the NMR data of proteins in the native state if electrostatic forces are fully taken into account (see Materials and Methods) and if the length of the MD trajectory is not too long (say up to 200 ns) (31).
As for the dihedral angles, the values of [α,Dα] of the N-H bonds are governed by the shape of the FEP V(θ,φ), where (θ,φ) are the polar coordinates of the N-H vector in a frame attached to the molecule (Fig. S1). The FEP V(θ,φ) was computed over the whole MD trajectory by using V(θ,φ) = -kT ln[P(θ,φ)], where P(θ,φ) is the 2D PDF of the vector u in the direction of the N-H bond (Fig. S1). Typical FEPs V(θ,φ) are shown in Fig. 1B, Inset. As for the dihedral angles γn, the constant Dα has a tendency toward smaller values in stiffer (harmonic) FEPs [as V(θ11,φ11)] (Dα ≈ 50 deg2 ps-α) and large values (Dα ≈ 90–100 deg2 ps-α) in wide FEPs [as V(θ20,φ20)] and in multiple-minima FEPs [as V(θ39,φ39)], which are located in loops except V(θ26,φ26) with two minima [as V(γ26)] (see Fig. 3B, Inset) and in a helix. The largest exponents α are found for the N-H bonds within the loop between residues 37 and 40 (Fig. S7a), which possess both multiminima V(θ,φ) and V(γ) (Fig. S3) (see Fig. 1B, Inset, and Fig. 3B, Inset), and the lowest exponents α are found in helix (Fig. S7a).
The constant Dα of the (N-H)n bond is generally larger than the one of the corresponding dihedral angle γn, which means that the amplitude of the displacement of the bond is larger than the one of the four virtual bonds Cα-Cα defining the dihedral angle, as expected (Fig. S7b). The exponents α of the N-H bonds at short time scale are in general lower than the ones of the dihedral angles because the RCFs 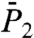 in general converge more rapidly to a constant stationary value than the RCFs
in general converge more rapidly to a constant stationary value than the RCFs 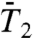 . The profiles of [α,Dα] extracted from
. The profiles of [α,Dα] extracted from 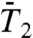 and
and 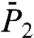 are thus only qualitatively similar (Fig. S7). It is worth noting, for example, that (N-H)9 and (N-H)30, respectively, at the N and C terminals of a helix have large Dα (they move in soft FEP), whereas the constant Dα of γ9 and γ30 located in a helix pertains to the smallest Dα (they move in a stiff FEP shown in Fig. S3) (Fig. S7a).
are thus only qualitatively similar (Fig. S7). It is worth noting, for example, that (N-H)9 and (N-H)30, respectively, at the N and C terminals of a helix have large Dα (they move in soft FEP), whereas the constant Dα of γ9 and γ30 located in a helix pertains to the smallest Dα (they move in a stiff FEP shown in Fig. S3) (Fig. S7a).
Three different typical types of behaviors of the RCF 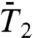 and
and 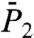 at long time were observed, as shown in Fig. 1 C and D, respectively. Similar types were found in MD simulations of
at long time were observed, as shown in Fig. 1 C and D, respectively. Similar types were found in MD simulations of 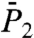 of backbone N-H bonds of other proteins (11, 12). We found that they correspond to three different types of FEPs: for a single-minimum stiff FEP [γ11 and (N-H)11] and soft FEP [γ20 and (N-H)20 and (N-H)35],
of backbone N-H bonds of other proteins (11, 12). We found that they correspond to three different types of FEPs: for a single-minimum stiff FEP [γ11 and (N-H)11] and soft FEP [γ20 and (N-H)20 and (N-H)35], 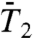 and
and 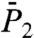 exhibit a very fast drop at short times and converge to a constant value on the 1-ns time scale close to 1.0 for a stiff harmonic FEP and around 0.8–0.9 for a soft FEP. For multiple-minima FEP [γ35, γ39 and (N-H)39],
exhibit a very fast drop at short times and converge to a constant value on the 1-ns time scale close to 1.0 for a stiff harmonic FEP and around 0.8–0.9 for a soft FEP. For multiple-minima FEP [γ35, γ39 and (N-H)39], 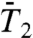 and
and 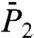 have a strong decay with no convergence to a plateau up to 1 ns (Fig. 1 C and D and Fig. S4).
have a strong decay with no convergence to a plateau up to 1 ns (Fig. 1 C and D and Fig. S4).
