

Abstract
Nonalcoholic fatty liver disease (NAFLD), which occurs in approximately 17 to 40% of Americans, encompasses progressive stages of liver damage ranging from steatosis to nonalcoholic steatohepatitis (NASH). Inflammation and oxidative stress are known characteristics of NAFLD; however, the precise mechanisms occurring during disease progression remain unclear. The purpose of the current study was to determine whether the expression or function of enzymes involved in the antioxidant response, NAD(P)H:quinone oxidoreductase 1 (NQO1), glutathione transferase (GST), and glutamate cysteine ligase, are altered in the progression of human NAFLD. Human livers staged as normal, steatotic, NASH (fatty), and NASH (not fatty) were obtained from the Liver Tissue Cell Distribution System. NQO1 mRNA, protein, and activity tended to increase with disease progression. mRNA levels of the GST isoforms A1, A2, A4, M3, and P1 increased with NAFLD progression. Likewise, GST A and P protein increased with progression; however, GST M protein levels tended to decrease. Of interest, total GST activity toward the substrate 1-chloro-2,4-dinitrobenzene decreased with NAFLD progression. GSH synthesis does not seem to be significantly dysregulated in NAFLD progression; however, the GSH/oxidized glutathione redox ratio seemed to be reduced with disease severity, indicating the presence of oxidative stress and depletion of GSH throughout progression of NAFLD. Malondialdehyde concentrations were significantly increased with disease progression, further indicating the presence of oxidative stress. Nuclear immunohistochemical staining of nuclear factor E2-related factor 2 (Nrf2), an indicator of activation of the transcription factor, was evident in all stages of NAFLD. The current data suggest that Nrf2 activation occurs in response to disease progression followed by induction of specific Nrf2 targets, whereas functionality of specific antioxidant defense enzymes seems to be impaired as NAFLD progresses.
Introduction
Nonalcoholic fatty liver disease (NAFLD) is recognized as an epidemic of modern, industrialized countries. Reports estimate that approximately 17 to 40% of the United States population is affected by NAFLD (McCullough, 2006; Ali and Cusi, 2009). It is a complex disease consisting of various stages characterized by progressive levels of hepatocellular damage. NAFLD originates as an infiltration of triglycerides within hepatocytes (Angulo, 2002) known as simple fatty liver (steatosis). Steatosis is generally considered a benign condition, although it renders the liver susceptible to further damage (Adams and Lindor, 2007; Marra et al., 2008). The initial accumulation of lipids within hepatocytes is described as the “first hit” in NAFLD pathogenesis and is believed to be associated with increased sensitivity to oxidative stress (Nagata et al., 2007). Steatosis can progress to the more severe nonalcoholic steatohepatitis (NASH), which is associated with an increase in inflammation, fibrosis, oxidative stress, and more widespread damage (Kleiner et al., 2005; Marra et al., 2008).
Although the mechanisms are not entirely clear, the presentation of oxidative stress is regarded as the “second hit” in NAFLD pathogenesis (Nagata et al., 2007). In addition, the development of inflammation and fibrosis, which cause impairments in liver function, are important factors in the differentiation of steatosis from NASH (Marra et al., 2008). Furthermore, activation of inflammatory cells and mitochondrial dysfunction are believed to be significant contributors to the perpetuation of reactive oxygen species (ROS) production in the progression from steatosis to NASH (Marra et al., 2008). NASH, which is considered to be the end stage of NAFLD, is estimated to affect 5.7 to 17% of the United States population and can further progress to cirrhosis and liver failure (McCullough, 2006). Approximately 15 to 25% of patients with NASH are estimated to progress to cirrhosis with 30 to 40% eventually dying of a liver disease-associated cause (McCullough, 2006).
Oxidative stress is a cellular state in which the generation of harmful molecules such as ROS, peroxidized lipids, reactive quinones, and electrophiles is disproportionate to the ability of the cell to remove these destructive agents. The capacity of a cell to manage oxidative stress is primarily mediated through antioxidant response elements (AREs), which are largely under the control of the transcription factor nuclear factor E2-related factor 2 (Nrf2) (Itoh et al., 1997; Jaiswal, 2004). Nrf2 is primarily a cytosolic protein due to proteasomal degradation regulated by Keap1 (Jaiswal, 2004; Zhang, 2006). When a cell is undergoing oxidative stress, Nrf2 escapes Keap1-mediated degradation, translocates to the nucleus, binds to ARE sequences within the promoters of target genes, and induces expression of phase I and II metabolizing enzymes and transporters pivotal to the maintenance of oxidative stress-inducing molecules (Jaiswal, 2004; Nakata et al., 2006; Zhang, 2006). Such enzymes include NAD(P)H:quinone oxidoreductase-1 (NQO1), glutathione transferase (GST) isoforms, and glutamate cysteine ligase (GCL) subunits (Itoh et al., 1997; Nguyen et al., 2000; Nakata et al., 2006).
NQO1 is a cytosolic protein that catalyzes the two-electron reduction of reactive quinones, which are capable of producing ROS and initiating redox cycling (Jaiswal, 2000; Ross and Siegel, 2004; Talalay and Dinkova-Kostova, 2004). Human GSTs are a large family of detoxifying enzymes that catalyze the conjugation of GSH to reactive electrophiles (Hayes et al., 2005). GSTs can be found in the cytosol, mitochondria, and microsomal membranes of most tissues throughout the body (Hayes et al., 2005). The cytosolic GST isoforms consist of several families, some of which are included in the Alpha, Mu, and Pi classes, each of which have multiple members (Pastore et al., 2003; Hayes et al., 2005). The Alpha and Mu classes have received significant attention as Nrf2 target genes, and specific Alpha isoforms are important for the inactivation of oxidative stress-inducing lipid peroxides. GCL is the rate-limiting step in the synthesis of GSH and is composed of two subunits, catalytic (GCLC) and modifier (GCLM) (Lu, 2009). Because GSH is a cofactor for GST isoforms, disruptions in GSH synthesis have the potential to severely disturb the detoxifying capacity of hepatic GSTs. However, reductions in GSH concentration do not always correlate with GCL expression or activity, e.g., during periods of significant depletion of GSH pools (Pastore et al., 2003; Lu, 2009).
