

Abstract
Objective
The purpose is to analyze DNA methylation profiling among different types of ovarian serous neoplasm, a task that has not been performed.
Study Design
The Illumina beads array was used to profile DNA methylation in enriched tumor cells isolated from 75 benign and malignant serous tumor tissues and six tumor-associated stromal cell cultures.
Results
We found significantly fewer hypermethylated genes in high-grade serous carcinomas than in low-grade serous carcinoma and borderline tumors, which in turn had fewer hypermethylated genes than serous cystadenoma. Unsupervised analysis identified that serous cystadenoma, serous borderline tumor and low-grade serous carcinoma tightly clustered together and were clearly different from high-grade serous carcinomas. We also performed supervised analysis to identify differentially methylated genes that may contribute to group separation.
Conclusion
The findings support the view that low-grade and high-grade serous carcinomas are distinctly different with low-grade, but not high-grade serous carcinomas, related to serous borderline tumor and cystadenoma.
Keywords: ovarian carcinoma, methylation, classification
Introduction
DNA methylation is an important mechanism in the regulation of gene function and serves as an epigenetic marker for tumor detection, classification and prognostication1, 2. DNA hypermethylation refers to the addition of a methyl group to the cytosine ring of those cytosines that precede a guanosine (referred to as CpG dinucleotides) to form methyl cytosine (5-methylcytosine). CpG dinucleotides are found at increased frequency in the promoter region of many genes. These regions have been termed “CpG islands”, and, with the exception of genes on the inactive X chromosome and imprinted genes, CpG islands are protected from methylation in normal cells2. This protection is critical, since methylation of CpG islands is usually associated with loss of expression of that particular gene. It has been demonstrated that the decreased expression of tumor suppressor genes and DNA repair genes are associated with promoter hypermethylation which is a common feature in human cancer, and serves as an alternative mechanism for loss of their function. For example, hypermethylation in the promoters of CDKN2 (p16), VHL, WT1 and MLH1 occurs in many solid tumors and is associated with loss of its expression1, 3, 4, indicating that aberrant DNA methylation is one of the main mechanisms in the pathogenesis of human cancer. To this end, 5-azacitidine has been shown to demethylate DR4 resulting in its re-expression, which leads in turn to enhanced sensitivity of platinum-resistant ovarian cancer cells to carboplatin through induction of apoptosis5.
Aberrant methylation of multiple CpG islands is frequently observed in ovarian carcinoma compared with normal ovarian surface epithelium or benign ovarian neoplasms6, 7. For example, frequent CpG island hypermethylation in BRCA1, RASSF1A, and OPCML is detected in ovarian carcinoma and plays a role in tumor development8–10. Different histologic subtypes of ovarian cancer including serous, endometrioid and clear cell carcinoma demonstrate distinct DNA methylation profiles11. Moreover, DNA methylation in certain gene promoters has been found to be a reliable epigenetic marker to predict treatment outcome in several types of human cancer including ovarian carcinoma6, 12–18. Thus, it has been suggested that DNA methylation changes have implications for ovarian cancer diagnosis, prognostication and treatment19. Although promoter methylation has been studied in ovarian carcinoma, a comprehensive analysis of methylation profiles has not yet been performed in different types of benign and malignant ovarian serous neoplasms including serous cystadenoma, serous borderline tumor (SBT), low-grade (LG) and high-grade (HG) serous carcinoma.
Recently, the Cancer Genome Atlas (TCGA) has performed the methylome profiles in a relatively large number of ovarian carcinoma but the consortium only focuses on primary high-grade serous carcinomas. Accordingly, recurrent high-grade serous carcinoma, low-grade serous carcinoma, SBT and benign serous cystadenomas were not analyzed. In this study, we applied the Illumina bead array to profile DNA methylation and compare their patterns in high-grade serous carcinoma, LG serous carcinoma, SBT, and benign cystadenoma together with normal stromal cells. Our results provide new evidence demonstrating that low-grade serous carcinoma is epigenetically distinct from high-grade serous carcinoma and is more closely associated to SBT and serous cystadenoma providing, further support to the dualistic model of ovarian serous carcinogenesis20. Therefore, the current study complements the TCGA database and offers a unique opportunity to investigate the biological significance of gene methylation in the pathogenesis of different types of ovarian serous neoplasms.