A Simple Model of the Nonexponential Decay of the RCF.
As demonstrated in SI Text Sec. 4, the stretched exponentials reproducing the RCF computed in MD (see Fig. 1), namely 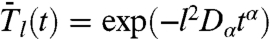 for diffusion on a circle and
for diffusion on a circle and 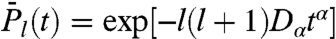 for diffusion on a sphere, are RCFs of rank l of a free-diffusion equation with an effective time-dependent diffusion coefficient D(t) ≡ αDαtα-1 on both a circle and a sphere. The coefficient D(t) is the instantaneous diffusion constant at time t (26), i.e., nD(t) = ∂MSD(t)/∂t [n = 2 (4) for diffusion on a circle (sphere)]. A power-law time dependence of D(t) means that the velocity–velocity correlation function Cv(t) (32) decays slowly approximately as Cv(t) ≈ α2(α - 1)Dαtα-2 (see details in SI Text Sec. 7).
for diffusion on a sphere, are RCFs of rank l of a free-diffusion equation with an effective time-dependent diffusion coefficient D(t) ≡ αDαtα-1 on both a circle and a sphere. The coefficient D(t) is the instantaneous diffusion constant at time t (26), i.e., nD(t) = ∂MSD(t)/∂t [n = 2 (4) for diffusion on a circle (sphere)]. A power-law time dependence of D(t) means that the velocity–velocity correlation function Cv(t) (32) decays slowly approximately as Cv(t) ≈ α2(α - 1)Dαtα-2 (see details in SI Text Sec. 7).
The quantity F(Δγ; t)dγ is the probability that the vector u, representing a dihedral angle γ (Fig. S1), is rotated by an angle Δγ after a time t > 0 [with F(Δγ; 0) = δ(Δγ)]. For a free-diffusion equation on a circle (Eq. S15 in SI Text) with diffusion coefficient D(t), we find that the PDF F(Δγ; t) is a series of Tchebychev polynomials (see Eq. S17 in SI Text). The product F(Δξ; t)dξ is the probability that the vector u, representing an N-H bond (Fig. S1), is rotated by an angle Δξ after a time t > 0 [with F(Δξ; 0) = δ(Δξ)]. For a free-diffusion equation on a sphere (Eq. S25 in SI Text) with diffusion coefficient D(t), we show that F(Δξ; t) is a series of Legendre polynomials (see Eqs. S24 and S26 in SI Text). The analytical solutions F(Δγ; t) (Eq. S17 in SI Text] and F(Δξ; t) (Eqs. S24 and S26 in SI Text) agreed quite well with the PDF computed by MD, as shown in Fig. S8 a–f for different residues and typical values of [α,Dα]. This demonstrates that the internal motions of a protein can be interpreted (to a very good approximation) as free random walks with a diffusion coefficient D(t) ≡ αDαtα-1.
Self-Similarity of the PDF.
Replacing the time-independent diffusion constant by D(t) ≡ αDαtα-1 in the diffusion equations preserves the self-similarity property of the Brownian motion (26): The PDF solutions of the rotational diffusion equation on a circle (Eq. S17 in SI Text) and on a sphere (Eqs. S24 and S26 in SI Text) are indeed self-similar under the scaling transformations t → ht and ΔΦ → ΔΦ/hα/2; i.e.,
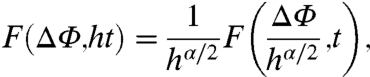 |
[4] |
where ΔΦ = Δγ (with F given by Eq. S17 in SI Text) or ΔΦ = Δξ (with F given by Eqs. S24 and S26 in SI Text). The PDFs computed from MD indeed obey the scaling property Eq. 4 very well, as shown in Fig. 2 for the N-H bond fluctuations and in Fig. S8 g–j for the dihedral angles γ fluctuations. In addition, Eq. 4 can be derived analytically from the PDF of the dihedral angles (see SI Text Sec. 5). Consequently, to a very good approximation from 1 ps to 1,000 ps studied here, the stochastic motions γ(t) and ξ(t) resemble each other in a statistical sense: The fluctuations of each of these angles at short time scales are similar to their fluctuations at large time scales. For example, choosing t = 10 ps and h = 100 in Eq. 4 provides the PDF at 1 ns (left-hand side of Eq. 4) from the scaled PDF observed at 10 ps (right-hand side of Eq. 4).