Because of the increasing prevalence of NAFLD, elucidating the mechanisms of oxidative stress-induced injury within the liver is vital to understanding the pathogenesis of NAFLD and susceptibility to further damage. The aim of the current study was to evaluate the expression and activity of NQO1, GST isoforms, and GSH synthesis in the livers of adult patients with progressive stages of NAFLD.
Materials and Methods
Materials.
Tris-HCl, EDTA, KCl, phenylmethanesulfonyl fluoride, 2,6-dichlorophenol-indophenol (DCPIP), NAD(P)H, 1-chloro-2,4-dinitrobenzene (CDNB), GSH, 3,3′diaminobenzidine tetrahydrochloride (DAB), and H2O2 were obtained from Sigma-Aldrich (St. Louis, MO).
Human Liver Samples and Tissue Preparations.
Frozen and formalin-fixed, paraffin-embedded adult human liver tissue was obtained from the Liver Tissue Cell Distribution System at the University of Minnesota, Virginia Commonwealth University, and the University of Pittsburgh. All samples were scored and categorized by a medical pathologist at the Liver Tissue Cell Distribution System according to the NAFLD Activity Score system developed by Kleiner et al. (2005) as part of the service provided by the agency to National Institutes of Health-funded investigators. Diagnosis was later confirmed by histological examination at the University of Arizona. Donor information, including age and gender, has been published previously (Fisher et al., 2009). The samples were diagnosed as normal (n = 20), steatotic (n = 12), NASH with fatty liver (NASH fatty, n = 11), and NASH without fatty liver (NASH not fatty, n = 11). Samples exhibiting >10% fatty infiltration of hepatocytes were considered steatotic. NASH (fatty) samples were diagnosed on the basis of >5% fatty infiltration of hepatocytes with significant inflammation and fibrosis. NASH (not fatty) samples were differentiated from NASH (fatty) samples by a reduction in fatty deposits within the hepatocytes (<5%). Furthermore, NASH (not fatty) samples presented more marked inflammation and fibrotic branching. Total RNA was isolated from human liver tissue using RNAzol B reagent (Tel-Test Inc., Friendswood, TX), according to the manufacturer's recommendations. RNA concentrations were determined by UV spectrophotometry. Integrity of the RNA was confirmed by ethidium bromide staining after agarose gel electrophoresis. Cytosolic fractions of human liver tissue were prepared by homogenization in buffer containing 50 mM Tris-HCl (pH 7.4), 1 mM EDTA, and 154 mM KCl with 0.5 ml/100 ml phenylmethanesulfonyl fluoride (100 mM) at 4°C. Homogenates were centrifuged at 10,000g for 30 min, and the supernatant was transferred to clean microcentrifuge tubes. Samples were then centrifuged at 100,000g for 70 min, and the supernatant was retained as the cytosolic fraction. Protein concentrations were determined using a Pierce BCA Protein Quantitation Assay (Thermo Fisher Scientific, Waltham, MA) per the manufacturer's protocol.
Branched DNA Assay.
Specific oligonucleotide probes (Supplemental Table 1) for NQO1, GCLC, and GCLM were diluted in lysis buffer supplied by the Quantigene HV Signal Amplification Kit (Genospectra, Fremont, CA). Substrate solution, lysis buffer, capture hybridization buffer, amplifier, and label probe buffer used in the analysis were all obtained from the Quantigene Discovery Kit (Genospectra). The assay was performed in a 96-well plate in which total RNA (1 μg/μl; 10 μl) was added to the capture hybridization buffer with 50 μl of the diluted probe set. The total RNA was then allowed to hybridize to the probe set overnight at 53°C. Hybridization steps were performed the following day per the manufacturer's protocol. Luminescence of the samples was measured with a Quantiplex 320 bDNA luminometer interfaced with Quantiplex Data Management Software (version 5.02).
Real-Time Reverse Transcriptase-Polymerase Chain Reaction.
cDNA was reverse-transcribed from total RNA using the Transcriptor First Strand cDNA Synthesis Kit (Roche Diagnostics, Indianapolis, IN) per the manufacturer's protocol. Specific primers for GST isoforms A1, A2, A4, M1, M2, M3, M4, and P1 (Supplemental Table 2) were synthesized by Integrated DNA Technologies, Inc. (Coralville, IA). Reactions were performed using the LightCycler 480 Probes Master Kit (Roche Diagnostics) and Universal ProbeLibrary Probes (Roche Diagnostics) according to the manufacturer's protocol. In brief, the reaction was performed as follows: one denaturation cycle (95°C for 2 min), 45 amplification cycles (95°C for 10 s and 60°C for 30 s), and one cooling period (40°C for 30 s). Relative mRNA levels were normalized to glyceraldehyde-3-phosphate dehydrogenase.
NQO1 Activity Assay.
NQO1 activity was determined by measuring the colorimetric reduction of DCPIP at 600 nm for an interval of 1 min as described previously (Aleksunes et al., 2006a). The reaction was performed in triplicate in a 1-ml format containing 200 μM NAD(P)H, 40 μM DCPIP, and liver cytosol in a Tris-HCl buffer (25 mM Tris, pH 7.4, and 0.7 mg/ml bovine serum albumin). Likewise, inhibition reactions were performed with the addition of 20 μM dicumarol to the reaction mixture. The activity was determined by the difference in absorbance rates of the uninhibited and inhibited reaction rates and was then normalized to total cytosolic protein content.
GST Activity Assay.
GST activity was quantified by spectrophotometry according to established methods using CDNB, which is a substrate for the GST isoforms A1, A2, A4, M1, M2, M3, M4, and P1 (Mannervik and Jemth, 1999). In particular, 1-ml reactions of 1 mM CDNB (prepared in 95% ethanol), 1 mM GSH, 0.1 M sodium phosphate buffer (pH 6.5), and liver cytosol were measured at 340 nm for a period of 1 min. All reactions were performed in triplicate. Enzyme activity was calculated as the difference of the rate of a sample reaction from a simultaneous blank reaction devoid of liver cytosol and was normalized to total cytosolic protein content.
Immunoblot Protein Analysis.
