

Abstract
Knowledge of the biologically relevant components of human tissues has enabled the invention of numerous clinically useful diagnostic tests, as well as non-invasive ways of monitoring disease and its response to treatment. Recent use of advanced MS-based proteomics revealed that the composition of human urine is more complex than anticipated. Here, we extend the current characterization of the human urinary proteome by extensively fractionating urine using ultra-centrifugation, gel electrophoresis, ion exchange and reverse-phase chromatography, effectively reducing mixture complexity while minimizing loss of material. By using high-accuracy mass measurements of the linear ion trap-Orbitrap mass spectrometer and LC-MS/MS of peptides generated from such extensively fractionated specimens, we identified 2362 proteins in routinely collected individual urine specimens, including more than 1000 proteins not described in previous studies. Many of these are biomedically significant molecules, including glomerularly filtered cytokines and shed cell surface molecules, as well as renally and urogenitally produced transporters and structural proteins. Annotation of the identified proteome reveals distinct patterns of enrichment, consistent with previously described specific physiologic mechanisms, including 336 proteins that appear to be expressed by a variety of distal organs and glomerularly filtered from serum. Comparison of the proteomes identified from 12 individual specimens revealed a subset of generally invariant proteins, as well as individually variable ones, suggesting that our approach may be used to study individual differences in age, physiologic state and clinical condition. Consistent with this, annotation of the identified proteome by using machine learning and text mining exposed possible associations with 27 common and more than 500 rare human diseases, establishing a widely useful resource for the study of human pathophysiology and biomarker discovery.
Keywords: Bioinformatics, Biomarker, Proteome profilling
1 Introduction
Knowledge of the biologically relevant components of human tissues has enabled the invention of numerous clinically useful diagnostic tests, as well as non-invasive ways of monitoring disease and its response to treatment. By virtue of tissue perfusion, blood serum is the most useful material for the discovery of such biomarkers in general. However, the relatively high concentration of serum proteins, as well as their wide range of concentrations, spanning at least nine orders of magnitude, often limit the study of serum biomarkers [1], though several recent approaches are promising [2–4].
On the other hand, of the biological fluids amenable to routine clinical evaluation, urine has the advantage of being frequently and non-invasively available, abundant, and as a result of being a filtrate of serum, relatively simple in its composition. Consequently, detection of urinary proteins has been used to identify markers of disease affecting the kidney and the urogenital tract [5, 6], as well as distal organs such as the brain and the intestine [7, 8]. However, our understanding of the human urinary proteome is incomplete, specifically with respect to its overall composition and dynamics, not to mention the identity of variable components that may be dependent on physiologic state and disease.
Several approaches have been used to characterize the human urinary proteome. Initial studies using electrophoresis and immunoblotting were able to identify tens of abundant and rare urinary proteins [9]. Recently, Pisitkun et al., [10] applied ultracentrifugation and LC-MS/MS to identify 295 highly abundant unique proteins isolated from urinary exosomes, recently expanded to identify more than 1000 proteins [11]. Sun et al., [12] identified 226 soluble proteins by using multidimensional LC-MS/MS. For an overview, see Pisitkun et al. [13]. Recently, Adachi et al., [14] identified more than 1500 unique proteins from ultrafiltered urine with a high degree of accuracy by using a hybrid linear ion trap-Orbitrap (LTQ-Orbitrap) mass spectrometer. Finally, by using capillary electrophoresis coupled to MS, several thousand peptides were recently detected using a platform designed for clinical peptidomic assessments [15, 16], including a variety of clinical conditions, reviewed in [17].
Here, we extend the current characterization of the human urinary proteome by extensively fractionating urine using ultracentrifugation, gel electrophoresis, ion exchange and reverse-phase chromatography, effectively reducing mixture complexity while minimizing loss of material. By using high-accuracy mass measurements of the LTQ-Orbitrap mass spectrometer and LC-MS/MS of peptides generated from such extensively fractionated specimens, we identified over 2000 unique proteins in routinely collected individual urine specimens. We provide assessments of the physical and tissue origins of the urinary proteome, as well as dependence of its detection on experimental and individual variables. Usage of this approach in a separate study of urinary markers of acute appendicitis allowed the discovery and validation of several novel markers with superior diagnostic performance, including those enriched in diseased appendices and filtered from serum. Finally, by using text mining and machine learning, we annotate the observed urinary proteome with respect to 27 common and more than 500 rare human diseases, thereby establishing a widely useful resource for the study of human pathophysiology and biomarker discovery.
2 Materials and methods
2.1 Sample collection
Urine was collected as clean catch, mid-stream specimens as part of routine evaluation of 12 children and young adults (ages 1–18 years) presenting with acute abdominal pain in the Children's Hospital Boston's Emergency Department. Upon obtaining informed consent, urine was frozen at −80°C in 12 mL aliquots in polyethylene tubes. Because ultimately we sought to identify medically useful urinary proteins, we obtained urine as routinely collected clean catch, mid-stream urine specimens, collected at the time of clinical evaluation. We examined urines of 12 children and young adults, including three healthy and asymptomatic controls, six patients with acute appendicitis and patients who were evaluated for abdominal pain without evidence of appendicitis. Complete demographic information about these patients is part of the accompanying manuscript that sought to identify urine markers of appendicitis [18]. All urines exhibited normal profiles without evidence of renal disease or infection, as assessed by using clinical urinalysis (data not shown). All urine specimens were frozen within 6h of collection, consistent with earlier temporal analysis of whole urine specimens which indicated that no detectable degradation occurred for as long as 24 h of 4°C refrigerated storage with subsequent freezing at −80°C [19–21]. This is expected given the fact that urine is stored in situ for many hours in the bladder, reaching a physiologic equilibrium prior to collection.