The self-similarity of F(Δξn,t) has been tested for all 195 PDFs computed for the 39 residues and 5 MD trajectories. Typical results, representative of all the data, are shown in Fig. 2 for residues 11 and 35. The PDFs were scaled according to Eq. 4 with ΔΦ = Δξ and t = 100 ps (Fig. 2A) and t = 1 ns (Fig. 2B) (see caption). There is a very good superposition of all the rescaled PDFs in all the panels in Fig. 2. Similar results were found for F(Δγn,t) as shown in SI Text Sec. 6.
Limits of 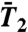 and
and 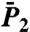 as t → ∞ and the FEP.
as t → ∞ and the FEP.
In NMR relaxation studies, the RCF C2(t) of each backbone 15N-H amide bond can be recorded by measuring the 15N spin relaxation of labeled proteins (4–6). The RCF is not measured in the time domain but is recorded as its Fourier transform in the frequency domain ω, instead, as the so-called spectral density JNH(ω) (4–6, 33). Because JNH(ω) is usually recorded at a few frequencies (4–6), the interpretation of the spectral density in terms of physically meaningful parameters requires a model for the rotational diffusion of the local probe un (15). The internal RCFs are extracted from the RCFs measured in the laboratory frame by assuming that the relaxation time of the overall tumbling of the whole protein [typically larger than 2 ns (15)] is much larger than the relaxation time of the motions of the N-H bonds (15). Under this assumption, the RCF is generally represented by the MF approach (13–15); i.e., 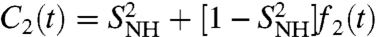 , where f2(t) describes the decay of the RCF toward its value at large times with f2(0) = 1 and f2(t → ∞) = 0 (1, 5, 11, 12). The limit
, where f2(t) describes the decay of the RCF toward its value at large times with f2(0) = 1 and f2(t → ∞) = 0 (1, 5, 11, 12). The limit 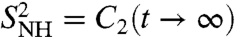 , named the generalized order parameter (1, 5, 6, 11, 12), is an important parameter used to characterize protein dynamics along the sequence of the amino acids in NMR. Because it represents the equilibrium value reached by the RCFs, it is related to the FEPs (13, 34). The generalized order parameters
, named the generalized order parameter (1, 5, 6, 11, 12), is an important parameter used to characterize protein dynamics along the sequence of the amino acids in NMR. Because it represents the equilibrium value reached by the RCFs, it is related to the FEPs (13, 34). The generalized order parameters 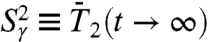 and
and 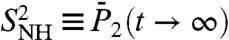 were computed exactly from the FEP V(γ) and V(θ,φ), respectively (see Eqs. S54 and S67, SI Text Sec. 8). In order to evaluate the time necessary for the RCFs of each dihedral angle γn and each (N-H)n bond to converge toward its equilibrium values at large times, in Fig. 3 we compared the values of
were computed exactly from the FEP V(γ) and V(θ,φ), respectively (see Eqs. S54 and S67, SI Text Sec. 8). In order to evaluate the time necessary for the RCFs of each dihedral angle γn and each (N-H)n bond to converge toward its equilibrium values at large times, in Fig. 3 we compared the values of 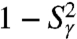 (Fig. 3A) and
(Fig. 3A) and 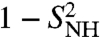 (Fig. 3B), computed from the FEP (open circles for a single-minimum and filled circles for multiple-minima FEPs) to the values of the
(Fig. 3B), computed from the FEP (open circles for a single-minimum and filled circles for multiple-minima FEPs) to the values of the 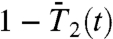 and
and 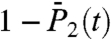 computed by MD at t = 1 ns (time chosen arbitrarily) (open squares).
computed by MD at t = 1 ns (time chosen arbitrarily) (open squares).