Cytosolic proteins (20 μg/well) were separated by SDS-polyacrylamide electrophoresis and transferred to polyvinylidene difluoride membranes. The following mouse monoclonal antibodies were obtained from Abcam, Inc. (Cambridge, MA): 1:1000 NQO1 [A180], 1:500 GST A, and 1:500 GST P ([F2C2]. GCL protein was detected using mouse monoclonal antibodies from Abnova (Taipei City, Taiwan): 1:8000 GCLC (3H1) and 1:3000 GCLM (2B8). Levels of GST M (1:3000; Abcam, Inc.) and total ERK (1:1000 C-16 and C-14; Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were measured using goat polyclonal antibodies. Quantification of relative protein expression was determined using image processing and analysis with ImageJ software (National Institutes of Health, Bethesda, MD) and normalized to total ERK.
GSH Assay.
GSH concentrations were determined using a Glutathione Assay Kit (BioVision Research Products, Mountain View, CA) per the manufacturer's recommendations in a 96-well format. In brief, ∼40 mg of tissue was homogenized and preserved with GSH buffer and 6 N perchloric acid. Samples were then precipitated with 3 N potassium hydroxide and centrifuged for 2 min at 13,000g. To detect reduced GSH, 5 μl of the neutralized sample was incubated with the o-phthalaldehyde probe and GSH buffer for 40 min. To detect oxidized glutathione (GSSG), 1 μl of neutralized sample was incubated with GSH buffer and 10 μl of GSH quencher for 10 min, followed by reducing agent and o-phthalaldehyde for 40 min. Fluorescence readings were captured using a SPECTRAmax GEMINI XS (Molecular Devices, Sunnyvale, CA) fluorescence reader.
TBARS Assay.
Malondialdehyde (MDA) concentrations were determined using the TBARS Assay Kit (Cayman Chemical, Ann Arbor, MI) per the manufacturer's recommendations for colorimetric measurement of MDA. In brief, ∼25 mg of tissue was sonicated on ice in radioimmunoprecipitation assay lysis buffer containing 1% NP-40, 1% w/v sodium deoxycholate, 0.1% SDS, 0.15 M NaCl, 0.01 M sodium phosphate, pH 8.0, 2 mM EDTA, and 1 Protease Inhibitor Cocktail Tablet per 25 ml (Roche Diagnostics). Samples were then centrifuged at 1600g for 10 min at 4°C, and the supernatant was preserved for analysis. To 100 μl of each sample and standard curve solutions, 100 μl of provided SDS solution was added followed by 4 ml of color reagent. Samples and standard curve solutions were boiled for 1 h, and the reaction was terminated by incubation on ice for 10 min followed by centrifugation at 1600g for 10 min at 4°C. Then 150 μl of samples and standard curve solutions were loaded in duplicate onto a 96-well plate, and absorbance was read at 540 nm in a Powerwave HT (BioTek Instruments, Winooski, VT) spectrophotometer.
Immunohistochemistry.
Immunohistochemical staining for Nrf2 was performed on formalin-fixed paraffin-embedded human liver samples. In brief, tissue sections were deparaffinized in xylene and rehydrated in ethanol, followed by antigen retrieval in citrate buffer (pH 6.0). Endogenous peroxidase activity was blocked with 0.3% (v/v) H2O2 in methanol for 20 min. All samples were incubated with a 1:75 solution of Nrf2 (C-20) antibody (Santa Cruz Biotechnology, Inc.) overnight at 4°C. Antibody binding was detected with biotinylated goat anti-rabbit IgG (Vector Laboratories, Burlingame, CA) and an avidin-biotin complex (Vector Laboratories). Color development was performed with a solution of 0.5% DAB in 0.015% H2O2 (0.01 M phosphate-buffered saline, pH 7.2). All slides were imaged with a Nikon Eclipse E4000 microscope and a Sony Exwave DXC-390 camera.
Quantification of Nrf2 Nuclear Translocation.
Hepatocyte nuclei positive for Nrf2 staining and hepatocyte nuclei negative for Nrf2 staining were quantified within a 20× field of vision with a total of 10 fields assessed for each sample. The number of total hepatocytes was calculated from the positive and negative nuclei counts and was used to determine the percentage of positive nuclei within a field. The average number of positive nuclei, negative nuclei, total nuclei, and percent positive nuclei was assessed for each diagnostic category.
Statistical Analysis.
The samples within this study can be categorized by disease progression/severity: normal < steatosis < NASH (fatty) < NASH (not fatty). All data collected in this study were continuous outcomes but exhibited a skewed distribution. Therefore, median values rather than mean values were compared, and the results are presented as box and whisker plots. Data were analyzed by a nonparametric trend analysis with a significance level of p ≤ 0.05 to determine significant increases or decreases with disease progression/severity. All analyses were performed with Stata10 software (Stata Corporation, College Station, TX).
Results
Human NAFLD Histology.
Representative images of hematoxylin and eosin staining of human liver samples staged as normal, steatotic, NASH (fatty), and NASH (not fatty) are shown in Fig. 1. A diagnosis of steatosis was made for samples exhibiting >10% fatty infiltration of hepatocytes. Samples exhibiting >5% fatty infiltration of hepatocytes with significant inflammation and fibrosis were staged as NASH (fatty). Those samples presenting with a reduction in fatty deposits within the hepatocytes to <5% with increased inflammation and branching fibrosis were diagnosed as NASH (not fatty). Microvesicular lipid deposits are readily apparent in the steatotic samples, and sinusoidal structure remains somewhat intact. The NASH (fatty) stage is characterized by macrovesicular fatty deposits within hepatocytes and the development of inflammation and fibrosis. As the liver progresses from NASH (fatty) to NASH (not fatty), the infiltration of fatty deposits is greatly reduced; however, inflammation increases, as does fibrosis.
Fig. 1.

Human NAFLD histology. Representative hematoxylin and eosin staining of human liver samples diagnosed as normal, steatotic, NASH (fatty), and NASH (not fatty) are shown. Original magnification, 20×. Steatosis was diagnosed as >10% fatty infiltration, NASH (fatty) as >5% fatty infiltration with significant inflammation and fibrosis, and NASH (not fatty) as <5% fatty infiltration with greater inflammation and fibrosis.
NQO1 Expression and Activity in Human NAFLD Progression.