Materials and Methods
Tissue samples
Eighty-one samples including 6 normal tissues (tumor adjacent stromal cells), 12 serous cystadenomas, 9 SBTs, 8 LG serous carcinomas and 46 HG serous carcinomas (26 primary tumors, 13 recurrent tumors and 7 specimens collected from primary ascites) were obtained from the Johns Hopkins Hospital except the tumor ascites which were obtained from the Norwegian Radium Hospital. The HG serous carcinomas were all advanced stage (IIIC and IV). Samples were collected after appropriate review board approval according to the institutional guidelines. For LG and HG serous tumors, the tumor cells were enriched by Epi-CAM-conjugated magnetic beads using the method previously described21. The epithelial cells from SBT and cystadenoma were isolated by carefully scrapping the tumor surface on fresh tissues.
DNA preparation and methylation beads array
Genomic DNA was extracted using a Qiagen DNA extraction kit. Fully methylated DNA was prepared by treating genomic DNA with the SssI methylase (New England Biolabs) as the positive control. DNA prepared by whole genomic amplification was used as the unmethylated DNA control. Bisulfite conversion was performed using EZ DNA Methylation Kit from Zymo Research (Orange, CA) and the Illumina Golden Gate beads arrays which contained probes for 1505 CpG sites were used to detect methylation profiles in different types of ovarian serous neoplasms. The methylation level was determined by the fluorescence intensity of methylated versus unmethylated alleles.
Clustering analysis
For unsupervised sample clustering, the methylation data was normalized or standardized for each CpG locus to mean and one standard deviation across all samples. The hierarchical sample clustering method was carried out based on all CpG loci using the Hierarchical Clustering Explorer tool22, where the Pearson correlation coefficient was chosen as the distance metric and the clusters were merged using the average linkage criterion.
Comparative analysis
Significant Analysis of Microarrays (SAM) analysis23 was implemented on two groups of methylation data for identifying the statistically significant hypermethylated or hypomethylated loci. In this study a threshold cutoff of q-value, 0.05, was used to define the number of significant loci. To assess the alterations in the overall methylation pattern among different clusters of tumor groups, we calculated the number of genes showing hypomethylation and hypermethylation in each sample. This gene count was based on the p-value< 0.05 in an individual gene as compared to the group of normal cells. To identify the differentially methylated genes between recurrent HG and primary HG serous carcinoma and between LG serous carcinoma and SBT, we used a supervised method to group samples under specific diagnosis because unsupervised clustering failed to distinguish them. For each comparison, we carried out a significance test and selected the genes with q value< 0.05 using SAM. For each comparison, based on the genes previously selected, we tested the methylation level and calculated the mean and standard deviation of the normal samples for each selected gene. Then we used a threshold of1.65*standard deviation which was equivalent to the p-value= 0.05 in one tailed test. For each sample, we define those genes with a z-score larger than +1.65 as hypermethylated and those with z-score smaller than −1.65 as hypomethylated. Finally, the genes, which were significantly hypomethylated or hypermethylated, were counted for each sample.
Results
Unsupervised hierarchical clustering was performed to determine the similarity in methylation patterns among all the samples (Fig. 1). The results demonstrated that the majority of serous cystadenomas, SBTs and LG serous carcinomas clustered under a major branch while the normal tissues clustered in another distinct group. The majority of HG serous carcinomas, including primary and recurrent tumors as well tumor ascites, clustered into two separate branches and one additional minor branch embedded within the normal tissue branch. The morphological features were similar in HG serous carcinomas in these different groups. Only two of 8 LG serous carcinomas, and 3 of 9 SBTs did not fall within the cystadenoma/SBT/LG carcinoma group, whereas the remaining 24 (83%) of 29 cystadenoma/SBT/LG carcinoma samples clustered into one group. For HG serous carcinoma, only one specimen did not fall into one of the three main HG carcinoma clusters whereas the remaining 45 (98%) of 46 HG serous carcinomas grouped into three clusters (Fig. 1). Interestingly, the HG serous carcinoma that was “misclassified” into the cystadenoma/SBT/LG carcinoma group developed from a pre-existing LG serous carcinoma.
Figure 1. Unsupervised clustering analysis in all 81 tumor serous neoplasms and normal cells.