For most of the residues (mainly in helix), the RCFs 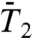 at 1 ns have already converged to their limit values. However, the RCFs of the following dihedral angles γn did not clearly converge on the 1-ns time scale n = 2, 18, 19, 25, 26, 30, 32–41, and 44. The largest deviations from
at 1 ns have already converged to their limit values. However, the RCFs of the following dihedral angles γn did not clearly converge on the 1-ns time scale n = 2, 18, 19, 25, 26, 30, 32–41, and 44. The largest deviations from 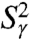 at 1 ns are observed for γ2, γ26, γ32-γ41, and γ44. All these dihedral angles have free-energy profiles with marked multiple minima (Fig. S3 and Fig. 3A, Inset) except γ32, γ36 (anharmonic FEP with no actual metastable minimum) and γ40 and γ44 (wide potentials). Approximation of the RCFs of the dihedral angles at larger time (t = 10 ns, chosen arbitrarily) (open diamonds in Fig. 3) using a stretched exponential fitted to the RCFs up to 1 ns (Fig. 1C) shows that only
at 1 ns are observed for γ2, γ26, γ32-γ41, and γ44. All these dihedral angles have free-energy profiles with marked multiple minima (Fig. S3 and Fig. 3A, Inset) except γ32, γ36 (anharmonic FEP with no actual metastable minimum) and γ40 and γ44 (wide potentials). Approximation of the RCFs of the dihedral angles at larger time (t = 10 ns, chosen arbitrarily) (open diamonds in Fig. 3) using a stretched exponential fitted to the RCFs up to 1 ns (Fig. 1C) shows that only 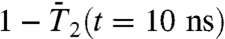 for γ36, γ38 converges to
for γ36, γ38 converges to 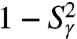 on this longer time scale. As for γn, the RCFs of a series of (N-H)n bonds, n = 2, 3, 4, 21–29, 34–45, did not converge to
on this longer time scale. As for γn, the RCFs of a series of (N-H)n bonds, n = 2, 3, 4, 21–29, 34–45, did not converge to 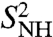 on the 1-ns time scale. The FEPs of the (N-H)2, (N-H)26, (N-H)30, and (N-H)37-(N-H)40 have clear multiple minima as shown, for example, for (N-H)26 and (N-H)37 in Fig. 3B, Inset. Only the RCF of (N-H)44 converges to
on the 1-ns time scale. The FEPs of the (N-H)2, (N-H)26, (N-H)30, and (N-H)37-(N-H)40 have clear multiple minima as shown, for example, for (N-H)26 and (N-H)37 in Fig. 3B, Inset. Only the RCF of (N-H)44 converges to 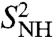 on a 10-ns time scale as shown by extrapolating the RCFs of the N-H bonds up to 10 ns using a SE fitted to the RCFs up to 1 ns (Fig. 1D).
on a 10-ns time scale as shown by extrapolating the RCFs of the N-H bonds up to 10 ns using a SE fitted to the RCFs up to 1 ns (Fig. 1D).
The two types of behavior found in Fig. 3, the convergence of the RCFs 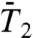 and
and 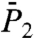 toward
toward 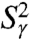 and
and 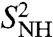 on a 1-ns time scale for residues in a single-minimum stiff FEP (located in helix; see also ref. 34), and a slow convergence toward these limits for residues in multiple-minima and soft anharmonic FEPs (located mainly in loops), correspond very well to the two types of behavior observed at different time scales in helix and loops by NMR in protein G (35). We emphasize here that the convergence of the RCFs toward the order parameters depends on the anharmonicity of the FEP and on the existence of multiple substates (see Fig. 3 Insets) rather than by the type of secondary structure. The residues with multiple-minima free-energy profiles are mainly in loops but not necessarily, as for CYS26 of VA3 (Fig. 3) located in a helix.
on a 1-ns time scale for residues in a single-minimum stiff FEP (located in helix; see also ref. 34), and a slow convergence toward these limits for residues in multiple-minima and soft anharmonic FEPs (located mainly in loops), correspond very well to the two types of behavior observed at different time scales in helix and loops by NMR in protein G (35). We emphasize here that the convergence of the RCFs toward the order parameters depends on the anharmonicity of the FEP and on the existence of multiple substates (see Fig. 3 Insets) rather than by the type of secondary structure. The residues with multiple-minima free-energy profiles are mainly in loops but not necessarily, as for CYS26 of VA3 (Fig. 3) located in a helix.