Gene expression of antioxidant response enzymes is indicative of Nrf2 transcriptional activation of the ARE. NQO1 gene expression was measured in human liver samples by assessing the levels of NQO1 mRNA using the bDNA assay, shown in Fig. 2A. There was an increasing trend in NQO1 mRNA expression with progression of NAFLD. An immunoblot analysis of NQO1 protein levels in human cytosolic liver samples was conducted and normalized to total ERK. Representative immunoblots with two samples from each diagnostic category and densitometry results of NQO1 in each stage of NAFLD are shown in Fig. 2, B and A, respectively. Densitometric analysis of relative NQO1 protein levels in all 54 human liver samples revealed an increasing trend in NQO1 protein with disease progression. The enzymatic activity of NQO1 protein was measured in human liver cytosolic samples using DCPIP as the substrate. Enzymatic activity of NQO1 was significantly increased with progression of NAFLD.
Fig. 2.

NQO1 expression and activity. Messenger RNA levels, densitometric analysis of NQO1 protein levels, and NQO1 enzymatic activity (A) in human liver samples staged as normal, steatotic, NASH (fatty), and NASH (not fatty). mRNA levels were measured by the bDNA assay and expressed as relative light units (RLU) per 10 μg of total RNA. Protein levels were determined by immunoblot analysis and expressed relative to control protein (Total ERK). A representative blot of NQO1 and total ERK is shown using two samples from each diagnostic category (B). NQO1 activity was measured by the colorimetric reduction of DCPIP and is expressed as nanomoles of DCPIP reduced per minute per milligram of protein. Arrows indicate an increasing or decreasing trend. *, statistically significant trend as determined by nonparametric trend analysis (p ≤ 0.05).
GST Isoform Expression and Pan-GST Activity in Human NAFLD Progression.
The expression of various GST isoforms is also dependent on the ARE and, thus, on transcriptional activation by Nrf2. Because of significant homology among the GST isoforms, mRNA levels could not be measured by bDNA analysis, which requires the use of multiple oligonucleotides to identify specific mRNA molecules. Therefore, relative mRNA levels of the GST isoforms A1, A2, A4, M1, M2, M3, M4, and P1 were measured by reverse transcriptase-polymerase chain reaction in each stage of NAFLD. As shown in Fig. 3A, mRNA levels of the GST A4, A2, A4, M3, and P1 isoforms exhibited a significant increasing trend with disease progression. GST protein levels in cytosolic liver fractions were assessed for each isoform family (GST A, GST M, and GST P) and normalized to total ERK. Protein levels were measured for all 54 liver specimens in the study. Representative immunoblots depicting two samples from each diagnostic category and densitometry results are shown in Fig. 3, C and B, respectively. There were increasing trends in expression for both GST A and GST P protein. In contrast, GST M protein levels were decreased with NAFLD progression. The functional capability of the GST isoforms to effectively conjugate GSH to electrophilic substrates was determined spectrophotometrically in human liver cytosolic samples using CDNB as a pan-GST substrate. As shown in Fig. 3D, a significant decreasing trend in GST enzymatic activity was observed with NAFLD progression.
Fig. 3.

GST isoform expression and activity. Messenger RNA (A), densitometric analysis of protein levels of GST isoforms (B), representative immunoblots of GST families (C), and pan-GST enzymatic activity (D) in human liver samples diagnosed as normal, steatotic, NASH (fatty), and NASH (not fatty). A, mRNA levels for specific GST isoforms (GST A1, A2, A4, M1, M2, M3, M4, and P1) were measured by reverse-transcriptase polymerase chain reaction and expressed as relative mRNA expression to glyceraldehyde-3-phosphate dehydrogenase. B, protein levels of GST families (GST A, GST M, and GST P) were determined by immunoblot analysis and expressed as relative to control protein (Total ERK). C, a representative immunoblot of GST A, GST M, and GST P monomer and dimer with total ERK is shown using two samples from each diagnostic category. D, GST activity was determined spectrophotometrically using CDNB as a pan-GST substrate and is expressed as micromoles per minute per milligram protein. Arrows indicate an increasing or decreasing trend. *, statistically significant trend as determined by nonparametric trend analysis (p ≤ 0.05).
GCL Expression.
mRNA and protein levels of GCL, the rate-limiting enzyme in the synthesis of GSH, were assessed by the bDNA assay and immunoblot analysis in human liver samples. mRNA results of GCLC, the catalytic subunit of GCL, and GCLM, the modifier subunit, are shown in Fig. 4A. No significant alterations were observed in the mRNA levels of GCLC; however, GCLM mRNA levels exhibited an increasing trend as NAFLD severity increased. The densitometric results of GCLC and GCLM protein levels in all 54 human liver samples, along with representative immunoblots depicting two samples from each diagnostic category, are shown in Fig. 4, A and B, respectively. No significant alterations in GCL protein were observed.
Fig. 4.

GCLC and GCLM expression. Messenger RNA (A) and densitometric analysis of protein levels (B) of GCLC and GCLM in human liver samples staged as normal, steatotic, NASH (fatty) and NASH (not fatty). mRNA levels were measured by the bDNA assay and expressed as relative light units (RLU) per 10 μg of total RNA. Protein levels were determined by immunoblot analysis and are expressed as relative to control protein (Total ERK). Arrows indicate an increasing or decreasing trend. *, statistically significant trend as determined by nonparametric trend analysis (p ≤ 0.05).
GSH Concentration and Redox Status in Human NAFLD.
GSH and GSSG concentrations were determined fluorometrically using the BioVision Glutathione Assay Kit. The results are presented in Fig. 5, along with the GSH/GSSG ratio (redox status). There were decreasing trends in GSH and GSSG concentrations throughout disease progression. The redox status (GSH/GSSG ratio), an estimation of the oxidative state in cells and tissues, is shown in Fig. 5. The redox status exhibited a significant decreasing trend with disease progression.
Fig. 5.