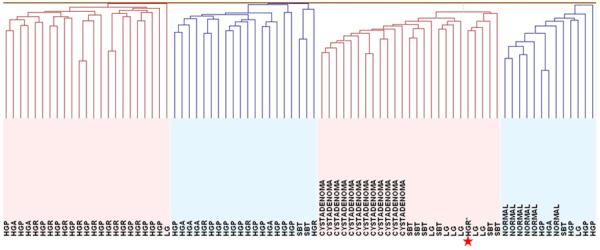
The majority of serous cystadenomas, serous borderline tumors (SBTs) and low-grade serous carcinomas (LG) were clustered under a major branch while the normal tissues were clustered in another distinct group. The majority of high-grade serous carcinomas including primary (HGP), recurrent (HGR) and ascites (HGA) tumors were clustered into two separate branches and one additional minor branch embedded within the normal tissue branch. HGR* represents the high-grade carcinoma that is associated with a prior LG serous carcinoma.
The overall methylation pattern demonstrated that more genes were hypermethylated in the cystadenoma/SBT/LG serous carcinoma group than in the HG serous carcinoma group (p< 0.0001) (Fig. 2). There was a statistically significant increase in the number of hypermethylated genes in serous cystadenoma as compared to SBT and LG serous carcinoma. The number of hypermethylated genes was similar between SBT and LG serous carcinoma (p= 0.89) but both had more hypermethylated genes than HG serous carcinoma (p= 0.0001). There were a number of genes showing aberrant methylation in HG serous carcinomas (both primary and recurrent tumors) as compared to other samples (Fig. 2). Based on SAM analysis, we identified 397 genes with significant differential methylation between high-grade serous carcinoma and other types of serous tumors and normal stromal cells (q<0.05). The 5 top differentially hypermethylated genes in high-grade serous carcinomas included GFAP, TAL1, IPF1, AREG, HOXA9 and the top 5 hypomethylated genes included SLC6A8, BGN, TIMP1, WEE1, and NCL (supplemental Table 1). SAM analysis was also used to identify genes with differential methylation between HG and LG serous carcinomas and there were 21 genes and 4 genes that were hypomethylated and hypermethylated in high-grade serous carcinomas (q<0.05; Fig. 3). Those genes were listed in the supplemental Table 2.
Figure 2. Supervised analysis of genes that separate HG serous carcinomas from the other groups.
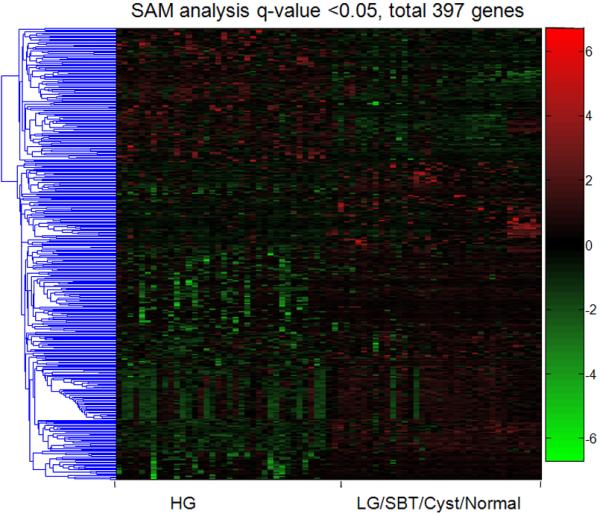
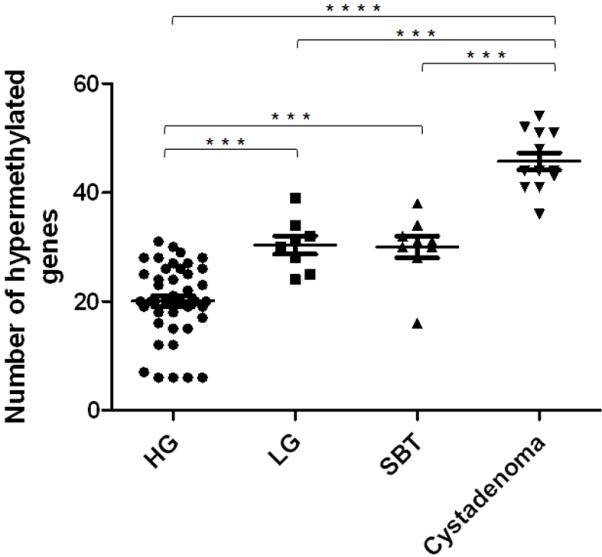
A: As compared to other groups, HG serous carcinomas contained 279 hypermethylated genes and 118 hypomethylated genes based on SAM q-value <0.05. Each column represents an individual specimen. The pseudocolor gradient in the heat map indicates the methylation levels from high (red) to low (blue). B: Further analysis demonstrates the number of hypermethylated genes in each tumor group. Overall, the number of hypermethylated gene in the cystadenoma/SBT/LG serous carcinoma group is larger than the number in the HG serous carcinoma group. The aberrantly methylated genes were defined as the comparison to the mean of normal stromal cells. The p values were calculated using Mann-Whitney test. Horizontal bars: medium ± one standard error. **** p<0.0001; *** p<0.001
Figure 3. Supervised analysis of genes that distinguish HG serous carcinomas from LG serous carcinomas.