Consequences of Anomalous Diffusion for the Interpretation of NMR Experimental Data.
Deviations of the time evolution of the RCFs from a simple exponential decay (free diffusion) were observed for a few residues along the sequence of proteins (14) (9 out 155 in Ribonuclease A in ref. 1). They were interpreted as due to slow conformational fluctuations, i.e., jumps of the N-H bonds between different orientations (14), which correlate with the bond orientation on a slower time scale τs. For these residues, the RCF is represented as a sum of a fast and a slow exponential decay; i.e., f2(t) = af exp(-t/τf) + (1 - af) exp(-t/τs) with af a parameter weighting the fast motions in the MF approach (see SI Text Sec. 9). The MF approach with a monoexponential decay (af = 1) (Eq. S70 in SI Text) and biexponential decay (af ≠ 1) (Eq. S71 in SI Text) were fitted to the RCFs computed from MD up to 1 ns (see SI Text Sec. 9 and Table S1). The relaxation time τf (14) typically varies between about 30 ps to about 300–500 ps in the monoexponential model and τf < 12 ps in the biexponential model with 170 < τs < 580 ps (see Table S1). The generalized order parameter 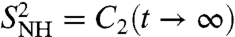 computed by fitting the MF approach to the RCFs computed by MD up to 1 ns (SI Text Sec. 9 and Table S1) are compared to those computed using a SE in Fig. 3B. There is very good agreement between the two approaches for all the amide bonds with small differences for the residues in loop 37-40, which have multiple-minima FEPs. However, the values of the parameters
computed by fitting the MF approach to the RCFs computed by MD up to 1 ns (SI Text Sec. 9 and Table S1) are compared to those computed using a SE in Fig. 3B. There is very good agreement between the two approaches for all the amide bonds with small differences for the residues in loop 37-40, which have multiple-minima FEPs. However, the values of the parameters 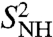 in the MF approach are very close to the values of the RCFs at 1 ns (compare the values shown in Fig. 3 with the values in Fig. 1D) and, therefore, the MF approach fails to predict the nonconvergence of the RCFs to
in the MF approach are very close to the values of the RCFs at 1 ns (compare the values shown in Fig. 3 with the values in Fig. 1D) and, therefore, the MF approach fails to predict the nonconvergence of the RCFs to 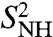 observed for bonds having strong anharmonic FEP (filled circles in Fig. 3).
observed for bonds having strong anharmonic FEP (filled circles in Fig. 3).
A (multi)exponential model is not easily interpreted. Even if a sum of exponentials could fit the RCF as well as a stretched exponential, a (multi)exponential model cannot explain anomalous diffusion. Because an exponential decay of an RCF corresponds to a diffusion equation with a time-independent diffusion constant, the probability distribution of the angular displacement solution of this diffusion equation is predicted to scale linearly with time (α = 1) according to Eq. 4, which is not the case as can be seen in Fig. 2. Therefore the (multi)exponential model, which assumes normal diffusion, cannot describe the SE decay (anomalous diffusion) found in MD at short time scale as can be seen in Fig. 1D. At short times (t ≪ τs), the RCFs of the (multi)exponential MF approach decay linearly as predicted by Eq. 1 with an effective diffusion constant DMF proportional to (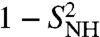 ) (Eq. S74 in SI) much lower than Dα (as shown by the comparison between Table S1 and Fig. S6b). It is, however, interesting to note that the quantity (
) (Eq. S74 in SI) much lower than Dα (as shown by the comparison between Table S1 and Fig. S6b). It is, however, interesting to note that the quantity (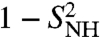 ) in the (multi)exponential MF approach can be interpreted as a diffusion constant that varies along the sequence of the amino acids as Dα (as shown by the comparison of Fig. 3B and Fig. S6b).
) in the (multi)exponential MF approach can be interpreted as a diffusion constant that varies along the sequence of the amino acids as Dα (as shown by the comparison of Fig. 3B and Fig. S6b).