2.2 Reagents
All reagents were of highest purity available and purchased from Sigma Aldrich unless specified otherwise. HPLC-grade solvents were purchased from Burdick and Jackson.
2.3 Urine sedimentation
Aliquots were thawed and centrifuged at 17 000×g for 15 min at 10°C to sediment cellular fragments [10]. Absence of intact cells in the sediment was confirmed by light microscopy (data not shown). Subsequently, supernatant was centrifuged at 210 000×g for 60 min at 4°C to sediment vesicles and high-molecular-weight complexes. Resultant pellets were resuspended in 0.5mL of 0.1×Laemmli buffer, concentrated 10-fold to 0.05mL by vacuum centrifugation and stored at −80°C.
2.4 Cation exchange chromatography
Supernatant remaining after ultracentrifugation was diluted fivefold with 0.1M acetic acid, 10% v/v methanol, pH 2.7 (Buffer A) and incubated with 1mL 50% v/v slurry of SP Sephadex (40–120 μm beads, Amersham) for 30 min at 4°C to adsorb peptides that are <30 kDa molecular weight. Upon washing the beads twice with Buffer A, peptides were eluted by incubating the beads in 5 mL of 0.5 M ammonium acetate, 10% v/v methanol, pH 7, for 30 min at 4°C. Eluted peptides were purified by reverse-phase chromatography by using PepClean C-18 spin columns, according to manufacturer's instructions (Pierce). Residual purification solvents were removed by vacuum centrifugation and small proteins and peptides were resuspended in aqueous 50mM ammonium bicarbonate buffer (pH 8.5).
2.5 Protein precipitation
Proteins remaining in solution after cation exchange were precipitated by adding trichloroacetic acid (TCA) to 20% w/v, with deoxycholate to 0.02% w/v and Triton X-100 to 2.5% v/v as carriers, and incubating the samples for 16 h at 4°C. Precipitates were sedimented at 10 000×g for 15 min at 4°C and pellets were washed twice with neat acetone at 4°C with residual acetone removed by air drying. Dried pellets were resuspended in 0.1mL of 1×Laemmli buffer.
2.6 Gel electrophoresis
Laemmli buffer suspended fractions (from 17 000×g and 210 000×g centrifugation, and from protein precipitation) were incubated at 70°C for 15 min and separated by using NuPage 10% polyacrylamide bis-Tris gels according to manufacturer's instructions (Invitrogen). Gels were washed three times with distilled water, fixed with 5% v/v acetic acid in 50% v/v aqueous methanol for 15 min at room temperature, and stained with Coomassie. Each gel lane was cut into six fragments and each fragment was cut into roughly 1mm3 particles, which were subsequently washed three times with water and once with ACN.
2.7 Protein reduction, alkylation and trypsinization
Protein containing gel particles and cation exchange-purified proteins were reduced with 10mM dithiotreitol in 50mM ammonium bicarbonate (pH 8.5) at 56°C for 45min. They were subsequently alkylated with 55mM iodoacetamide in 50mM ammonium bicarbonate (pH 8.5) at room temperature in darkness for 30 min. Gel particles were washed three times with 50mM ammonium bicarbonate (pH 8.5) prior to digestion. Alkylated peptides were purified by using PepClean C-18 spin columns, as described in Section 2.4, to remove residual iodoacetamide from the cation exchange fraction. They were then digested with 12.5 ng/μL sequencing grade bovine trypsin in 50mM ammonium bicarbonate (pH 8.5) at 37°C for 16h. Tryptic products were purified by using PepClean C-18 spin columns as described in Section 2.4, vacuum centrifuged and stored at −80°C.
2.8 LC and MS
Fractions containing tryptic peptides dissolved in aqueous 5% v/v ACN and 0.1% v/v formic acid were resolved and ionized by using nanoflow HPLC (nanoLC, Eksigent) coupled to the LTQ-Orbitrap hybrid mass spectrometer (Thermo Scientific). Nanoflow chromatography and electrospray ionization were accomplished by using a 15 cm fused silica capillary with 100μm inner diameter, in-house packed with Magic C18 resin (200Å, 5μm, Michrom Bio-resources). Peptide mixtures were injected onto the column at a flow rate of 1000 nL/min and resolved at 400 nL/min using 45 min linear ACN gradients from 5 to 40% v/v aqueous ACN in 0.1% v/v formic acid. Mass spectrometer was operated in data-dependent acquisition mode, recording high-accuracy and high-resolution survey Orbitrap spectra using the lock mass for internal mass calibration, with the resolution of 60 000 and m/z range of 350–2000. Six most intense multiply charged ions were sequentially fragmented by using collision induced dissociation, and spectra of their fragments were recorded in the linear ion trap; all precursors selected for dissociation were dynamically excluded for 60 s.
2.9 Spectral processing and peptide identification
Custom-written software was used to extract the 200 most intense peaks from each MS/MS spectrum and to generate mascot generic format files. Peak lists were searched against the human International Protein Index database (version 3.36, http://www.ebi.ac.uk/IPI) by using MASCOT (version 2.1.04; Matrix Science), allowing for variable formation of N-pyroglutamate for N-terminal Gln, Asn and Gln deamidation, N-terminal N-acetylation and methionine oxidation, requiring full trypsin cleavage of identified peptides with two possible miscleavages, and mass tolerances of 5ppm and 0.8Da for the precursor and fragment ions, respectively. Searches allowing semi-tryptic peptides did not affect overall search yields (data not shown). Spectral counts were calculated by summing the number of fragment ion spectra assigned to each unique precursor peptide.