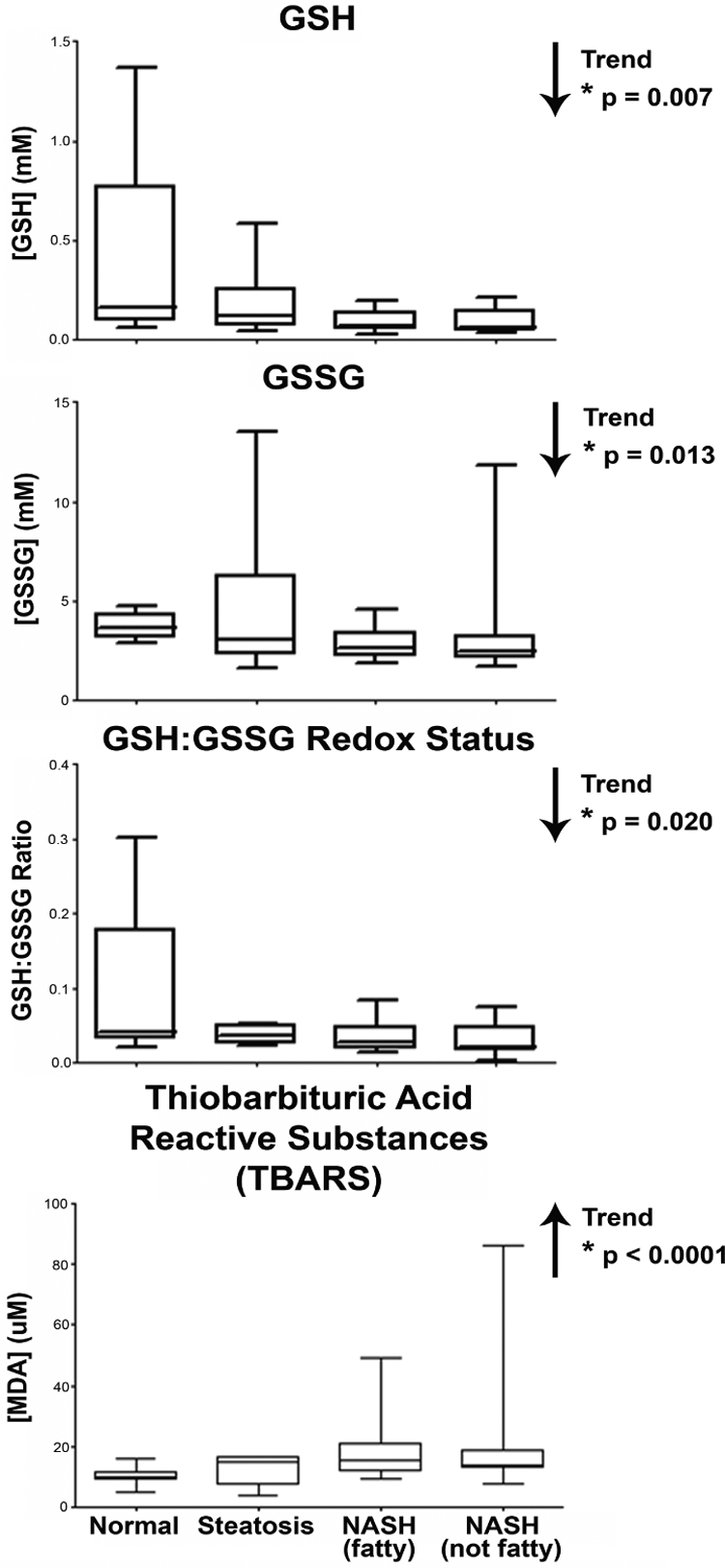
GSH and GSSG concentrations, redox status, and MDA concentration. GSH, GSSG, and MDA concentrations in human liver samples diagnosed as normal, steatotic, NASH (fatty) and NASH (not fatty). GSH and GSSG concentrations were determined fluorometrically using o-phthalaldehyde as the probe, normalized to weight of tissue, and are expressed as millimolar concentration. GSH and GSSG concentrations were then used to determine redox status of the tissue and represented as the GSH/GSSG ratio. MDA concentrations were measured colorimetrically; concentrations were determined by standard curve analysis and are expressed as micromolar concentrations. Arrows indicate an increasing or decreasing trend. *, statistically significant trend as determined by nonparametric trend analysis (p ≤ 0.05).
MDA Concentration in Human NAFLD.
MDA is a product of lipid peroxidation and is often used as an indicator of oxidative stress in tissues and cells. MDA concentration was measured colorimetrically using the Cayman Chemical TBARS Assay Kit. The results, as shown in Fig. 5, reveal a significant increasing trend in MDA concentration with disease progression.
Nrf2 Activation in Human NAFLD.
Induction of ARE genes is dependent on the escape of Nrf2 from Keap1-mediated degradation in the cytosol, enabling its translocation to the nucleus where it may bind to ARE promoters. To determine whether Nrf2 had translocated into the nuclei of hepatocytes and was therefore activated, formalin-fixed, paraffin-embedded human liver samples were immunohistochemically stained for Nrf2. Representative images are shown in Fig. 6. Positive nuclear staining of Nrf2 (indicated by open arrows) can be seen in all stages of NAFLD. Nuclei negative for Nrf2 staining are indicated by closed arrows. Quantification of Nrf2 translocation revealed that in normal human liver samples an average of 2.06% of hepatocytes within a 20× field of vision exhibited positive Nrf2 nuclear staining. In contrast, steatotic, NASH (fatty), and NASH (not fatty) samples presented on average with 24.67, 22.60, and 22.34%, respectively, of hepatocytes bearing positive Nrf2 nuclear staining.
Fig. 6.

Immunohistochemical staining of Nrf2. Representative immunohistochemical staining of Nrf2 in human liver samples staged as normal, steatotic, NASH (fatty) and NASH (not fatty). Antibody binding was detected by the avidin-biotin complex method using DAB for color development. Original magnification, 20×. Open arrows indicate nuclei positive for Nrf2 staining, and closed arrows indicate negative nuclear staining.
Discussion
NAFLD, like obesity, is becoming an increasingly significant health concern. Accumulation of triglycerides within hepatocytes, insulin resistance, inflammation, chronic oxidative stress levels, and fibrosis in combination make this a complex disease. The specific mechanisms by which each of these parameters presents in the progression of NAFLD are not certain. With the exception of cytochrome P450 enzymes (Fisher et al., 2009), the metabolic capacity toward administered pharmaceutical agents in patients with NAFLD has not been well characterized. This is of serious concern as patients with NAFLD are often medicated for symptoms of the metabolic syndrome. Therefore, there is a great need for investigation into the pathogenic process of NAFLD as it relates to oxidative stress and the capacity to metabolize pharmaceutical agents.