The diffusion equation with a time-dependent coefficient D(t) is a compact (approximate) model with only two parameters: Dα, which quantifies the width of the probability distribution of the angular displacements at 1 ps, and α, which quantifies the self-similarity of this distribution. It remains a challenge to extract from NMR relaxation data these parameters characterizing the angular MSD of the N-H bonds. The values [α,Dα] could be extracted by fitting the RCFs measured on a 100-ps time scale to a SE (Fig. 1 and Fig. S6 b and c) or by using a MF approach with a SE decay (Eq. S75 and Fig. S6 in SI Text). This objective will be pursued in the near future and will require accurate values of JNH(ω) for a model protein.
Conclusions
By using MD, we demonstrated that the RCFs of two local probes of the backbone dynamics (coarse-grained dihedral angles and backbone N-H bonds) decay as stretched exponentials at least up to 1 ns (Fig. 1). They are characterized by a constant Dα and an exponent α varying along the amino acid sequence of the protein according to the anharmonicity of the FEP. Comparison with the MF approach used in NMR shows that the constant Dα of the local probes varies along the amino acid sequence like (1 - S2). The exponent α measures the “speed” of the exploration of the conformational space and was always found to be smaller than 1 (subdiffusion). Anomalous subdiffusion of the local probes was described by a rotational diffusion equation with a power-law diffusion coefficient that accounts for the self-similarity of the PDF of the angular displacements of the probes (Fig. 2). The larger exponents α of the local probes were found for residues moving on multiple-minima free-energy profiles for which the RCF converges slowly to the generalized order parameter S2 (Fig. 3). Measurement of the parameters [α,Dα] for backbone 15N-H bonds at short time scale and of the P2(t) on different time scales should enable one to detect multiple substates of the segments of the backbone of a protein. Such conformational substates may be related to protein function and protein folding, which involve both small reorganization of the backbone segments and large conformational changes occurring on different time scales.
Materials and Methods
Five MD simulations of VA3, each of a duration of 80 ns, were carried out at 300 K in explicit water (Simple Point Charge force field) with the GROMACS package (36) and the all-atom Optimized Potentials for Liquid Simulations force field (37) with no cutoff of the electrostatic interactions as this improves the accuracy of the force field compared to NMR data (31). The structure of VA3 was taken from the NMR model 1 of ref. 21. Details of all MD simulations are given in SI Materials and Methods. The coordinates of VA3 were saved every 1 ps. Consequently, the different initial times t′ in Eqs. 2 and 3 were sampled every picosecond. For each t′, the vector un(t′) = { cos[γn(t′)], sin[γn(t′)]} was computed for each dihedral angle γn. For each time, the direction un(t′) of each (N-H)n bond (Fig. S1) was computed after the overall translation and rotation of the protein had been eliminated by aligning the structure at time t′ on the initial structure. The averages in Eqs. 2 and 3 were calculated for the whole trajectory at different times t. The fits of the stretched exponentials to the RCFs computed from MD were made with a root-mean-square algorithm, and the PDFs shown in Fig. 2 and Fig. S8 were computed from MD by sampling Δγ and Δξ with a resolution of 1 °.
Supplementary Material
Acknowledgments.
Y.C. thanks the Centre National de la Recherche Scientifique (CNRS) and the Conseil Regional de Bourgogne for a PhD fellowship. P.S. thanks the CNRS for sabbatical funding. This research was supported by grants from the National Institutes of Health (GM-14312) and the National Science Foundation (MCB05-41633) and was conducted by using the resources of the 736-processor Beowulf cluster at the Baker Laboratory of Chemistry and Chemical Biology, Cornell University, and the resources of the Centre de Calcul de l’Université de Bourgogne.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1013674107/-/DCSupplemental.