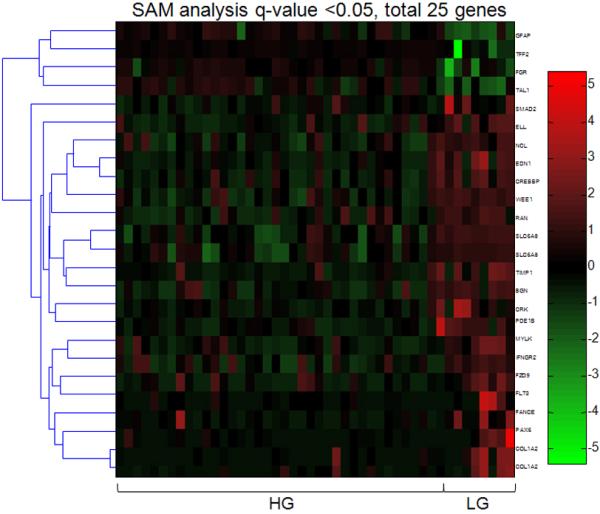
Using a SAM q-value <0.05, there are 21 hypermethylated and 4 hypomethylated genes in HG serous carcinoma as compared to LG serous carcinoma. Each column represents an individual specimen and the genes are listed in the right column.
Although unsupervised clustering and SAM analyses failed to separate primary and recurrent high-grade serous carcinomas based on global methylation profiles, a Student's t test was used to identify genes that were differentially methylated between these two groups. As expected, only a small number of genes demonstrated different methylation patterns (Fig. 4). Those genes were listed in the supplemental Table 3. Similarly, unsupervised clustering and SAM analyses did not distinguish low-grade serous carcinoma from SBT; therefore, a supervised analysis based on Student's t test was used to identify genes with different methylation patterns between low-grade serous carcinoma and SBT in an effort to characterize the molecular mechanisms underlying tumor progression from SBT to low-grade serous carcinoma. As expected, there were only a few differentially methylated genes detected between those two tumor types (Fig. 5). These genes represented only 44 (3%) of 1505 genes analyzed. As compared to SBT, the hypermethylated genes in low-grade serous carcinoma included MAPK4, HOXA9, AATK, WNT5A, GFI1 and the hypomethylated genes in low-grade serous carcinoma included TUBB3, TSG101, HDAC6, DBC1 and GPATC3 (supplemental Table 4).
Figure 4. Supervised analysis of genes that are differentially methylated in recurrent and primary HG serous carcinomas.
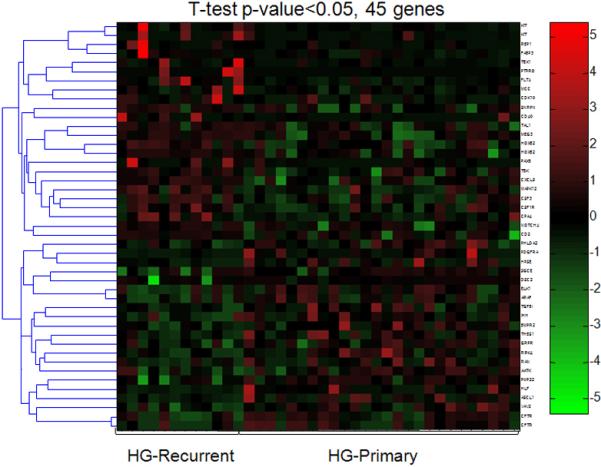
Using a Student's t test with p-value <0.05, there are 21 hypermethylated and 24 hypomethylated genes in primary carcinoma as compared to recurrent carcinoma. Each column represents an individual specimen and the genes are listed in the right column. The pseudocolor gradient in the heat map indicates the methylation levels from high (red) to low (blue).