2.10 Data analysis
Assessment of identification accuracy was carried out by searching a decoy database composed of reversed protein sequences of the target IPI database. Frequency of apparent false-positive identifications was calculated by merging individual target and decoy searches for each sample. An initial estimate of the apparent false-positive rate was obtained by dividing the number of peptide identifications with a MASCOT score greater than the identity score obtained from the target search by the number of peptide identifications with a score higher than the identity score threshold extracted from the decoy search [22]. Only proteins identified on the basis of two or more peptides were included in the comparison. Parsimonious protein grouping was performed by remapping all peptide identifications onto their corresponding proteins as listed in the IPI. This step was necessary to generate a minimal, non-redundant list of proteins that explain all of the identified peptides, while excluding proteins that could not be unambiguously unidentified. This parsimonious list of proteins was used for comparisons of various samples at the protein level. For Gene Ontology (GO) annotation, we used GO slim terms version 1.8, accessed by using GOfact (http://www.hupo.org.cn/GOfact). For annotation of tissue expression of detected proteins, we used version 2 of the GNF gene expression atlas (http://expression.gnf.org), accessed by using BioMart (http://www.biomart.org).
2.11 Disease annotations
We linked proteins found in the urine proteome to published articles that associate a protein with a human disease, as well articles that associate a disease with a protein. For the former, we derived sets of diseases from OMIM (Online Mendelian Inheritance in Man) [23], MeSH (http://www.nlm.nih.gov/mesh/), and a short list of common diseases of interest not described in OMIM or MeSH (Additional Files, http://www.childrenshospital.org/research/steenlab). We extracted disease names from MeSH by selecting MeSH concepts with DescriptorRecord DescriptorClass=1, and marked by Semantic-TypeName “Disease or Syndrome”. Synonym disease names were obtained from the content of Term or TermList elements for the main concept. For OMIM, documents matching an OMIM entry were obtained by searching Medline with a query of the form (Term1 OR Term2… Termk), where Termk include the 100 lowest frequency terms in a given OMIM entry. These OMIM disease queries were executed by using Twease with the BM25EC scorer against abstracts in Medline [24], accessed July 7, 2008. Documents that matched the query with a BM25EC score above a Z-score of 10 were considered matching the OMIM disease [25]. Each MeSH disease name and synonyms were expressed as a query of the form (“disease name” | ”alias 1” | “alias 2” |…). Common disease names were expressed as a single phrase query.
To determine diseases that are associated with a given protein, we queried BioMart by using IPI identifiers for proteins in the urine proteome to obtain corresponding protein descriptions and gene names. Queries of the form (IPI-id | “description” | GeneName) were generated for each protein, where IPI-id is the IPI identifier, and description is the description phrase retrieved from BioMart. These queries were run against Medline by using Twease with the slider parameter set to 0. Lists of documents matching protein names were stored and overlapped with lists of documents matching diseases. Pairs of disease-associated proteins that matched less than five documents were discarded (manual examination indicated that this level of overlap frequently happens as an artifact of the search procedure). To further increase stringency of the protein disease literature associations, we estimated the odds that the number of overlapping documents found between a given disease and protein could occur by chance, considering the number of documents matching either the disease or the proteins in Medline. Only protein name/disease name pairs with odds ratio greater than 2000 were reported. Lists of overlapping documents were formatted in HTML files organized in hierarchies of diseases or proteins.
3 Results
3.1 Exhaustive protein capture from routinely collected human urine
Urine is a complex mixture with abundant proteins such as albumin and uromodulin obscuring the identification of less concentrated, biologically more informative proteins such as secreted cytokines and hormones, for example. Thus, we adopted a fractionation method that reduced mixture complexity while minimizing loss of material by first ultracentrifugating to fractionate urinary exosomes and other high-molecular-weight complexes from soluble peptides and proteins, subsequently capturing the latter by using size exclusion cation exchange chromatography and TCA precipitation, respectively, which has been shown to capture more than 95% of proteins under similar conditions [26, 27].
Secondary and tertiary fractionations of thus captured proteins and peptides were achieved by using one-dimensional SDS-PAGE of the ultracentrifugation and precipitation fractions, and LC of the tryptic peptides of SDS-PAGE-resolved proteins, respectively. As a result, high-abundance-proteins such as albumin and uromodulin, which would otherwise comprise more than 99% of the mixture, can be separated effectively from the bulk of the proteome (Fig. 1). Though the composition and concentration of urine vary with physiologic state, there was less than 10±10% (mean±standard deviation) difference in total protein abundance among individual specimens, as ascertained by using gel image densitometry (Fig. 1), similar to earlier studies of urine of children [28–30].
Figure 1.