The aim of the current study was to investigate the expression and functionality of enzymes, such as NQO1, GST, and GCL, involved in the antioxidant response. Of these enzymes, the GST isoforms, in particular, are known to play a significant role in the phase II metabolism of endogenous molecules and administered pharmaceutical agents, whereas NQO1 is primarily responsible for the reduction of oxidative stress-inducing quinones. Increased expression and/or activity of NQO1 has been shown to be an important protective mechanism against oxidative stress in cholestasis (Aleksunes et al., 2006b), coronary heart disease (Martin et al., 2009), acetaminophen-induced hepatotoxicity (Moffit et al., 2007), and primary biliary cirrhosis (Aleksunes et al., 2006a). Although not traditionally considered a drug-metabolizing enzyme, NQO1 is capable of reducing N-acetyl-p-benzoquinone imine, the toxic quinone metabolite of acetaminophen (Moffit et al., 2007). Furthermore, induction of NQO1 activity leads to protection against acetaminophen-induced hepatotoxicity (Moffit et al., 2007). We have observed an increase in NQO1 mRNA levels, protein, and activity with the progression of NAFLD. These findings are similar to observations made in animal models of steatosis and NASH. We previously found that Sprague-Dawley rats fed a methionine- and choline-deficient diet to induce NASH exhibited significant elevation of NQO1 mRNA and enzymatic activity (Lickteig et al., 2007). In a separate study, rats fed the methionine- and choline-deficient diet exhibited significant induction of NQO1 mRNA levels, and, when treated with oltipraz, an Nrf2 activator, were still able to induce NQO1 to an even greater level (Fisher et al., 2008). Together with our previous animal studies, the current data suggest that as NAFLD progresses, increased NQO1 expression and activity levels may confer a greater capacity to protect patients with NASH from oxidative stress-induced and pharmaceutical-induced quinone redox cycling.
In contrast, GSTs are responsible for protecting against electrophilic molecules such as those generated during oxidative stress or administration of pharmaceutical agents. The expression and activity of specific GST isoforms have been closely associated with the severity of multiple diseases. For instance, GST M1 and T1 polymorphisms imparting an impairment of enzyme functionality impose a greater risk for coronary artery disease in patients with type II diabetes (Manfredi et al., 2009). Furthermore, GST M1 functionality has been shown to be important in protection against oxidative stress-induced DNA damage in patients undergoing maintenance hemodialysis (Lin et al., 2009). Traditionally, the induction of GST isoforms has been associated with protective mechanisms because of their ability to catalyze the conjugation of GSH to various ROS molecules (Hayes et al., 2005). Although several GST isoforms are inducible by Nrf2, there is generally a low, constitutive expression of GST A3, A4, M1, M3, and M4 (Aleksunes and Manautou, 2007), whereas GST A1 and A2 are believed to be more highly expressed in liver (Coles and Kadlubar, 2005). Moreover, GST A4 expression is believed to be induced during periods of prolonged oxidative stress (Coles and Kadlubar, 2005). GST P genes, on the other hand, are considered to be inducible, but not constitutively expressed within the liver (Henderson and Wolf, 2005). At present, we have observed an increasing trend in GST A1, A2, A4, M3, and P1 mRNA expression throughout progression of NAFLD. The induction of GST P1 mRNA has previously been recognized as a marker for hepatocellular carcinogenesis in the rat but may or may not be associated with hepatocellular carcinoma in humans (Higashi et al., 2004; Sakai and Muramatsu, 2007). Therefore, the induction of GST P1 shown in the present study may be an indicator of a predisposition to hepatocellular carcinoma but is not definitive. We have observed increases in GST A and P protein with disease progression. In contrast with GST A and P protein, there was a significant downward trend in GST M protein levels. These data suggest that the stability of each GST protein family may be differentially regulated throughout progression of NAFLD.
Because of the increased expression of multiple GST isoforms and induction of protein levels of the GST A and P families, GST enzymatic activity could be expected to similarly increase with the progression of NAFLD. However, in the current study, we observed a gradual decline in GST activity levels. These results suggest that as NAFLD advances, patients may have a reduced capacity to combat the oxidative insults of electrophilic compounds. A similar phenomenon has been observed in a murine model of ulcerative colitis (Clapper and Szarka, 1998). Note that this downward trend in GST activity parallels the expression of GST M protein within our samples. GST A isoforms, on the other hand, are traditionally considered the dominant isoforms expressed in the liver (Coles and Kadlubar, 2005). These data indicate that there may be a more complicated mechanism of regulating GST isoforms during NAFLD progression.
Studies in human hepatic cirrhosis and hepatocellular carcinoma tissues have presented a complex interplay in GSH-dependent enzyme activities. These studies have shown that GST activity is significantly increased in cirrhotic livers compared with that of malignant hepatic tissue (Czeczot et al., 2006). Further analysis in this study identified a significant reduction in GSH within cirrhotic hepatic tissue compared with both malignant and normal tissues (Czeczot et al., 2006). In the current study, we observed a similar reduction in hepatic GSH levels during disease progression, and, although GSSG levels exhibited a significant downward trend, GSSG concentrations were much higher than those of GSH. Whereas this can sometimes be due to the insensitivity of the analytical method used or the labile nature of GSH in tissues, previous reports indicated that GSSG levels may indeed be much higher than GSH levels in NAFLD (Pastore et al., 2003). Using the GSH and GSSG concentrations measured in each sample, we calculated the GSH/GSSG redox ratio, which is an indicator of oxidative stress within the tissue. The reduction in the GSH/GSSG redox ratio observed during NAFLD progression indicates that oxidative stress is increasing with the severity of disease. An additional indication of the presence of oxidative stress during NAFLD progression was observed by the increasing concentration of MDA with disease severity. MDA is a product of lipid peroxidation that occurs under oxidative stress conditions. Increasing levels of MDA confirm the conclusion of increased oxidative stress occurring during NAFLD made during the measurement of GSH levels. Nevertheless, the decrease in GST activity seen with NAFLD progression cannot be simply attributed to the measured reductions in GSH concentration. In this activity assay, GSH is supplemented in the reaction, thereby reducing the confounding effect of depletion of GSH pools in the sample, therefore leading to the conclusion that additional factors must play a role in the conjugating capabilities of GST isoforms during NAFLD progression. One possible explanation is the adduction of 4-hydroxy-2-nonenal (HNE) to specific GST isoforms. Studies have identified GST P as a target of HNE adduction in human colorectal carcinoma cells (Codreanu et al., 2009). Inhibition studies of GST P1 have indicated that cysteine 47, which is associated with the active site of the enzyme, is the target of covalent binding by HNE (van Iersel et al., 1997). In the present study, we have observed a significant increase in expression of GST P protein that, if inhibited by HNE may, in conjunction with GST M protein expression, explain the observed depletion in GST enzymatic activity. Further investigations are necessary to identify the specific mechanism by which GST activity is inhibited in NAFLD.