Figure 5. Supervised analysis of genes that distinguish LG serous carcinomas from SBT.
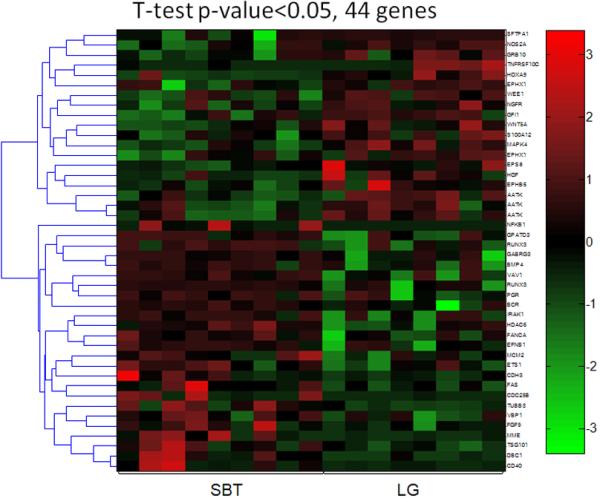
Using a Student's t test with p-value <0.05, there are 19 hypermethylated and 25 hypomethylated genes in LG serous carcinoma as compared to SBT. Each column represents an individual specimen and the genes are listed in the right column. The pseudocolor gradient in the heat map indicates the methylation levels from high (red) to low (blue).
Comment
It has been well established that distinct clinical, histopathological and molecular features characterize different histologic types of ovarian neoplasms24. Among them, serous carcinoma is the most common type, accounting for more than half of ovarian cancers. Previous clinicopathological and molecular studies have shown that ovarian serous carcinoma is composed of HG (conventional) and LG serous carcinoma20, 25, 26. LG carcinomas appear to develop in a stepwise fashion from SBTs as histological transitions can be found between them and SBTs and LG serous carcinomas share similar molecular genetic changes27, 28. These tumors are indolent and have a better prognosis than HG serous carcinomas. Molecular genetic and gene expression analyses have demonstrated that SBT/LG serous carcinomas develop along a pathway that is distinctly different from HG serous carcinoma. SBTs/LG carcinomas typically display sequence mutations in KRAS/BRAF/ERBB2, but rarely harbor TP53 mutations. In contrast, HG serous carcinomas are highly aggressive, rarely harbor mutations in KRAS/BRAF/ERBB2 but have a high frequency of TP53 mutations. The chromosomal instability level as reflected by DNA copy number changes is significantly higher in HG compared to LG serous carcinoma29.
As previously discussed, molecular genetics and gene expression have been studied in different types of ovarian serous neoplasms but their DNA methylation profiles have not yet been fully characterized. In this report, we provide new epigenetic evidence that LG and HG serous carcinomas have distinct DNA methylation patterns and that unlike HG serous carcinoma the methylation profile of LG serous carcinoma is closely related to SBT and serous cystadenoma. The differential clustering of HG and LG/SBT serous tumor based on methylation profiling provides further support for the dualistic model of serous carcinogenesis. Specifically, LG and HG serous carcinomas are characterized by distinct molecular genetic changes as we previously have described27, 28, 30. The observation that an unsupervised method fails to separate LG serous carcinoma from SBT and serous cystadenoma based on their global DNA methylation patterns is consistent with the view that STB and LG serous carcinoma are closely associated25. In a previous study we suggested that SBTs develop from serous cystadenomas based on mutational analysis of small SBTs associated with serous cystadenomas31. In the current study, we found that SBTs and serous cystadenomas have a very similar DNA methylation profile. Therefore, the molecular genetic and epigenetic studies strongly suggest that intra-cystic SBTs develop from serous cystadenomas.
