

Abstract
Methuosis is a unique form of non-apoptotic cell death triggered by alterations in the trafficking of clathrin-independent endosomes, ultimately leading to extreme vacuolization and rupture of the cell. Methuosis can be induced in glioblastoma cells by expression of constitutively active Ras. This study identifies the small GTPases, Rac1 and Arf6, and the Arf6 GTPase-activating-protein, GIT1, as key downstream components of the signaling pathway underlying Ras-induced methuosis. The extent to which graded expression of active H-Ras(G12V) triggers cytoplasmic vacuolization correlates with the amount of endogenous Rac1 in the active GTP state. Blocking Rac1 activation with the specific Rac inhibitor, EHT 1864, or co-expression of dominant-negative Rac1(T17N), prevents the accumulation of vacuoles induced by H-Ras(G12V). Coincident with Rac1 activation, H-Ras(G12V) causes a decrease in the amount of active Arf6, a GTPase that functions in recycling of clathrin-independent endosomes. The effect of H-Ras(G12V) on Arf6 is blocked by EHT 1864, indicating that the decrease in Arf6-GTP is directly linked to activation of Rac1. Constitutively active Rac1(G12V) interacts with GIT1 in immunoprecipitation assays. Ablation of GIT1 by shRNA prevents the decrease in active Arf6, inhibits vacuolization, and prevents loss of cell viability in cells expressing Rac1(G12V). Together the results suggest that perturbations of endosome morphology associated with Ras-induced methuosis are due to downstream activation of Rac1, combined with reciprocal inactivation of Arf6. The latter appears to be mediated through Rac1 stimulation of GIT1. Further insights into this pathway could suggest opportunities for induction of methuosis in cancers that are resistant to apoptotic cell death.
Keywords: Ras, Rac, Arf6, Macropinocytosis, Cell Death, Vacuoles, Methuosis, Necrosis, Glioblastoma
Introduction
The reduced sensitivity of many types of cancer cells to apoptotic cell death has stimulated interest in identifying alternative non-apoptotic cell death pathways that might be used to kill tumor cells in a therapeutic context. Several different forms of non-apoptotic cell death have been described, based on specific cellular or molecular criteria. These include autophagy-associated cell death (1–3), paraptosis (4;5), oncosis (6–8), necroptosis (9;10), entosis (11), and programmed necrosis (12;13).
A decade ago Chi et al. (14) reported a unique form of non-apoptotic cell death that can be induced in glioblastoma and gastric carcinoma cells by constitutive stimulation of Ras signaling pathways. We recently determined that this form of cell death is morphologically and mechanistically distinct from the various types of cell death noted above (15). It entails a stimulation of macropinocytosis (cell drinking), combined with defects in clathrin-independent endocytic vesicle trafficking, ultimately resulting in cellular disruption as large vacuoles expand to fill the cytoplasmic space. We have termed this form of cell death ‘methuosis’ from the Greek methuo, to drink to intoxication. Morphologically methuosis resembles type IIIA non-lysosomal necrotic cell death (16), originally observed in vacuolated cartilage during mineralization (17). It also bears some similarity to the extensive endosomal vacuolation noted in gastric epithelial cells exposed to the H. pylori VacA cytotoxin (18;19) and in regions of liver necrosis induced by furosemide (20). Despite these superficial similarities, it remains unclear whether these forms of cell death share a common molecular mechanism. Although it is not yet known if methuosis occurs in normal physiological contexts, it is possible that understanding the molecular signals that drive this non-conventional form of cell death may prove useful for devising new approaches to eliminate cancer cells that are refractory to apoptosis.
We have found that stimulation of vacuolization in glioblastoma cells by activated forms of H- and K-Ras does not depend on conventional Ras effector pathways such as the Raf-MEK-ERK kinase cascade or the phosphatidylinositide 3’-kinase (PI3K)-Akt pathway (21). In considering alternative mechanisms, we noted that previous studies had implicated the Rac1 GTPase as a positive regulator of macropinocytosis (22), phagocytosis (23), and cellular vacuolization that occurs in response to the VacA cytotoxin (18). Since downstream targets of Ras include guanine nucleotide exchange factors that can stimulate activation of Rac1 (24), we hypothesized that Ras might work through Rac to trigger methuosis. Consistent with this possibility, we observed that expression of constitutively active Rac1 (15), but not the related GTPases, Cdc42 or RhoA (21), could mimic the effects of Ras in glioblastoma cells. Based on these findings, we initiated the present study to test the hypothesis that Rac1 is an essential downstream mediator of Ras-induced methuosis. The results clarify the pathway for Ras-induced methuosis by establishing that the underlying perturbations in macropinosome/endosome trafficking that lead to cell vacuolization require both activation of Rac1 and a coordinate Rac1-dependent decrease in the pool of active Arf6, a GTPase implicated in endocytic trafficking. The latter is mediated through an interaction between the active form of Rac1 and an Arf GTPase activating protein (GAP), GIT1.