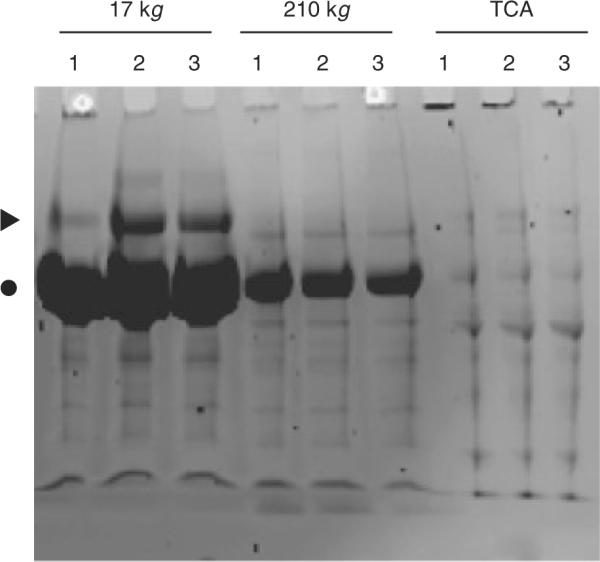
Representative SDS-PAGE separation of 17 000×g, 210 000×g, and TCA fractions of three urine specimens (1, 2, 3), demonstrating small differences in total protein abundance among different urine specimens, and preferential fractionation of albumin (●) and uromodulin (▶) in the 17 000×g fraction, enabling improved detection of the remaining urinary proteins. The majority of albumin and uromodulin appears to sediment at 17 000×g, suggesting that they exist in high-molecular-weight complexes, consistent with uromodulin's ability to polymerize in urine.
3.2 Accurate and comprehensive identification of urinary proteomes
In order to maximize detection sensitivity while minimizing identification errors, we used the recently developed hybrid LTQ-Orbitrap mass spectrometer for tryptic peptide sequencing of the above-fractionated proteomes. A representative set of tandem mass spectra is shown in Fig. 2, achieving mass errors of less than 2ppm for the majority of the LC-MS runs as judged from analysis of trypsin autolysis peptides (Fig. 3). Peptide sequences were identified from tandem mass spectra by using probability-based MASCOT searches of the human IPI database (Section 2). By carrying out simultaneous searches of the data against a decoy database containing reversed protein sequences, and rejecting (false) identifications of spectra that matched decoy sequences, as well as excluding proteins identified on the basis of single peptides, we were able to achieve an apparent false-positive protein identification frequency of less than 1%.
Figure 2.

Representative mass spectra. Relative ion intensity as a function of m/z values of precursor ions (MS, top), with the doubly charged peptide LDITAEILAVR from plunc labeled by arrow, and its fragmentation spectrum with fragment ions labeled as y- and b-series fragment ions (MS/MS, bottom).
Figure 3.

Apparent mass accuracy error of the LTQ-Orbitrap, as assessed by comparison of observed masses of the trypsin autolysis peptide VATVSLPR, as compared with its expected monoisotopic mass, indicating that most peptides have apparent mass errors of less than 2 ppm. Histogram on the bottom; cumulative probability on the top.
As a result, we were able to identify with high degree of accuracy 12 126 unique peptides, corresponding to 2362 proteins; the median number of unique peptides per identified protein was 10. These proteins include 891 proteins identified in an earlier high-accuracy study of the human urine proteome [14], as well as 575 proteins recently identified in human urinary exosomes [11], and more than 1000 additional proteins identified for the first time (Fig. 4). These data, including a list of all identified proteins and peptides, are provided as Additional Files, and can be accessed publicly from our server (http://www.childrenshospital.org/research/steenlab).
Figure 4.
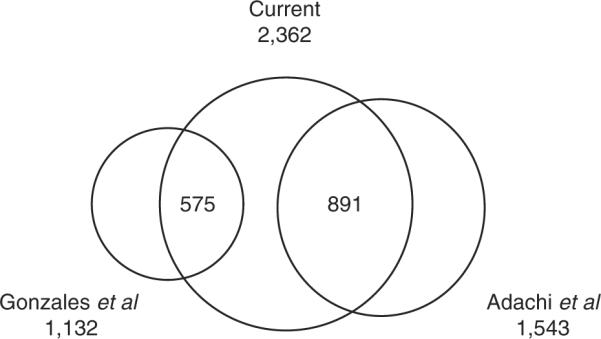
Venn diagram of the comparisons of the observed aggregate urine proteome with those published by Adachi et al. [14], and Gonzales et al. [11], demonstrating high concordance with the previous studies of human urine, as well as discovery of not previously observed proteins.
3.3 Origin of the human urinary proteome
The composition of the identified proteomes was characterized with respect to GO-annotated biological function, apparent physical origin and predicted tissue expression. As compared with the entire list of IPI entries, analysis of GO-annotated biological function revealed saturation of cellular components such as the cytoplasm, endoplasmic reticulum, Golgi, lysosome and the plasma membrane. Proteins from the nucleus were relatively under-represented, consistent with the general absence of intact cells in human urine. Similar to Adachi et al. [14], we observed a relative enrichment of hydrolases, peptidases, carbohydrate and lipid-binding proteins, and a relative under-representation of nucleic acid-binding proteins.
By comparing whether identified proteins sedimented in the 17 000×g versus 210 000×g ultracentrifugation fractions, were adsorbed onto size exclusion ion exchange resin or were TCA precipitated, we defined them as large or small complexes, and soluble peptides or proteins, respectively. The fractions of proteins identified uniquely from these physical states were 14, 20, 3 and 9%, respectively, suggesting that individual proteins or their variants exist in multiple physical states, with the caveat that the apparent physical states are subject to physiologic processes such as proteolysis and aggregation that may occur in situ or during storage. For example, components of the urinary exosomes including the endosomal sorting complex (ESCRT-I), BRO1/ALIX and VPS4, were detected as both small complexes and soluble proteins. Similarly, insulin-like growth factor binding proteins that are low-molecular-weight-circulating hormones were detected as soluble proteins, peptides and in small complexes. Though the size-excluded ion exchange fraction contributed only 3% to the total unique protein identifications, it was substantially enriched for biomedically significant molecules which would not be detected otherwise, including circulating hormones such as hepcidin and chromogranin [31, 32], and shed cell surface molecules such as Ly-6 and platelet glycoproteins [33, 34].