Although the unsupervised clustering analysis clearly separates LG and HG serous tumors, there are a few outliers which are “misclassified” with other tumor types but interestingly, reviewing their clinicopathological data resolves this apparent discrepancy. For example, the HG serous carcinoma that was “misclassified” into the cystadenoma/SBT/LG carcinoma group was from a patient who had a previous diagnosis of LG serous carcinoma. Thus, this HG carcinoma, unlike the other HG serous carcinomas in this study, very likely progressed from the previous LG serous carcinoma, a rare event in serous carcinogenesis in our experience32. Accordingly, it appears that when a HG serous carcinoma develops from a LG serous carcinoma, it retains a DNA methylation profile similar to the LG carcinoma; however, more cases like these need to be analyzed to confirm this conclusion. Another intriguing finding was that HG serous carcinomas cluster into 3 different groups. The diagnosis of HG serous carcinoma is based on morphological features and a wide spectrum of neoplasms fall within that category. The methylation data showing three different clusters for HG serous carcinoma suggests that there may be molecularly distinct tumors in this category which cannot be distinguished by their morphological features. Another interesting finding was that progression from primary to recurrent disease is not associated with a change in the global methylation pattern as SAM analysis did not separate primary and recurrent high-grade serous carcinomas.
This study also provides new clues about how aberrant promoter methylation participates in the pathogenesis of ovarian serous neoplasms. For example, one of the most frequently hypomethylated genes in HG serous carcinoma is WEE1 encoding a tyrosine kinase that regulates the G2/M cell cycle transition and is involved in DNA damage repair. In ovarian cancer, WEE1 expression prevents cancer cells from undergoing apoptosis in response to DNA damage, contributing to a high level of chromosomal instability in HG serous carcinoma33. Based on the supervised analysis, we identified several genes whose methylation patterns are associated with the development of recurrent HG serous carcinoma. Interestingly, several of them have been shown to play a role in human neoplastic diseases. For example, hypermethylation in the promoters of FLT4 and KIT and hypomethylation in the promoters of TGF-β1, gastrin-releasing peptide receptor occur more frequently in recurrent than primary untreated HG serous carcinomas. Similarly, hypermethylation in the promoters of genes including BMP4, TSG101 and FAS were observed more frequently in LG serous carcinoma than in SBT while hypermethylation in the promoter of WEE134 occurred more frequently in SBT than in LG serous carcinoma. Accordingly, these differentially methylated genes deserve further investigation to determine their biological roles in the development of recurrent HG serous carcinoma and in the progression from SBT to LG serous carcinoma.
In summary, the current study is the first comprehensive analysis of DNA methylation in all types of ovarian serous neoplasms. Our findings indicate that the global methylation profile of LG serous carcinomas is distinct from HG serous carcinoma and is closely related to SBTs and serous cystadenomas. We also identified several candidate hypomethylated and hypermethylated genes that may be involved in the development of recurrent HG serous carcinoma and in the progression from SBT to LG serous carcinoma. These findings describe a unique epigenetic landscape which complements previous molecular genetic studies and provides new insights into the molecular pathogenesis of serous carcinoma.
Clinical Implications.
Ovarian carcinoma is a heterogeneous group of neoplastic diseases and even in the serous ovarian tumor group, there are two types with unique clinicopathological features: high-grade (conventional) and low-grade serous carcinoma.
The current study provides new molecular evidence that high-grade serous carcinoma is distinct from low-grade serous carcinoma which is closely related to serous borderline tumor and serous cystadenoma.
This data together with previous studies indicate that high-grade and low-grade serous carcinomas are different ovarian tumors. This fact may have implications in studying the pathogenesis of ovarian cancer and serves as the foundation for future clinical-oriented studies on ovarian serous neoplasms.