Materials and Methods
Cell Lines for Conditional Expression of Ras and Rac1 Constructs
U251 human glioblastoma cells were purchased from the DCT Tumor Repository (National Cancer Institute, Frederick, MD, USA). U20S human osteosarcoma cells were obtained from the American Type Culture Collection (Rockville, MD). HEK293T cells were obtained from Systems Biosciences (Mountain View, CA). Unless stated otherwise, cell lines were grown in Dulbecco’s modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS), at 37°C with 5% CO2.
Generation of the U251-C18 cell line, which expresses myc-H-Ras(G12V) under the control of a tetracycline (tet)-inducible promotor, was described previously (15). The U251-C18 cells were maintained in DMEM supplemented with 10% tetracycline-free FBS (Clontech, Mountain View, CA ), 200 μg/ml G418 and 200 μg/ml hygromycin. A stable U251 cell line that expresses myc-Rac1(G12V) under control of a tet-inducible promoter, herein designated U251-Rac1(G12V)tet was generated as described previously (15).
To generate new stable cell lines that conditionally express activated Ras or dominant negative Rac1 or both, U251 cells were infected with virus-enriched medium containing the regulatory vector pRetroX-Tet-On (Clontech), which encodes the reverse tet-regulated transactivator. Cells were selected in medium containing 200 μg/ml G418, and clonal cell lines were tested in transient transfection assays to determine which ones gave the tightest doxycycline (Dox)-regulated gene expression. These were designated as U251 Tet-On. cDNAs encoding myc-H-Ras(G12V) and FLAG-Rac1(T17N) were subcloned into the NotI/MluI sites of the expression vector, pRetroX-Tight-Pur (Clontech). Retrovirus was produced by transfecting these constructs together with pRetroX-VSVG-Env into 293 GP2 packaging cells (Clontech) using Lipofectamine Plus reagent. Cells were maintained in DMEM + 10% heat-inactivated FBS, using Biosafety Level 2 precautions. Virus-enriched medium was passed through a 0.45 μm filter and used to infect the U251Tet-On cells. Cells were maintained in medium containing 1μg/ml puromycin and 200 μg/ml G418. For the expression of the genes of interest 1 μg/ml Dox was added to the medium.
Nucleofection and Retroviral Expression of Proteins
Myc-tagged H-Ras(G12V) and H-Ras(G12V, C186S) were generated and cloned into the pCMV5 expression vector as described previously (21). pCMV5-myc-Rac1(G12V) was provided by A.L. Wilson-Delfosse, Case Western Reserve University, Cleveland, OH. U251 cells were nucleofected with an Amaxa Nucleofector II system, using Solution T and Program T30, according to the manufacturer’s protocol (Lonza, Walkersville, MD). For each reaction 5 μg of plasmid DNA was introduced into 3 x 106 cells that had been harvested in RPMI medium 1640 supplemented with 10% heat inactivated FBS. Following nucleofection, cells were plated and analyzed 18–20 hours later.
For constitutive expression of myc-Rac1(G12V) or myc-H-Ras(G12V) in various cell lines, the cDNAs encoding the latter constructs were cloned into the pQCXI-H retroviral vector (Clontech) using the NotI and BamHI sites. Retrovirus was produced in 293 GP2 packaging cells (Clontech) co-transfected with pQCXI-H-Rac1(G12V) and pRetroX-VSVG-Env (Clontech), using Lipofectamine Plus reagent. At 48 h after transfection, the virus-enriched medium was collected, filtered and used to infect the cells. After 24 h, the virus was removed and the cells were maintained in DMEM with 10% FBS, 1 μg/ml puromycin and 200 μg/ml hygromycin until they were examined.
shRNA-Mediated Gene Silencing
For silencing expression of Tiam1, oligonucleotides corresponding to nucleotides 5263–5283 (shRNA#1, 5’-CCGTAGAGAATGTGTGTAGAT-3’) in the 3’ UTR of huTiam1 (accession number U16296) or 3157–3177 of the coding region (shRNA#2, 5’-CGCACCTACGTGAAGGATTTA-3’) were used. Each oligonucleotide was followed by a 12-base loop region (CTTCCTGTCAGA) and the reverse complement of the 21-bp gene-specific sequence. The hairpin sequences were flanked by BamH1 and EcoR1 restriction sites. The oligonucleotides were phosphorylated and annealed to their respective reverse complementary sequences, then ligated into the pSIH1-H1-Puro lentivector (System Biosciences, Mountain View, CA).
shRNA constructs to suppress expression of Eps8 or GIT1 were made as described above except with different oligonucleotides: For Eps8, bases 1543–1563 (5’-CCAACTTCTAATCGCCATATA-3’) of huEps8 (accession number NM_004447). For GIT-1, bases 816–834 (5’-GCACACCCATTGACTATGC-3’) and 1070–1090 (5’-GAGGTGGATCGAAGAGAAAAT-3’) of huGIT1 (accession number NM_014030) (25). Control shRNA constructs utilized oligonucleotides targeting Chiridius poppei GFP (5’-GGAGGAGCTGCACAGCAA-3’) or Aequorea victoria GFP (5’-GCAAGCTGACCCTGAAGTTC-3’), neither of which match sequences in the human genome.
shRNA lentivirus was produced by simultaneous transfection of 293TN cells with shRNA-pSIH1-H1-Puro vector, pPackH1-Rev, pPackH1-Gag and pPackVSV-G (System Biosciences), using Lipofectamine Plus reagent (Invitrogen, Carlsbad, CA). Virus collected between 24–48 h after transfection was passed through a 0.45 μm filter and used to infect U251 C3-18 cells that were approximately 50% confluent. Cells were exposed to the virus for 2–3 days. One day after removal of the virus Dox was added to the medium (1 μg/ml) to induce myc-H-Ras(G12V) expression, and cells were analyzed 2–3 days later by phase contrast microscopy or western blotting.