We assessed the probable tissue origin of the identified proteome by comparing it with published tissue expression atlases. As expected, 90% of the proteins detected in the urinary proteome have tissue expression profiles that include organs of the urogenital tract, such as the kidneys and the bladder, from which they likely originate. In addition to these proximal organs, the urinary proteome contains a substantial number of proteins that appear to originate from distal tissues. Among them are 336 proteins that are uniquely expressed in distal tissues such as the nervous system, heart and vasculature, lung, blood and bone marrow, intestine, liver and other intra-abdominal viscera, suggesting that a substantial portion of the urinary proteome is formed as a result of their systemic circulation and serum filtration. For example, the urinary proteome includes angiopoietin-2, involved in angiogenesis and vascular homeostasis, and is expressed by the vascular endothelium [35].
3.4 Individual urinary proteomes
By virtue of studying individual urinary proteomes, we were able to assess the extent of similarities and differences among them. For the 12 specimens studied here, we detected 1124±292 (mean±standard deviation) proteins per individual proteome, with the average concordance of 68%, as calculated over all binary comparisons. Highly abundant proteins, as measured by using spectral counting [36], common to all individual proteomes include molecules involved in renotubular trafficking (uromodulin, cubilin and megalin/LRP2), serum-filtered enzymes and carriers (bikunin/AMBP, aminopeptidase N, ceruloplasmin, apolipoproteins and immunoglobulins), extracellular structural components (perlecan, glial fibrillary acidic proteins), as well as a variety of other secreted molecules such as CD44, tetraspanin and lysosomal-associated membrane proteins. Many of these have been detected in human urine previously, and many were identified for the first time. Examples of the latter include claudin, a regulator of tight junctions involved in the maintenance of glomerular and tubular integrity [37], collectrin, a novel homolog of the angiotensin-converting enzyme-related carboxypeptidease implicated in renal failure and the pathogenesis of polycystic kidney disease [38], SLC5A2, a tubular sodium-glucose transporter which causes autosomal recessive renal glucosuria when defective [39], and numerous other proteins with poorly understood functions such as peflin and trefoil factor 2.
In large part, the variability observed among individual proteomes appears to be multifactorial in origin, as suggested by the multimodal distribution of the coefficients of variation of proteins' apparent detectability, as measured by using spectral counting [36] (Fig. 5; representative proteins are labeled). Proteins with high degree of apparent variability included complement factors, α1-anti-trypsin, protein C inhibitor, galectin (LGALS3BP), CD59, CD14, α-enolase, α2-macroglobulin, gelsolin, haptoglobin, hemopexin, intelectin, fibrinogen, arylsulfatase, serum amyloid A2, cystatin C, angiotensin and resistin, among others. Many of these proteins are components of the acute-phase response [40], consistent with the collection of some of the studied specimens from patients with acute abdominal pain. Other differences among proteomes included components of seminal fluid and other sex-specific proteins such as semenogelin. The investigation of the origin of these and other differences such as age are important directions of future work.
Figure 5.
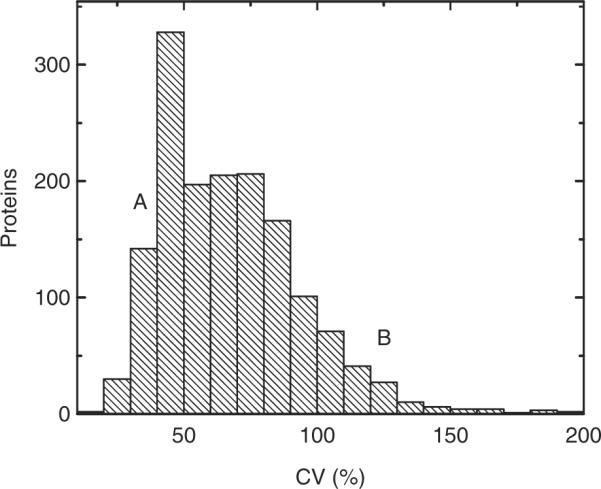
Variability in the composition of individual urine proteomes, as assessed by the coefficients of variation of their proteins' spectral counts, demonstrating a broad distribution, including proteins that are relatively invariant (A: Albumin, cubilin and megalin), and those that appear to vary among individual proteomes (B: α1-anti-trypsin, fibrinogen, α2-macro-globulin).
3.5 Urine proteomics for profiling of human disease
As part of a separate study of acute appendicitis [18], we carried out specific comparisons of the detected urinary proteomes to identify candidate markers of acute appendicitis, insofar as six of the 12 examined proteomes were collected from patients with histologically proven appendicitis. Candidate markers were identified by using relative enrichment ratios, class pattern recognition and comparisons with gene expression profiles of diseased appendices [18]. In all, 57 candidate markers were identified, including several with superior diagnostic performance, such as calgranulin A (S100-A8), α-1-acid glycoprotein 1 (orosomucoid), and leucine-rich α-2-glycoprotein, with the receiver operating curve areas of 0.84 (95% CI 0.72–0.95), 0.84 (0.72–0.95) and 0.97 (0.93–1.0), respectively. In particular, leucine-rich α-2-glycoprotein was enriched in diseased appendices as confirmed by immunohistochemistry and its abundance correlated with severity of appendicitis [18].