References
- 1.Mandel AM, Akke M, Palmer AG. Backbone dynamics of Escherichia coli ribonuclease HI: Correlations with structure and function in an active enzyme. J Mol Biol. 1995;246:144–163. doi: 10.1006/jmbi.1994.0073. [DOI] [PubMed] [Google Scholar]
- 2.Pletneva EV, Gray HB, Winkler JR. Many faces of the unfolded state: Conformational heterogeneity in denatured yeast cytochrome c. J Mol Biol. 2005;345:855–867. doi: 10.1016/j.jmb.2004.10.085. [DOI] [PubMed] [Google Scholar]
- 3.Korzhnev DM, Kay LE. Probing invisible, low-populated states of protein molecules by relaxation dispersion NMR spectroscopy: An application to protein folding. Acc Chem Res. 2008;41:442–451. doi: 10.1021/ar700189y. [DOI] [PubMed] [Google Scholar]
- 4.Palmer AG, Williams J, McDermott A. Nuclear magnetic resonance studies of biopolymer dynamics. J Phys Chem. 1996;100:13293–13310. [Google Scholar]
- 5.Kay LE. Protein dynamics from NMR. Nat Struct Biol. 1998;5:S512–S517. doi: 10.1038/755. [DOI] [PubMed] [Google Scholar]
- 6.Jarymowycz VA, Stone MJ. Fast time scale dynamics of protein backbones: NMR relaxation methods, applications, and functional consequences. Chem Rev. 2006;106:1624–1671. doi: 10.1021/cr040421p. [DOI] [PubMed] [Google Scholar]
- 7.Bonvin AMJJ, Rullmann JAC, Lamerichs RMJN, Boelens R, Kaptein R. “Ensemble” iterative relaxation matrix approach: A new NMR refinement protocol applied to the solution structure of crambin. Proteins. 1993;15:385–400. doi: 10.1002/prot.340150406. [DOI] [PubMed] [Google Scholar]
- 8.Prompers JJ, Brüschweiler R. General framework for studying the dynamics of folded and nonfolded proteins by NMR relaxation spectroscopy and MD simulation. J Am Chem Soc. 2002;124:4522–4534. doi: 10.1021/ja012750u. [DOI] [PubMed] [Google Scholar]
- 9.Nederveen AJ, Bonvin AMJJ. NMR relaxation and internal dynamics of ubiquitin from a 0.2 μs MD simulation. J Chem Theory Comput. 2005;1:363–374. doi: 10.1021/ct0498829. [DOI] [PubMed] [Google Scholar]
- 10.Showalter SA, Brüschweiler R. Validation of molecular dynamics simulations of biomolecules using NMR spin relaxation as benchmarks: Application to the AMBER99SB force field. J Chem Theory Comput. 2007;3:961–975. doi: 10.1021/ct7000045. [DOI] [PubMed] [Google Scholar]
- 11.Trbovic N, Kim B, Friesner RA, Palmer AG. Structural analysis of protein dynamics by MD simulations and NMR spin-relaxation. Proteins. 2008;71:684–694. doi: 10.1002/prot.21750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindorff-Larsen K, Best RB, DePristo MA, Dobson CM, Vendruscolo M. Simultaneous determination of protein structure and dynamics. Nature. 2005;433:128–132. doi: 10.1038/nature03199. [DOI] [PubMed] [Google Scholar]
- 13.Lipari G, Szabo A. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 14.Clore GM, et al. Deviations from the simple two-parameter model-free approach to the interpretation of Nitrogen-15 nuclear magnetic relaxation of proteins. J Am Chem Soc. 1990;112:4989–4991. [Google Scholar]
- 15.Halle B. The physical basis of model-free analysis of NMR relaxation data from proteins and complex fluids. J Chem Phys. 2009;131:224507. doi: 10.1063/1.3269991. [DOI] [PubMed] [Google Scholar]
- 16.Nishikawa K, Momany FA, Scheraga HA. Low-energy structures of two dipeptides and their relationship to bend conformations. Macromolecules. 1974;7:797–806. doi: 10.1021/ma60042a020. [DOI] [PubMed] [Google Scholar]