Cell Morphology and Viability
To determine the percentage of cells with cytoplasmic vacuoles, phase contrast images of live cells were captured with an Olympus IX70 microscope equipped with a digital camera and SPOT imaging software (Diagnostics Instruments, Inc. Sterling Heights, MI). For each culture multiple photographs were taken of different fields of cells and at least 100 cells were scored for vacuolization. Cells containing three or more phase-lucent vacuoles with diameters > 0.5 μm or >10 smaller vacuoles (0.1–0.5 μm) were counted as positive.
Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay, as described previously (26). PARP cleavage in the presence or absence of zVAD-fmk (Bachem Biosciences, King of Prussia, PA) was determined by western blot analysis.
Where replicate assays were performed, statistical significance of changes in cell morphology or viability was assessed by Student’s t-test.
Western Blot Analysis
The 9E10 monoclonal antibody against the myc epitope (Calbiochem/EMD Biosciences, San Diego, CA) and the antibody to GIT1 (BD Biosciences, San Jose, CA) were purchased from the indicated sources. Antibody against α-tubulin was obtained from Sigma (St. Louis, MO). The antibody against PARP was obtained from BD Biosciences. Methods for protein determination, SDS-PAGE and western blot analysis have been described (27). Chemiluminescence signals were quantified using an Alpha Innotech FluorChem HD2 imaging system.
Immunofluorescence
Cells were prepared for immunofluorescence as described previously (28). Myc-tagged proteins were detected with a rabbit polyclonal antibody (EMD Biosciences, San Diego, CA, USA) conjugated to FITC. FLAG-tagged proteins were detected with a mouse monoclonal antibody, M2, from Sigma (St Louis, MO), followed by goat-anti-mouse IgG conjugated with Alexa Fluor™ 568 (Invitrogen, Carlsbad, CA). Cells were examined with a Nikon Eclipse TE2000-U fluorescence microscope equipped with a digital camera and NIS-Elements AR software (Nikon Instruments, Inc., Melville, NY), and the percentage of vacuolated cells was determined by counting at least 70 cells in photomicrographs from randomly selected fields. Criteria for counting cells as vacuolated were the same as those noted above for phase contrast micrographs.
Uptake of Fluid Phase Tracers
Endocytic compartments were labeled with the fluid phase tracer, Lucifer Yellow, as described previously (15). For labeling cells with dextran-Alexa Fluor 568 (AF568), U251 C3-18 cells were grown in 60 mm dishes for 3 days in phenol red-free medium in the presence or absence of 1.0μg/ml Dox and 20 μM EHT1864, with medium replacement each day. To quantify dextran uptake, cells were incubated with 0.5 mg/ml dextran-AF 568 (Invitrogen) for 15 min, washed with HBSS, and harvested by trypsinization. Cells were suspended in a final volume of 500 μl, and 100 μl of cell suspension was transferred to a black-walled, black-bottom microplate and analyzed on a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA) with 568-nm excitation and 603 nm emission filters. Background fluorescence was subtracted using values obtained from cells incubated in the absence of dextran-AF568.
Regulated Ras Expression with the ProteoTuner® System
The Lenti-X ProteoTuner System was purchased from Clontech. Myc-tagged H-Ras cDNA constructs (G12V or G12V,C186S) were subcloned into the EcoR1-BamH1 restriction sites of the lentiviral expression vector, pLVX- PTuner Green, placing the Ras constructs in frame with the 12 kDa destabilization domain (DD) and upstream from an IRES, which allows constitutive expression of ZsGreen1. Lentivirus was produced in HEK293T cells (ATCC, Manassas, VA) using Lipofectamine Plus reagents (Invitrogen), and pPack-H1 Lentiviral Packaging System (System Biosciences). Forty-eight hours after transfection, the medium containing the virus was collected and filtered through a 0.45 μm membrane. U251 glioblastoma cells were infected with the filtered virus to express DD-myc-H-Ras (G12V). Four days after infection, Shield1 was added to the medium at concentrations of 30 nM or 100 nM, in order to stabilize the DD tag and allow varying levels of Ras expression. Twenty four hours after addition of Shield1, overall infection efficiency in all cultures was determined to be greater than 85% by counting the percentage of green fluorescent cells. The amount of DD-myc-H-Ras expressed was determined by immunoblotting with anti-myc monoclonal antibody or anti-Ras antibody M-90 (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Cell morphology was assessed by phase contrast microscopy. The relative amount of activated Rac1 was assayed in cell lysates as described below.