Since the identified urinary proteomes may contain proteins previously reported to be associated with other human diseases, we annotated the identified urinary proteins with respect to possible associations with human disease by using machine learning and text mining of Medline abstracts. Annotations identified for the 26 common and more than 200 rare examined diseases are available in hypertext documents (Additional Files, http://www.childrenshospital.org/research/steenlab), with links to information about the identified proteins and original studies about their role in disease. They include common kidney diseases such as nephrotic syndrome (72 proteins) and nephritis (139), systemic illnesses such as sepsis (42), diseases of distal organs such as pneumonia (34), meningitis (22) and colitis (45). In addition, the identified proteins were annotated with respect to more than 500 rare diseases, including storage diseases such as Niemann–Pick disease, immune system disorders such as Wiskott–Aldrich syndrome, and diseases of the nervous system such as spinocerebellar ataxia. If the abundance levels of any of these proteins are affected by their respective diseases, our associations with the urinary proteome may be useful for the development of diagnostic tests or new approaches for the study and monitoring of disease progression.
4 Discussion
Recent advances in proteomics allowed for unprecedented discovery of the composition of human urine, and its application to the study of human physiology and disease. In the current study we extended this work by applying extensive fractionation and protein capture with high-accuracy LTQ-Orbitrap LC-MS/MS to identify 2362 proteins based on two or more unique peptides in routinely collected human urine. Consequently, the identified aggregate urinary proteome, as based on profiling of 12 different specimens, combined with those of previous studies [10–12, 14] [16], constitutes the most comprehensive characterization of human urine with respect to the generally present human urinary proteins (Fig. 4). Differences observed between current and earlier studies are likely due to differences in urine fractionation, as well as individual differences in age, physiologic state, clinical condition and genetic variation, all of which are important directions of future studies.
In agreement with previous studies, we observed a relative enrichment of the urinary proteome with respect to endosomal and lysosomal components, as well as enrichment with respect to soluble enzymes, consistent with the formation of urinary exosomes, and protein secretion and/or filtration, respectively. Likely as a result of extensive fractionation and protein capture, including size exclusion ion exchange chromatography, we were able to identify more than 1000 additional proteins, not detected in previous proteomic studies of urine. Many of these are biomedically significant molecules, including glomerularly filtered circulating hormones such as hepcidin and shed cell molecules such as platelet glycoproteins, and renally produced transporters and structural molecules such as claudin and collectrin, for example. Interestingly, most proteins were identified in multiple physical fractions, such as large or small complexes, and soluble peptides or proteins, suggesting multiple processing mechanisms and/or exchange upon their deposition in urine. Finally, though most of the urinary proteins have corresponding tissue expression profiles that include the urogenital tract and are likely formed by the kidneys or the bladder, a substantial fraction does not. Instead, these 336 proteins appear to be glomerularly filtered from serum and delivered from virtually every tissue, suggesting that urine may be used for studies of a wide variety of human organs and diseases.
In the spirit of this goal, we annotated the aggregate urinary proteome with respect to 27 common and more than 500 rare human diseases. These annotations, available in cross-referenced hypertext documents (Additional Files, http://www.childrenshospital.org/research/steenlab), provide a widely useful resource for the study of human pathophysiology and potential biomarker discovery. For example, platelet activating factor acetylhydrolase is an important mediator of allergy and anaphylaxis, and recently its serum levels were described to be predictive of severity of anaphylaxis [41]. Its detection in routinely collected urine suggests the potential of more accessible and convenient diagnostic tests, such as those that seek to assess disease burden of allergic asthma, for example. Similarly, detection of urinary matrix metalloproteinase 9 (gelatinase B) enables the possibility of its use to study nephritis in urine [42]. Even for rare diseases of distal organs such as the Niemann–Pick disease [43], detection of urinary acid sphingomyelinase may enable the development of screening tests based on much more abundant and easily accessible urine specimens. Usage of advanced MS methods for the study of human urine promises to offer significant insights into human physiology and its application for the diagnosis and treatment of human disease [17].
Acknowledgments
The authors are grateful to Bogdan Budnik, Yin Yin Lin and Zachary Waldon for technical assistance, Samuel Lux and Richard Lee for critical discussions, and to the staff of the Children's Hospital Boston's Emergency Medicine Department for help with specimen collection. Funded in part by the Frederick Lovejoy, Jr., M.D. Housestaff Research and Education grant, and by Children's Hospital Boston Houseofficer Development Award.
Abbreviations
- GO
Gene Ontology
- LTQ
linear ion trap
- OMIM
online Mendelian inheritance in man
- TCA
trichloroacetic acid
Footnotes
The authors have declared no conflict of interest.