Supplementary Material
Acknowledgement
This study is supported by NIH/NCI grants CA129080 and CA103937.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Herman Jg, Baylin Sb. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–54. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- 2.Baylin Sb, Ohm Je. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107–16. doi: 10.1038/nrc1799. [DOI] [PubMed] [Google Scholar]
- 3.Satoh Y, Nakagawachi T, Nakadate H, et al. Significant reduction of WT1 gene expression, possibly due to epigenetic alteration in Wilms' tumor. J Biochem. 2003;133:303–8. doi: 10.1093/jb/mvg041. [DOI] [PubMed] [Google Scholar]
- 4.Kaneuchi M, Sasaki M, Tanaka Y, et al. WT1 and WT1-AS genes are inactivated by promoter methylation in ovarian clear cell adenocarcinoma. Cancer. 2005;104:1924–30. doi: 10.1002/cncr.21397. [DOI] [PubMed] [Google Scholar]
- 5.Li Y, Hu W, Shen Dy, Kavanagh Jj, Fu S. Azacitidine enhances sensitivity of platinum-resistant ovarian cancer cells to carboplatin through induction of apoptosis. Am J Obstet Gynecol. 2009;200:177, e1–9. doi: 10.1016/j.ajog.2008.08.030. [DOI] [PubMed] [Google Scholar]
- 6.Wei Sh, Chen Cm, Strathdee G, et al. Methylation microarray analysis of late-stage ovarian carcinomas distinguishes progression-free survival in patients and identifies candidate epigenetic markers. Clin Cancer Res. 2002;8:2246–52. [PubMed] [Google Scholar]
- 7.Wiley A, Katsaros D, Chen H, et al. Aberrant promoter methylation of multiple genes in malignant ovarian tumors and in ovarian tumors with low malignant potential. Cancer. 2006;107:299–308. doi: 10.1002/cncr.21992. [DOI] [PubMed] [Google Scholar]
- 8.Sellar Gc, Watt Kp, Rabiasz Gj, et al. OPCML at 11q25 is epigenetically inactivated and has tumor-suppressor function in epithelial ovarian cancer. Nat Genet. 2003;34:337–43. doi: 10.1038/ng1183. [DOI] [PubMed] [Google Scholar]
- 9.Choi Yl, Kang Sy, Shin Yk, et al. Aberrant hypermethylation of RASSF1A promoter in ovarian borderline tumors and carcinomas. Virchows Arch. 2006;448:331–6. doi: 10.1007/s00428-005-0091-3. [DOI] [PubMed] [Google Scholar]
- 10.Ibanez De Caceres I, Battagli C, Esteller M, et al. Tumor cell-specific BRCA1 and RASSF1A hypermethylation in serum, plasma, and peritoneal fluid from ovarian cancer patients. Cancer Res. 2004;64:6476–81. doi: 10.1158/0008-5472.CAN-04-1529. [DOI] [PubMed] [Google Scholar]
- 11.Houshdaran S, Hawley S, Palmer C, et al. DNA methylation profiles of ovarian epithelial carcinoma tumors and cell lines. PLoS One. 5:e9359. doi: 10.1371/journal.pone.0009359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng Q, Deftereos G, Hawes Se, et al. DNA hypermethylation, Her-2/neu overexpression and p53 mutations in ovarian carcinoma. Gynecol Oncol. 2008;111:320–9. doi: 10.1016/j.ygyno.2008.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fiegl H, Windbichler G, Mueller-Holzner E, et al. HOXA11 DNA methylation--a novel prognostic biomarker in ovarian cancer. Int J Cancer. 2008;123:725–9. doi: 10.1002/ijc.23563. [DOI] [PubMed] [Google Scholar]
- 14.Chou Jl, Su Hy, Chen Ly, et al. Promoter hypermethylation of FBXO32, a novel TGF-beta/SMAD4 target gene and tumor suppressor, is associated with poor prognosis in human ovarian cancer. Lab Invest. doi: 10.1038/labinvest.2009.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hoque Mo, Begum S, Brait M, et al. Tissue inhibitor of metalloproteinases-3 promoter methylation is an independent prognostic factor for bladder cancer. J Urol. 2008;179:743–7. doi: 10.1016/j.juro.2007.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mandelker Dl, Yamashita K, Tokumaru Y, et al. PGP9.5 promoter methylation is an independent prognostic factor for esophageal squamous cell carcinoma. Cancer Res. 2005;65:4963–8. doi: 10.1158/0008-5472.CAN-04-3923. [DOI] [PubMed] [Google Scholar]
- 17.Wick W, Hartmann C, Engel C, et al. NOA-04 randomized phase III trial of sequential radiochemotherapy of anaplastic glioma with procarbazine, lomustine, and vincristine or temozolomide. J Clin Oncol. 2009;27:5874–80. doi: 10.1200/JCO.2009.23.6497. [DOI] [PubMed] [Google Scholar]