Rac1 and Arf6 Activation Assays
Rac1 activation assays were performed using the EZ-Detect Rac1 Activation Kit (Thermo Scientific Pierce, Rockford, IL). The assay employs a GST-fusion protein containing the p21-binding domain of p21-activated protein kinase 1 (PAK1) to selectively bind active Rac1 in whole cell lysates. The active Rac1 is then collected on glutathione agarose beads and quantified by immunoblot analysis using anti-Rac1 monoclonal antibody (Thermo Scientific Pierce). The value for active Rac1 collected on the beads was normalized to the concentration of protein in the cell lysate. On a separate immunoblot, the total Rac1 in a sample of the cell lysate was determined and normalized to the protein concentration. The normalized results were then expressed as the ratio of active Rac1 to total Rac1 in the sample. The same approach was used to quantify active Arf6, except that the assay employed GST-GGA3 (Golgi-associated gamma adaptin ear-containing Arf binding protein 3) to pull down active Arf6, and a monoclonal antibody against Arf6 for subsequent immunoblot analysis (Thermo Scientific Pierce). Where replicate assays were performed, statistical significance of changes in activation of Rac1 or Arf6 was assessed by Student’s t-test.
For analysis of Rac1 or Arf6 in stable cell lines, activation assays were performed on cells harvested on the third day after addition of Dox to induce expression of H-Ras(G12V). Controls cells were maintained without Dox. Where indicated, EHT 1864 (provided by Exonhit Therapeutics, Paris, France) was added to the culture medium at a concentration of 25 μM to block Rac1 activation. Both Dox and EHT 1864 were replaced each day after treatment until the assays were performed. For cell cultures transiently nucleofected with H-Ras constructs, the Rac1 or Arf6 activation assays were performed 18–24 h after nucleofection.
Immunoprecipitation
For immunoprecipitation of myc-Rac(G12V), cells were washed with Hanks balanced salt solution and homogenized in IP buffer: 10 mM Tris-HCl, pH 7.4, 1 mM MgCl2, 150 mM NaCl, 0.75% v/v NP-40, with Complete Mini Protease Inhibitors (Roche, Indianapolis, IN). Lysates were cleared by centrifugation at 10,000 x g for 30 min at 4°C, and 1/10th of the supernatant solution was reserved for checking expression of myc-Rac1(G12V) and endogenous GIT1. The remainder of the solution was incubated with rabbit anti-myc agarose beads (Sigma) for 2 h at 4°C. The beads were washed 3 times with IP buffer and the bound proteins were subjected to SDS-PAGE and immunoblot analysis for myc-Rac1(G12V) or GIT1, using monoclonal antibodies noted above.
Results
Constitutively Active Rac1 Mimics the Effect of Active H-Ras in Stimulating Non-Apoptotic Cell Death
Our previous studies showed that overexpression of constitutively active Rac1(G12V) in glioblastoma cells causes a striking accumulation of vacuoles that incorporate the fluid-phase marker, Lucifer yellow, similar to what we observed with H-Ras(G12V) (15). This is not a general effect of all Ras-related GTPases, as active Cdc42(G12V) and RhoA(G14V) did not cause vacuolization (21). The accumulation of vacuoles induced by Rac1(G12V) represents the first observable step in a pathway that ultimately leads to cell detachment from the substratum and disruption of the cell membrane. However, in these earlier studies we did not establish if the loss of cell viability caused by Rac1(G12V) was caspase-independent, as was the case for H-Ras(G12V) (15). To examine this possibility, we introduced myc-Rac1(G12V) into the U251 glioblastoma cell line using a retroviral vector. Within 48 h the expression of Rac1(G12V) caused extreme cytoplasmic vacuolization (Fig. 1A), and within 5–7 days there was a loss of cells from the substratum and decrease in the number of viable cells compared with cells infected with the empty viral vector (Fig. 1D). Although caspase activation could be detected by examining PARP cleavage in the detached cells (Fig. 1C), addition of the general caspase inhibitor, zVAD-fmk, at a concentration that completely blocked PARP cleavage, did not protect the cells from Rac1(G12V)-induced vacuolization and cell death (Fig. 1A,C&D). In this respect the effects of Rac1(G12V) were essentially identical to the cell death phenotype induced by activated Ras, which we have termed methuosis (15). Identical results were obtained when the same experiment was performed with a stable U251 cell line in which myc-Rac1(G12V) was conditionally expressed in response to doxycyline (Dox) (Supplementary Data, Fig. S1).
Figure 1.