5 References
- [1].Anderson NL, Anderson NG. The human plasma proteome: history, character, and diagnostic prospects. Mol. Cell. Proteomics. 2002;1:845–867. doi: 10.1074/mcp.r200007-mcp200. [DOI] [PubMed] [Google Scholar]
- [2].Adkins JN, Varnum SM, Auberry KJ, Moore RJ, et al. Toward a human blood serum proteome: analysis by multidimensional separation coupled with mass spectrometry. Mol. Cell. Proteomics. 2002;1:947–955. doi: 10.1074/mcp.m200066-mcp200. [DOI] [PubMed] [Google Scholar]
- [3].Stahl-Zeng J, Lange V, Ossola R, Eckhardt K, et al. High sensitivity detection of plasma proteins by multiple reaction monitoring of N-glycosites. Mol. Cell. Proteomics. 2007;6:1809–1817. doi: 10.1074/mcp.M700132-MCP200. [DOI] [PubMed] [Google Scholar]
- [4].Keshishian H, Addona T, Burgess M, Kuhn E, Carr SA. Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution. Mol. Cell. Proteomics. 2007;6:2212–2229. doi: 10.1074/mcp.M700354-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Woroniecki RP, Orlova TN, Mendelev N, Shatat IF, et al. Urinary proteome of steroid-sensitive and steroid-resistant idiopathic nephrotic syndrome of childhood. Am. J. Nephrol. 2006;26:258–267. doi: 10.1159/000093814. [DOI] [PubMed] [Google Scholar]
- [6].Oetting WS, Rogers TB, Krick TP, Matas AJ, Ibrahim HN. Urinary beta2-microglobulin is associated with acute renal allograft rejection. Am. J. Kidney. Dis. 2006;47:898–904. doi: 10.1053/j.ajkd.2006.01.034. [DOI] [PubMed] [Google Scholar]
- [7].Berger RP, Kochanek PM. Urinary S100B concentrations are increased after brain injury in children: a preliminary study. Pediatr. Crit. Care Med. 2006;7:557–561. doi: 10.1097/01.PCC.0000244426.37793.23. [DOI] [PubMed] [Google Scholar]
- [8].Propst A, Propst T, Herold M, Vogel W, Judmaier G. Interleukin-1 receptor antagonist in differential diagnosis of inflammatory bowel diseases. Eur. J. Gastroenterol. Hepatol. 1995;7:1031–1036. doi: 10.1097/00042737-199511000-00004. [DOI] [PubMed] [Google Scholar]
- [9].Laurell CB. Composition and variation of the gel electrophoretic fractions of plasma, cerebrosinal fluid and urine. Scand. J. Clin. Lab. Invest. Suppl. 1972;124:71–82. doi: 10.3109/00365517209102754. [DOI] [PubMed] [Google Scholar]
- [10].Pisitkun T, Shen RF, Knepper MA. Identification and proteomic profiling of exosomes in human urine. Proc. Natl. Acad. Sci. USA. 2004;101:13368–13373. doi: 10.1073/pnas.0403453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gonzales PA, Pisitkun T, Hoffert JD, Tchapyjnikov D, et al. Large-scale proteomics and phosphoproteomics of urinary exosomes. J. Am. Soc. Nephrol. 2008;20:363–379. doi: 10.1681/ASN.2008040406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Sun W, Li F, Wu S, Wang X, et al. Human urine proteome analysis by three separation approaches. Proteomics. 2005;5:4994–5001. doi: 10.1002/pmic.200401334. [DOI] [PubMed] [Google Scholar]
- [13].Pisitkun T, Johnstone R, Knepper MA. Discovery of urinary biomarkers. Mol. Cell. Proteomics. 2006;5:1760–1771. doi: 10.1074/mcp.R600004-MCP200. [DOI] [PubMed] [Google Scholar]
- [14].Adachi J, Kumar C, Zhang Y, Olsen JV, Mann M. The human urinary proteome contains more than 1500 proteins, including a large proportion of membrane proteins. Genome Biol. 2006;7:R80. doi: 10.1186/gb-2006-7-9-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mischak H, Coon JJ, Novak J, Weissinger EM, et al. Capillary electrophoresis-mass spectrometry as a powerful tool in biomarker discovery and clinical diagnosis: an update of recent developments. Mass Spectrom. Rev. 2008 doi: 10.1002/mas.20205. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Coon JJ, Zurbig P, Dakna M, Dominiczak A, et al. CE-MS analysis of the human urinary proteome for biomarker discovery and disease diagnostics. Proteomics Clin. Appl. 2008;2:964–973. doi: 10.1002/prca.200800024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Decramer S, Gonzalez de Peredo A, Breuil B, Mischak H, et al. Urine in clinical proteomics. Mol. Cell. Proteomics. 2008;7:1850–1862. doi: 10.1074/mcp.R800001-MCP200. [DOI] [PubMed] [Google Scholar]
- [18].Kentsis A, Lin YY, Kurek K, Colicchio M, et al. Discovery and validation of urine markers of acute pediatric appendicitis using high accuracy mass spectrometry. Ann. Emerg. Med. 2009 doi: 10.1016/j.annemergmed.2009.04.020. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Traum AZ, Wells MP, Aivado M, Libermann TA, et al. SELDI-TOF MS of quadruplicate urine and serum samples to evaluate changes related to storage conditions. Proteomics. 2006;6:1676–1680. doi: 10.1002/pmic.200500174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Zhou H, Yuen PS, Pisitkun T, Gonzales PA, et al. Collection, storage, preservation, and normalization of human urinary exosomes for biomarker discovery. Kidney Int. 2006;69:1471–1476. doi: 10.1038/sj.ki.5000273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lee RS, Monigatti F, Briscoe AC, Waldon Z, et al. Optimizing sample handling for urinary proteomics. J. Proteome Res. 2008;7:4022–4030. doi: 10.1021/pr800301h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- [23].McKusick VA. Mendelian Inheritance in Man. A Catalog of Human Genes and Genetic Disorders. 12th Edn. Johns Hopkins University Press; Baltimore: 1998. [Google Scholar]