- 17.Senet P, Maisuradze GG, Foulie C, Delarue P, Scheraga HA. How main-chains of proteins explore the free-energy landscape in native states. Proc Natl Acad Sci USA. 2008;105:19708–19713. doi: 10.1073/pnas.0810679105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liwo A, Khalili M, Scheraga HA. Ab initio simulations of protein-folding pathways by molecular dynamics with the united-residue model of polypeptide chains. Proc Natl Acad Sci USA. 2005;102:2362–2367. doi: 10.1073/pnas.0408885102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liwo A, et al. Modification and optimization of the united-residue (UNRES) potential-energy function for canonical simulations. I. Temperature dependence of the effective energy function and tests of the optimization method with single training proteins. J Phys Chem B. 2007;111:260–285. doi: 10.1021/jp065380a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korkuta A, Hendrickson WA. A force field for virtual atom molecular mechanics of proteins. Proc Natl Acad Sci USA. 2009;106:15667–15672. doi: 10.1073/pnas.0907674106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romagnoli S, et al. NMR structural determination of Viscotoxin A3 from Viscum album L. Biochem J. 2000;350:569–577. [PMC free article] [PubMed] [Google Scholar]
- 22.Perrin F. Etude mathématique du mouvement Brownien de rotation. (Mathematical study of rotational Brownian motion) Ann Sci Ecole Norm S. 1928;45:1–51. [Google Scholar]
- 23.Mandelbrot BB, Van Ness JW. Fractional Brownian motion, fractional noises and applications. SIAM Rev. 1968;10:422–437. [Google Scholar]
- 24.Bouchaud JP, Georges A. Anomalous diffusion in disordered media: Statistical mechanisms and physical applications. Phys Rep. 1990;195:127–293. [Google Scholar]
- 25.Lim SC, Muniandy SV. Self-similar Gaussian process for modelling anomalous diffusion. Phys Rev E. 2002;66:021114. doi: 10.1103/PhysRevE.66.021114. [DOI] [PubMed] [Google Scholar]
- 26.Wu J, Berland KM. Propagators and time-dependent coefficients for anomalous diffusion. Biophys J. 2008;95:2049–2052. doi: 10.1529/biophysj.107.121608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Min W, Luo G, Cherayil BJ, Kou SC, Xie XS. Observation of a power-law memory kernel for fluctuations within a single protein molecule. Phys Rev Lett. 2005;94:198302. doi: 10.1103/PhysRevLett.94.198302. [DOI] [PubMed] [Google Scholar]
- 28.Luo G, Andricioaei I, Xie XS, Karplus M. Dynamic distance disorder in proteins is caused by trapping. J Phys Chem B. 2006;110:9363–9367. doi: 10.1021/jp057497p. [DOI] [PubMed] [Google Scholar]
- 29.Kneller GR, Hinsen K. Fractional Brownian dynamics in proteins. J Chem Phys. 2004;121:10278–10283. doi: 10.1063/1.1806134. [DOI] [PubMed] [Google Scholar]
- 30.Lu CY, Vanden Bout BA. Effect of finite trajectory length on the correlation function analysis of single molecule data. J Chem Phys. 2006;125:124701. doi: 10.1063/1.2352748. [DOI] [PubMed] [Google Scholar]
- 31.Lange OF, van der Spoel D, de Groot BL. Scrutinizing molecular mechanics force fields on the submicrosecond timescale with NMR data. Biophys J. 2010;99:647–655. doi: 10.1016/j.bpj.2010.04.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McQuarrie DA. Statistical Mechanics. Sausalito, CA: University Science Books; 2000. pp. 512–522. Chap 21. [Google Scholar]
- 33.Farrow NA, Zhang O, Szabo A, Torchia DA, Kay LE. Spectral density function mapping using 15N relaxation data exclusively. J Biomol NMR. 1995;6:153–162. doi: 10.1007/BF00211779. [DOI] [PubMed] [Google Scholar]
- 34.Massi F, Palmer AG. Temperature dependence of NMR order parameters and protein dynamics. J Am Chem Soc. 2003;125:11158–11159. doi: 10.1021/ja035605k. [DOI] [PubMed] [Google Scholar]
- 35.Bouvignies G, et al. Identification of slow correlated motions in proteins using residual dipolar and hydrogen-bond scalar couplings. Proc Natl Acad Sci USA. 2005;102:13885–13890. doi: 10.1073/pnas.0505129102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hess B, Cutzner C, van der Spoel D, Lindahl E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4:435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 37.Jorgensen WL, Maxwell DS, Tirado-Rives J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J Am Chem Soc. 1996;118:11225–11236. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.