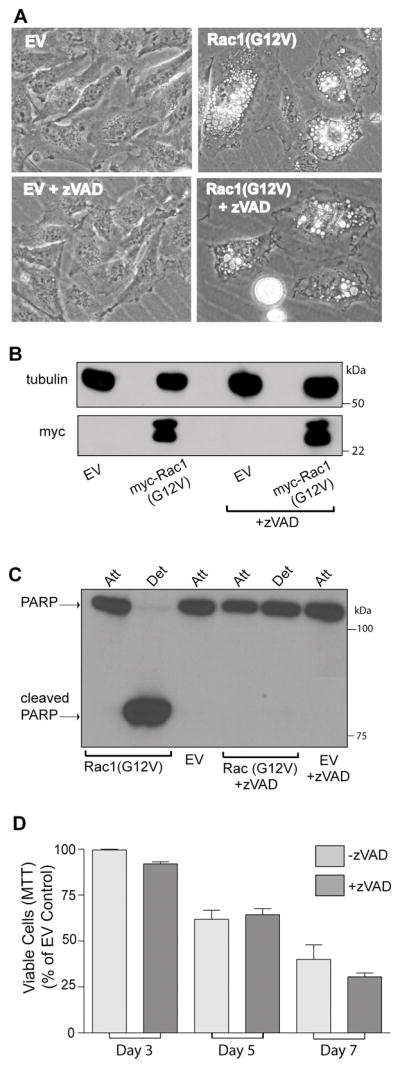
Expression of activated Rac1 induces caspase-independent death in U251 glioblastoma cells. A) Cells were infected with retrovirus encoding myc-Rac1(G12V) or virus without an cDNA insert (empty vector, EV). On the day after infection, 50 μM z-VAD-fmk was added to half of the cultures, and the others were maintained without the caspase inhibitor. On day-6 after infection A) live cells were examined by phase contrast microscopy, B) western blot analysis was performed to verify expression of myc-Rac1(G12V) and C) attached and detached cells were harvested for analysis of PARP cleavage. There were no detached cells in the EV controls. D) Cells expressing Rac1(G12V) were subjected to MTT assays over a period of 7 days to compare their viability with and without zVAD (the inhibitor was replenished every day). The results shown in the bar graph are from three independent experiments (mean ± SD). The decreases in viable cells at day 3 and day 7 (+/− zVAD) were significant at p 0.04 .
Induction of Methuosis by Activated H-Ras Depends on Activation of Endogenous Rac
The preceding observations, coupled with reports that Ras can activate Rac1, suggested that Rac lies downstream from Ras in the signaling pathway for methuosis. To begin testing this possibility, we asked if overexpression of constitutively active H-Ras(G12V) could stimulate activation of endogenous Rac in U251 glioblastoma cells. Cells were nucleofected with an expression vector encoding either myc-H-Ras(G12V) or the farnesylation-incompetent myc-H-Ras(G12V, C186S), which is relatively ineffective at inducing vacuolization (21) (Fig. 2, A&B). The pool of activated endogenous Rac GTPase was then measured by an established assay that takes advantage of the selective interaction of Rac-GTP with the p21 binding domain of the Rac effector, PAK1 (Fig. 2, C &D). The results of this experiment established that in cells overexpressing H-Ras(G12V), vacuolization was accompanied by a robust activation of endogenous Rac. Neither vacuolization nor activation of endogenous Rac occurred in cells expressing H-Ras(G12V) that contained a C-terminal cysteine substitution that prevents farnesylation and membrane association of the protein. Activation of Rac2 was not examined in glioblastoma cells, since Rac2 is expressed mainly in cells of hematopoietic lineage (29). Rac3 is strongly expressed in neuronal tracts of the brain (30) and in neuronal cell lines (31), but we were unable to detect Rac3 expression in glioblastoma cells, using an antibody specific for Rac3 (32) (data not shown). Therefore, for all further studies we inferred that Rac1 was the predominant form of Rac being measured in glioblastoma cells.
Figure 2.
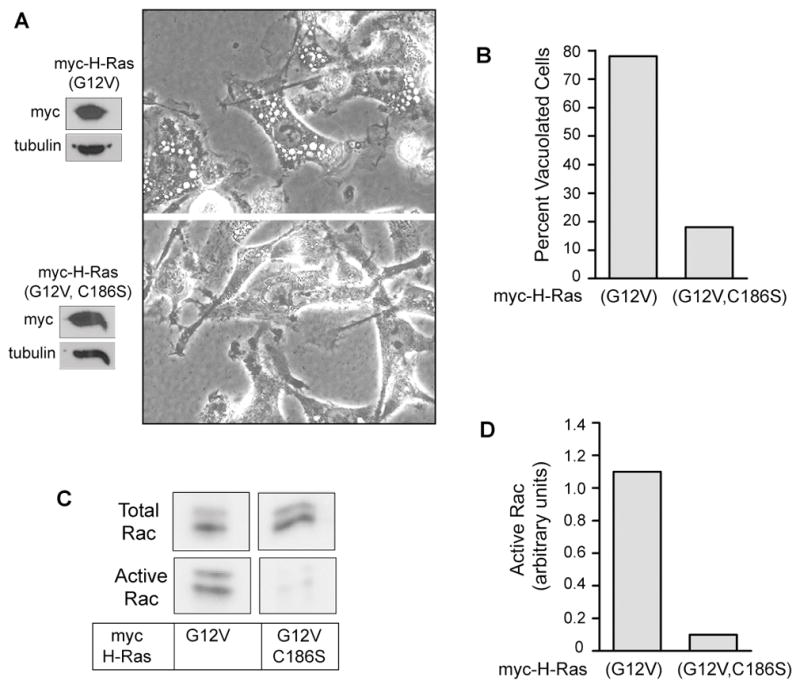
Expression of activated H-Ras causes an increase in the amount of active Rac1 in U251 glioblastoma cells. A) Cells were nucleofected with expression vectors encoding the indicated Ras constructs. After 24 h live cells were examined by phase contrast microscopy and harvested for western blot analysis to check expression of the myc-tagged Ras proteins. B) Cells were scored for vacuolization as described in Materials and Methods. C) Pull-down assays for activated Rac1 were performed on lysates from two pooled 10 cm dishes of cells. The lower blots show the active endogenous Rac1 that bound to the GST-PAK1, and the upper panels show the total Rac1 in an aliquot of the cell lysate. D) The immunoblot signals for active and total Rac1 were quantified and the units for active Rac1 were normalized to total Rac as described in Materials and Methods.