To identify whether the observed reductions in GSH concentration are due to a depletion of GSH pools or impairment of GSH synthesis, we investigated expression of the GCLC and GCLM subunits of GCL. GCL is responsible for the initial catalysis of glutamate and cysteine to produce γ-glutamyl-l-cysteine (Lu, 2009). Further conjugation with glycine is catalyzed by GSH synthetase (Lu, 2009). GCL is regulated by feedback inhibition through GSH concentration, and induction of GCL expression does correlate with increased GSH synthesis; however, induction of GSH synthetase does not lead to a subsequent increase in GSH levels (Lu, 2009). In the current study, we have identified an increasing trend in GCLM mRNA levels; however, there was no significant alteration of GCLM protein levels. In addition, no alterations to GCLC expression were observed. Together, these data provide no indication of an impairment of GSH synthesis, thereby leading to the conclusion that the observed reductions in GSH levels are most likely due to a depletion of GSH pools.
As stated previously, activation of the transcription factor Nrf2 leads to nuclear accumulation of Nrf2 protein and induction of downstream target ARE genes (Zhang, 2006). In the current study, we have identified nuclear accumulation of Nrf2 in each of the stages of NAFLD. Quantification of hepatocytes exhibiting positive Nrf2 nuclear staining revealed a stark increase in the percentage of total hepatocytes in which Nrf2 was activated among each of the disease states compared with normal. Activation of Nrf2 is a viable explanation for the observed induction of ARE target genes. The increase in enzymatic activity of NQO1 implies an enhanced ability to diminish oxidative damage due to quinone redox cycling in NAFLD. However, the observed decrease in GST enzymatic activity suggests that patients with NAFLD have a limited capacity to manage electrophilic compounds. The current study provides novel insight to the mechanisms of oxidative stress as they occur in the progression of human NAFLD.
Supplementary Material
Acknowledgments.
We thank the National Institutes of Health-funded Liver Tissue Cell Distribution System for continued assistance and procurement of liver tissue samples from patients with various stages of NAFLD. We also extend sincere appreciation to Marion Namenwirth (University of Minnesota), Melissa Thompson (Virginia Commonwealth University), and Dr. Stephen C. Strom and Kenneth Dorko (University of Pittsburgh).
This work was supported in part by the National Institutes of Health National Institute of Diabetes and Digestive and Kidney Diseases [Grant DK068039]; the National Institutes of Health National Institute of Environmental Health Sciences [Grant ES006694]; and the National Institutes of Health National Center for Complementary and Alternative Medicine [Grant AT002842]. The Liver Tissue Cell Distribution System was sponsored by the National Institutes of Health [Contract N01-DK-7-0004/HHSN267200700004C].
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
doi:10.1124/dmd.110.035006.
The online version of this article (available at http://dmd.aspetjournals.org) contains supplemental material.
- NAFLD
- nonalcoholic fatty liver disease
- NASH
- nonalcoholic steatohepatitis
- ROS
- reactive oxygen species
- ARE
- antioxidant response element
- Nrf2
- nuclear factor E2-related factor 2
- NQO1
- NAD(P)H:quinone oxidoreductase 1
- GST
- glutathione transferase
- GCL
- glutamate cysteine ligase
- GCLC
- glutamate cysteine ligase catalytic subunit
- GCLM
- glutamate cysteine ligase modifier subunit
- DCPIP
- 2,6-dichlorophenol indophenol
- CDNB
- 1-chloro-2,4-dinitrobenzene
- DAB
- 3,3′diaminobenzidine tetrahydrochloride
- ERK
- extracellular signal-regulated kinase
- GSSG
- oxidized glutathione
- TBARS
- thiobarbituric acid reactive substances
- MDA
- malondialdehyde
- HNE
- 4-hydroxy-2-nonenal.
References
- Adams LA, Lindor KD. (2007) Nonalcoholic fatty liver disease. Ann Epidemiol 17:863–869 [DOI] [PubMed] [Google Scholar]
- Aleksunes LM, Goedken M, Manautou JE. (2006a) Up-regulation of NAD(P)H quinone oxidoreductase 1 during human liver injury. World J Gastroenterol 12:1937–1940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksunes LM, Manautou JE. (2007) Emerging role of Nrf2 in protecting against hepatic and gastrointestinal disease. Toxicol Pathol 35:459–473 [DOI] [PubMed] [Google Scholar]
- Aleksunes LM, Slitt AL, Maher JM, Dieter MZ, Knight TR, Goedken M, Cherrington NJ, Chan JY, Klaassen CD, Manautou JE. (2006b) Nuclear factor-E2-related factor 2 expression in liver is critical for induction of NAD(P)H:quinone oxidoreductase 1 during cholestasis. Cell Stress Chaperones 11:356–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali R, Cusi K. (2009) New diagnostic and treatment approaches in non-alcoholic fatty liver disease (NAFLD). Ann Med 41:265–278 [DOI] [PubMed] [Google Scholar]
- Angulo P. (2002) Nonalcoholic fatty liver disease. N Engl J Med 346:1221–1231 [DOI] [PubMed] [Google Scholar]
- Clapper ML, Szarka CE. (1998). Glutathione S-transferases—biomarkers of cancer risk and chemopreventive response. Chem Biol Interact 111–112:377–388 [DOI] [PubMed] [Google Scholar]
- Codreanu SG, Zhang B, Sobecki SM, Billheimer DD, Liebler DC. (2009) Global analysis of protein damage by the lipid electrophile 4-hydroxy-2-nonenal. Mol Cell Proteomics 8:670–680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles BF, Kadlubar FF. (2005) Human alpha class glutathione S-transferases: genetic polymorphism, expression, and susceptibility to disease. Methods Enzymol 401:9–42 [DOI] [PubMed] [Google Scholar]