- 18.Ye Y, Mcdevitt Ma, Guo M, et al. Progressive chromatin repression and promoter methylation of CTNNA1 associated with advanced myeloid malignancies. Cancer Res. 2009;69:8482–90. doi: 10.1158/0008-5472.CAN-09-1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barton Ca, Hacker Nf, Clark Sj, O'brien Pm. DNA methylation changes in ovarian cancer: implications for early diagnosis, prognosis and treatment. Gynecol Oncol. 2008;109:129–39. doi: 10.1016/j.ygyno.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 20.Shih I-M, Kurman Rj. Ovarian tumorigenesis- a proposed model based on morphological and molecular genetic analysis. Am J Pathol. 2004;164:1511–1518. doi: 10.1016/s0002-9440(10)63708-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shih Ie M, Wang Tl. Apply innovative technologies to explore cancer genome. Curr Opin Oncol. 2005;17:33–8. doi: 10.1097/01.cco.0000147382.97085.e4. [DOI] [PubMed] [Google Scholar]
- 22.Seo J, Shneiderman B. Interactively exploring hierachical clustering results. IEEE Computer. 2002;35:80–86. [Google Scholar]
- 23.Tusher Vg, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001;98:5116–21. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cho Kr, Shih Im. Ovarian cancer. Annu Rev Pathol Mech Dis. 2009;4:287–313. doi: 10.1146/annurev.pathol.4.110807.092246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shih Ie M, Kurman Rj. Molecular pathogenesis of ovarian borderline tumors: new insights and old challenges. Clin Cancer Res. 2005;11:7273–9. doi: 10.1158/1078-0432.CCR-05-0755. [DOI] [PubMed] [Google Scholar]
- 26.Kurman Rj, Shih Ie M. Pathogenesis of ovarian cancer: lessons from morphology and molecular biology and their clinical implications. Int J Gynecol Pathol. 2008;27:151–60. doi: 10.1097/PGP.0b013e318161e4f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Singer G, Kurman Rj, Chang H-W, Cho Skr, Shih I-M. Diverse tumorigenic pathways in ovarian serous carcinoma. Am J Pathol. 2002;160:1223–1228. doi: 10.1016/s0002-9440(10)62549-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Singer G, Oldt R, 3rd, Cohen Y, et al. Mutations in BRAF and KRAS characterize the development of low-grade ovarian serous carcinoma. J Natl Cancer Inst. 2003;95:484–6. doi: 10.1093/jnci/95.6.484. [DOI] [PubMed] [Google Scholar]
- 29.Kuo K, Mao T, Feng Y, et al. DNA copy number profiles in affinity-purified ovarian clear cell carcinoma. Clin Cancer Res. 2010 doi: 10.1158/1078-0432.CCR-09-2105. April 1 2010 issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuo Kt, Guan B, Feng Y, et al. Analysis of DNA Copy Number Alterations in Ovarian Serous Tumors Identifies New Molecular Genetic Changes in Low-Grade and High-Grade Carcinomas. Cancer Res. 2009;69:4036–4042. doi: 10.1158/0008-5472.CAN-08-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ho C-L, Kurman Rj, Dehari R, Wang T-L, Shih I-M. Mutations of BRAF and KRAS precede the development of ovarian serous borderline tumors. Cancer Res. 2004;64:6915–6918. doi: 10.1158/0008-5472.CAN-04-2067. [DOI] [PubMed] [Google Scholar]
- 32.Dehari R, Kurman Rj, Logani S, Shih Im. The Development of High-grade Serous Carcinoma From Atypical Proliferative (Borderline) Serous Tumors and Low-grade Micropapillary Serous Carcinoma: A Morphologic and Molecular Genetic Analysis. Am J Surg Pathol. 2007;31:1007–1012. doi: 10.1097/PAS.0b013e31802cbbe9. [DOI] [PubMed] [Google Scholar]
- 33.Wang F, Zhu Y, Huang Y, et al. Transcriptional repression of WEE1 by Kruppel-like factor 2 is involved in DNA damage-induced apoptosis. Oncogene. 2005;24:3875–85. doi: 10.1038/sj.onc.1208546. [DOI] [PubMed] [Google Scholar]
- 34.Yoshida T, Tanaka S, Mogi A, Shitara Y, Kuwano H. The clinical significance of Cyclin B1 and Wee1 expression in non-small-cell lung cancer. Ann Oncol. 2004;15:252–6. doi: 10.1093/annonc/mdh073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.