Numerous studies have documented the ability of activated Ras to function as an oncogene, promoting cell proliferation and cell survival through activation of the Raf-MEK-ERK and PI3K-Akt signaling pathways (33;34). Our earlier results indicated that Ras-induced cytoplasmic vacuolization does not require activation of Raf or PI3K signaling pathways (21). This suggests that the induction of methuosis by H-Ras(G12V) might occur only when the level of activated Ras reaches a threshold sufficient to stimulate Rac1 activation. To examine this possibility we used the ProteoTunerR system to express myc-H-Ras(G12V) as a fusion protein with an N-terminal destabilizing domain (DD) domain that promotes rapid proteasomal degradation. By adding increasing amounts of the small molecule, Shield1, which binds to the DD domain and impairs degradation, we were able to obtain graded increases in the expression level of myc-H-Ras(G12V) in parallel cultures (Fig. 3A). Under these conditions, the extent of cellular vacuolization (Fig. 3C&D) correlated with the amount of Rac1 activation (Fig. 3B).
Figure 3.

Rac1 activation and cellular vacuolization correlate with increasing levels of H-Ras(G12V) expression in U251 glioblastoma cells. A) Cells were grown in the presence of increasing concentrations of Shield1 to obtain graded expression levels of DD-myc-H-Ras(G12V), using the ProteoTuner system. The blot shows the expressed Ras protein detected with the myc antibody. In a separate blot (not shown) an antibody against H-Ras was used to determine the ratio of expressed DD-myc-H-Ras(G12V) to endogenous H-Ras. The results were 1.4 with no Shield1, 3.0 with 30 nM Shield1, and 4.5 with 100 nM Shield1. B) One day after addition of Shield1, the amount of active endogenous Rac1, normalized to total Rac1, was determined as described in Materials and Methods. C &D) Cell morphology was assessed by phase contrast microscopy and the percentage of cells that were vacuolated was determined.
To verify a direct link between Ras-induced activation of Rac1 and the extensive cytoplasmic vacuolization that precedes methuosis, we utilized the compound EHT 1864, which has been established as a selective inhibitor of Rac GTPases (35). The compound acts by displacing bound nucleotide, thereby preventing interaction of Rac with its downstream effectors (35). The inhibitor studies were done with a stable glioblastoma cell line, U251-C18 (15), in which conditional expression of myc-H-Ras(G12V) is obtained in response to addition of Dox (Fig. 4A). In the absence of the Rac inhibitor, Dox-induced expression of H-Ras(G12V) was accompanied by a significant increase in activation of endogenous Rac1 (Fig. 4B), and this coincided with extensive vacuolization (Fig 4C&D). When the Rac inhibitor, EHT 1864, was added together with Dox, there was no diminution of myc-H-Ras(G12V) expression (Fig. 4A), but the Ras-dependent increase in active Rac1 was blocked (Fig. 4B). Most notably, inhibiting the activation of Rac1 strongly suppressed Ras-induced vacuolization (Fig. 4C&D) and significantly reduced cellular uptake of the fluid-phase marker, Dextran-AF568 (Fig. 4E).
Figure 4.

Blocking Rac1 activation in glioblastoma cells prevents Ras-induced vacuolization. A) The stable cell line, U251-C18, was incubated for 3d in the presence or absence of Dox (1 μg/ml), with or without the Rac1 inhibitor, EHT 1864 (25 μM). Inducible expression of myc-H-Ras(G12V) was verified by western blot analysis. B) Rac activation assays were performed on cells subjected to each condition. The results are the mean (± SD) of three independent determinations performed on separate cultures. The increase in active Rac1 in the cells expressing H-Ras(G12V) (+Dox alone) (*) was significant at p < 0.001compared with the cells not expressing Ras (−Dox). The decrease in active Rac1 in the cultures expressing H-Ras(G12V) in the presence of EHT 1864 (+Doc, +EHT 1864) (**) was significant at p < 0.001 compared with the corresponding control (+ Dox, −EHT 1864). The decrease in the basal level of active Rac1in the −Dox cultures treated with EHT1864 (***) was significant at p <0.05. C&D) At the same time that cultures were harvested for the Rac activation assays, parallel cultures were examined by phase contrast microscopy to determine the percentage of cells that were vacuolated. The results of three separate experiments (mean ± SD) are shown. The suppression of vacuolization by the Rac inhibitor in the cultures expressing H-Ras(G12V) (+Dox, +EHT 1864) (*) was significant at p < 0.0001 compared to the cells expressing H-Ras(G12V) without the Rac inhibitor (+Dox, −EHT 1864). E) Uptake of dextran-AF568 was measured in the +Dox cells expressing myc-H-Ras(G12V), with or without EHT1864. The results are the mean ± S.D. of separate determinations on three cultures. The decrease in dextran uptake in the cells treated with the Rac inhibitor (*) was significant at p < 0.04.