- Czeczot H, Scibior D, Skrzycki M, Podsiad M. (2006) Glutathione and GSH-dependent enzymes in patients with liver cirrhosis and hepatocellular carcinoma. Acta Biochim Pol 53:237–242 [PubMed] [Google Scholar]
- Fisher CD, Jackson JP, Lickteig AJ, Augustine LM, Cherrington NJ. (2008) Drug metabolizing enzyme induction pathways in experimental non-alcoholic steatohepatitis. Arch Toxicol 82:959–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher CD, Lickteig AJ, Augustine LM, Ranger-Moore J, Jackson JP, Ferguson SS, Cherrington NJ. (2009) Hepatic cytochrome P450 enzyme alterations in humans with progressive stages of nonalcoholic fatty liver disease. Drug Metab Dispos 37:2087–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes JD, Flanagan JU, Jowsey IR. (2005) Glutathione transferases. Annu Rev Pharmacol Toxicol 45:51–88 [DOI] [PubMed] [Google Scholar]
- Henderson CJ, Wolf CR. (2005) Disruption of the glutathione transferase pi class genes. Methods Enzymol 401:116–135 [DOI] [PubMed] [Google Scholar]
- Higashi K, Hiai H, Higashi T, Muramatsu M. (2004) Regulatory mechanism of glutathione S-transferase P-form during chemical hepatocarcinogenesis: old wine in a new bottle. Cancer Lett 209:155–163 [DOI] [PubMed] [Google Scholar]
- Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et al. (1997) An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236:313–322 [DOI] [PubMed] [Google Scholar]
- Jaiswal AK. (2000) Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic Biol Med 29:254–262 [DOI] [PubMed] [Google Scholar]
- Jaiswal AK. (2004) Nrf2 signaling in coordinated activation of antioxidant gene expression. Free Radic Biol Med 36:1199–1207 [DOI] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, et al. (2005) Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 41:1313–1321 [DOI] [PubMed] [Google Scholar]
- Lickteig AJ, Fisher CD, Augustine LM, Cherrington NJ. (2007) Genes of the antioxidant response undergo upregulation in a rodent model of nonalcoholic steatohepatitis. J Biochem Mol Toxicol 21:216–220 [DOI] [PubMed] [Google Scholar]
- Lin YS, Hung SC, Wei YH, Tarng DC. (2009) GST M1 polymorphism associates with DNA oxidative damage and mortality among hemodialysis patients. J Am Soc Nephrol 20:405–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC. (2009) Regulation of glutathione synthesis. Mol Aspects Med 30:42–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi S, Calvi D, del Fiandra M, Botto N, Biagini A, Andreassi MG. (2009) Glutathione S-transferase T1- and M1-null genotypes and coronary artery disease risk in patients with type 2 diabetes mellitus. Pharmacogenomics 10:29–34 [DOI] [PubMed] [Google Scholar]
- Mannervik B, Jemth P. (1999) Measurement of glutathione transferases, in Current Protocols in Toxicology, pp 6.4.1–6.4.10, John Wiley & Sons, New York: [DOI] [PubMed] [Google Scholar]
- Marra F, Gastaldelli A, Svegliati Baroni G, Tell G, Tiribelli C. (2008) Molecular basis and mechanisms of progression of non-alcoholic steatohepatitis. Trends Mol Med 14:72–81 [DOI] [PubMed] [Google Scholar]
- Martin NJ, Collier AC, Bowen LD, Pritsos KL, Goodrich GG, Arger K, Cutter G, Pritsos CA. (2009) Polymorphisms in the NQO1, GSTT and GSTM genes are associated with coronary heart disease and biomarkers of oxidative stress. Mutat Res 674:93–100 [DOI] [PubMed] [Google Scholar]
- McCullough AJ. (2006) Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol 40:S17–S29 [DOI] [PubMed] [Google Scholar]
- Moffit JS, Aleksunes LM, Kardas MJ, Slitt AL, Klaassen CD, Manautou JE. (2007) Role of NAD(P)H:quinone oxidoreductase 1 in clofibrate-mediated hepatoprotection from acetaminophen. Toxicology 230:197–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata K, Suzuki H, Sakaguchi S. (2007) Common pathogenic mechanism in development progression of liver injury caused by non-alcoholic or alcoholic steatohepatitis. J Toxicol Sci 32:453–468 [DOI] [PubMed] [Google Scholar]
- Nakata K, Tanaka Y, Nakano T, Adachi T, Tanaka H, Kaminuma T, Ishikawa T. (2006) Nuclear receptor-mediated transcriptional regulation in phase I, II, and III xenobiotic metabolizing systems. Drug Metab Pharmacokinet 21:437–457 [DOI] [PubMed] [Google Scholar]
- Nguyen T, Huang HC, Pickett CB. (2000) Transcriptional regulation of the antioxidant response element. Activation by Nrf2 and repression by MafK. J Biol Chem 275:15466–15473 [DOI] [PubMed] [Google Scholar]
- Pastore A, Federici G, Bertini E, Piemonte F. (2003) Analysis of glutathione: implication in redox and detoxification. Clin Chim Acta 333:19–39 [DOI] [PubMed] [Google Scholar]
- Ross D, Siegel D. (2004) NAD(P)H:quinone oxidoreductase 1 (NQO1, DT-diaphorase), functions and pharmacogenetics. Methods Enzymol 382:115–144 [DOI] [PubMed] [Google Scholar]
- Sakai M, Muramatsu M. (2007) Regulation of glutathione transferase P: a tumor marker of hepatocarcinogenesis. Biochem Biophys Res Commun 357:575–578 [DOI] [PubMed] [Google Scholar]
- Talalay P, Dinkova-Kostova AT. (2004) Role of nicotinamide quinone oxidoreductase 1 (NQO1) in protection against toxicity of electrophiles and reactive oxygen intermediates. Methods Enzymol 382:355–364 [DOI] [PubMed] [Google Scholar]
- van Iersel ML, Ploemen JP, Lo Bello M, Federici G, van Bladeren PJ. (1997) Interactions of α,β-unsaturated aldehydes and ketones with human glutathione S-transferase P1-1. Chem Biol Interact 108:67–78 [DOI] [PubMed] [Google Scholar]
- Zhang DD. (2006) Mechanistic studies of the Nrf2-Keap1 signaling pathway. Drug Metab Rev 38:769–789 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.