- [24].Dorff KC, Wood MJ, Campagne F. Twease at TREC 2006: Breaking and Fixing bm25 Scoring with Query Expansion, A Biologically Inspired Double Mutant Recovery Experiment. National Institutes of Standards; Gaithersburg, MD, USA: 2006. pp. 836–850. [Google Scholar]
- [25].Campagne F. Objective and automated protocols for the evaluation of biomedical search engines using No Title Evaluation protocols. BMC Bioinformatics. 2008;9:132. doi: 10.1186/1471-2105-9-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Renard C, Chappey O, Wautier MP, Nagashima M, et al. Recombinant advanced glycation end product receptor pharmacokinetics in normal and diabetic rats. Mol. Pharmacol. 1997;52:54–62. doi: 10.1124/mol.52.1.54. [DOI] [PubMed] [Google Scholar]
- [27].Gudehithlu KP, Pegoraro AA, Dunea G, Arruda JA, Singh AK. Degradation of albumin by the renal proximal tubule cells and the subsequent fate of its fragments. Kidney Int. 2004;65:2113–2122. doi: 10.1111/j.1523-1755.2004.00633.x. [DOI] [PubMed] [Google Scholar]
- [28].Cindik N, Baskin E, Agras PI, Kinik ST, et al. Effect of obesity on inflammatory markers and renal functions. Acta Paediatr. 2005;94:1732–1737. doi: 10.1111/j.1651-2227.2005.tb01845.x. [DOI] [PubMed] [Google Scholar]
- [29].De Palo EF, Gatti R, Lancerin F, Cappellin E, et al. The measurement of insulin-like growth factor-I (IGF-I) concentration in random urine samples. Clin. Chem. Lab. Med. 2002;40:574–578. doi: 10.1515/CCLM.2002.099. [DOI] [PubMed] [Google Scholar]
- [30].Skinner AM, Clayton PE, Price DA, Addison GM, Mui CY. Variability in the urinary excretion of growth hormone in children: a comparison with other urinary proteins. J. Endocrinol. 1993;138:337–343. doi: 10.1677/joe.0.1380337. [DOI] [PubMed] [Google Scholar]
- [31].Nemeth E, Tuttle MS, Powelson J, Vaughn MB, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- [32].Helle KB, Corti A, Metz-Boutigue MH, Tota B. The endocrine role for chromogranin A: a prohormone for peptides with regulatory properties. Cell. Mol. Life Sci. 2007;64:2863–2886. doi: 10.1007/s00018-007-7254-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Pflugh DL, Maher SE, Bothwell AL. Ly-6 superfamily members Ly-6A/E, Ly-6C, and Ly-6I recognize two potential ligands expressed by B lymphocytes. J. Immunol. 2002;169:5130–5136. doi: 10.4049/jimmunol.169.9.5130. [DOI] [PubMed] [Google Scholar]
- [34].Varga-Szabo D, Pleines I, Nieswandt B. Cell adhesion mechanisms in platelets. Arterioscler. Thromb. Vasc. Biol. 2008;28:403–412. doi: 10.1161/ATVBAHA.107.150474. [DOI] [PubMed] [Google Scholar]
- [35].Hato T, Tabata M, Oike Y. The role of angiopoietin-like proteins in angiogenesis and metabolism. Trends Cardiovasc. Med. 2008;18:6–14. doi: 10.1016/j.tcm.2007.10.003. [DOI] [PubMed] [Google Scholar]
- [36].Carvalho PC, Hewel J, Barbosa VC, Yates JR., III Identifying differences in protein expression levels by spectral counting and feature selection. Genet. Mol. Res. 2008;7:342–356. doi: 10.4238/vol7-2gmr426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Angelow S, Yu AS. Claudins and paracellular transport: an update. Curr. Opin. Nephrol. Hypertens. 2007;16:459–464. doi: 10.1097/MNH.0b013e32820ac97d. [DOI] [PubMed] [Google Scholar]
- [38].Zhang Y, Wada J. Collectrin, a homologue of ACE2, its transcriptional control and functional perspectives. Biochem. Biophys. Res. Commun. 2007;363:1–5. doi: 10.1016/j.bbrc.2007.08.136. [DOI] [PubMed] [Google Scholar]
- [39].Wright EM. Renal Na(1)-glucose cotransporters. Am. J. Physiol. Renal Physiol. 2001;280:F10–F18. doi: 10.1152/ajprenal.2001.280.1.F10. [DOI] [PubMed] [Google Scholar]
- [40].Ritchie RF, Palomaki GE, Neveux LM, Navolotskaia O, et al. Reference distributions for the positive acute phase serum proteins, alpha1-acid glycoprotein (orosomucoid), alpha1-antitrypsin, and haptoglobin: a practical, simple, and clinically relevant approach in a large cohort. J. Clin. Lab. Anal. 2000;14:284–292. doi: 10.1002/1098-2825(20001212)14:6<284::AID-JCLA7>3.0.CO;2-U. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Vadas P, Gold M, Perelman B, Liss GM, et al. Platelet-activating factor, PAF acetylhydrolase, and severe anaphylaxis. N. Engl. J. Med. 2008;358:28–35. doi: 10.1056/NEJMoa070030. [DOI] [PubMed] [Google Scholar]
- [42].Sanders JS, van Goor H, Hanemaaijer R, Kallenberg CG, Stegeman CA. Renal expression of matrix metalloproteinases in human ANCA-associated glomerulonephritis. Nephrol. Dial. Transplant. 2004;19:1412–1419. doi: 10.1093/ndt/gfh186. [DOI] [PubMed] [Google Scholar]
- [43].Schuchman EH. The pathogenesis and treatment of acid sphingomyelinase-deficient Niemann-Pick disease. J. Inherit. Metab. Dis. 2007;30:654–663. doi: 10.1007/s10545-007-0632-9. [DOI] [PubMed] [Google Scholar]