To obtain an independent line of evidence that Rac1 functions downstream from H-Ras for initiation of methuosis, we attempted to stably suppress the expression of endogenous Rac1 with short-hairpin RNA (shRNA). However, we found that loss of Rac1 adversely affected cell attachment and proliferation. As an alternative, we took advantage of the well known ability of the dominant-negative Rac1(T17N) to interfere with the function of endogenous Rac1 (22;36). For these studies, we used the pRetroX-Tight-Puro vector, in conjunction with the RetroX-Tet-On system to generate U251 cell lines capable of conditional expression of myc-H-Ras(G12V) alone, FLAG-Rac1(T17N) alone, or myc-H-Ras(G12V) + FLAG-Rac1(T17N). As shown in Fig. 5A, within 48h of adding Dox to the medium, phase contrast microscopy revealed that the cells expressing H-Ras(G12V) were extensively vacuolated and had begun to detach from the dish, whereas the cells expressing Rac1(T17N) showed a minimal level of vacuolization, similar to the cells incubated without Dox. Most notably, co-expressing the dominant-negative Rac1 construct together with active H-Ras markedly suppressed vacuolization, compared to cells expressing H-Ras(G12V) alone. To account for variations in transfection efficiency, immunofluorescence microscopy was performed to identify only those cells that were expressing myc-H-Ras(G12V), FLAG-Rac1(T17N), or the combination of both constructs (Fig. 5B). The percentage of vacuolated cells was then calculated using only the labeled cells. The results confirmed the qualitative impressions of cellular vacuolization based on the phase contrast microscopy of the live cells. Taken together with the preceding studies with the Rac1 inhibitor, these observations support the idea that activation of a Rac1-mediated pathway is necessary for the early perturbations of endocytic trafficking that lead to cellular vacuolization associated with methuosis.
Figure 5.
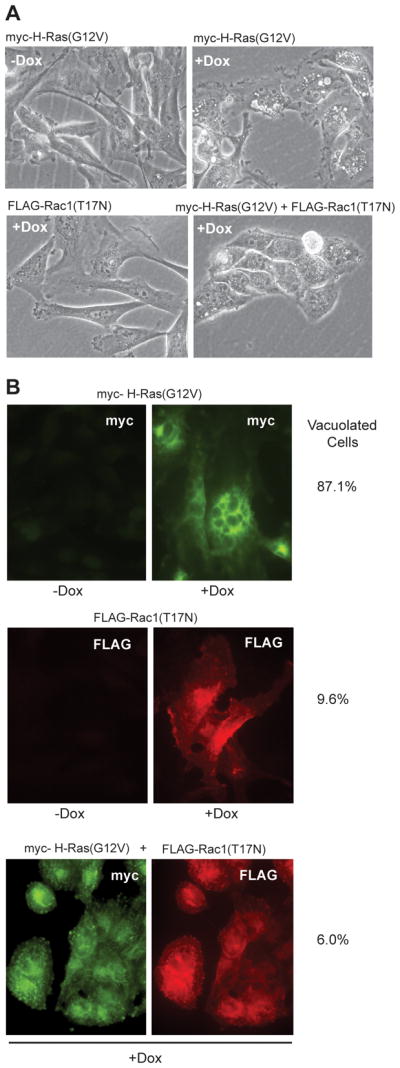
Expression of dominant-negative Rac1 blocks the induction of vacuolization by active H-Ras. U251 cell lines capable of conditional expression of myc-H-Ras(G12V), FLAG-Rac1(T17N) or both constructs, were generated using the pRetroX-Tight-Pur Tet-On system as described in Materials and Methods. A) Phase contrast microscopy of the cells after 2 days with or without Dox in the medium. B) Immunofluorescence microscopy demonstrates expression of myc-H-Ras(G12V) and/or FLAG-Rac1(T17N) only in the presence of Dox. The percentage of vacuolated cells in each culture was determined by scoring only the cells with visible expression of the myc and/or FLAG constructs. The results are based on counting a minimum of 70 cells.
Stimulation of Rac1 Activation by H-Ras(G12V) Does not Depend on Conventional Exchange Factors in Glioblastoma Cells
We next turned our attention to the question of how H-Ras(G12V) might promote the activation of Rac1 in the glioblastoma cells. A number of Rac guanine nucleotide exchange factors (GEFs) can be activated by PI(3,4,5)P3, the product of PI3K (37). Thus, one possibility is that Ras(G12V) stimulates Rac1 activation indirectly through positive modulation of PI3K. We believe that this is unlikely because our previous studies have demonstrated that over-expression of PTEN in U251 cells (where PTEN is mutated) suppresses PI3K signaling but does not prevent Ras-induced vacuolization (21). A more likely possibility is that at high expression levels H-Ras(G12V) can directly stimulate the activity of a Rac GEF. Among the members of the Rac GEF family, Tiam1 stands out as a candidate because it contains a Ras binding domain and has been reported to promote activation of Rac1 in a Ras-dependent manner (24).
To explore the possible contribution of Tiam1 to Ras-induced methuosis, we used a lentiviral vector to introduce anti-Tiam1 shRNA into the U251-C18 stable cell line. The shRNA suppressed Tiam1 to undetectable levels (Fig. 6A), but did not prevent Dox-inducible expression of myc-H-Ras(G12V) (Fig. 6B). Under these conditions, expression of H-Ras(G12V) continued to trigger the vacuolar phenotype that is characteristic of methuosis, despite the suppression of Tiam1 (Fig. 6B).
Figure 6.

Suppression of Tiam1 or Eps8 expression does not prevent Ras-induced vacuolization of glioblastoma cells. A) shRNA-mediated knockdown of Tiam1 or Eps8 expression was performed as described in Materials and Methods. Western blots of whole cell lysates show that the specific shRNAs reduced expression of Tiam1 or Eps8 by >90% compared with the respective controls. The loading controls, α-tubulin and LDH, confirm that comparable amounts of cell protein were applied to the lanes. B) When cells were grown in the presence of Dox to induce expression of myc-H-Ras(G12V) (lower blots), there was no decrease in vacuolization of the cells lacking Tiam1 or Eps8, compared with the control. Vacuolization was assessed by phase contrast microscopy and quantified as described in Materials and Methods.
Sos1 is another GEF that has the potential to respond to H-Ras(G12V) by activating Rac1. Although Sos1 is best known for its ability to form a complex with the adapter, Grb2, to facilitate activation of Ras in response to receptor tyrosine kinase signals (38), it can also behave as a Rac GEF when assembled into a tricomplex with Eps8 and a scaffolding protein, E3b1 (Abi-1) (39;40). To determine if such a complex might be responsible for mediating Ras-induced Rac1 activation and cellular vacuolization, we used shRNA directed against Eps8 to dramatically reduce the level of the protein in U251-C18 cells prior to inducing the expression of H-Ras(G12V) (Fig. 6A). Despite the near complete suppression of Eps8 expression, the morphological response of the knockdown cells was similar to that observed in the control cells with normal levels of Eps8 (Fig. 6B).
Taken together, the preceding results indicate that the two most obvious Ras-responsive Rac GEFs, Tiam1 and Sos1/Eps8/E3b1, are not essential for initiation of vacuolization in glioblastoma cells expressing H-Ras(G12V).
Ras-Induced Activation of Rac1 Causes a Decrease in the Amount of Active Arf6
As we considered possible molecular mechanisms whereby over-expression of H-Ras(G12V) and consequent activation of endogenous Rac1 might cause dysfunction of endocytic compartments, our attention was drawn to a series of studies that have defined functional connections between Rac1 and another small GTPase, Arf6. In its active state, Arf6 promotes membrane recycling from clathrin-independent endosomes (CIE) to the plasma membrane (PM) (41;42). Arf6 and Rac1 co-localize in these compartments, which also contain nucleotide exchange factors (ARNO and DOCK180/Elmo) and effectors (Arfaptin-2/POR1, PI4P 5-Kinase) for both proteins. Most interestingly, Arf6 is required for trafficking of Rac1 from endosomes to the PM, so that when active Rac1(Q61L) is expressed together with inhibitory Arf6(T27N), both proteins accumulate in vacuole-like structures in HeLa cells (36). A recent study has shown that Arf6 also plays a role in the trafficking of macropinosomes induced by Ras(G12V) in HeLa cells, and that perturbation of Arf6 function can lead to the accumulation of CIE-derived vacuoles that contain Ras (43). For these reasons, we hypothesized that cytoplasmic vacuolization induced by H-Ras(G12V) in glioblastoma cells might entail changes in the activation state of Arf6, in addition to activation of Rac1.
To test this hypothesis we used the U251-C18 cell line to determine if Dox-mediated induction of H-Ras(G12V) expression and cellular vacuolization were accompanied by changes in the amount of active Arf6. The latter was assessed in pull-down assays using a fusion protein, GST-GGA3, which interacts specifically with the GTP-bound form of Arf6. The results showed that the relative amount of active Arf6 decreased by about 50% in the cells expressing H-Ras (G12V) (Fig. 7A&B), contrasting with the increase in Rac1 activation observed under identical conditions (Fig. 4A&B). To determine if the decrease in active Arf6 in the cells expressing H-Ras(G12V) was specifically related to activation of Rac1, we added the Rac inhibitor, EHT 1864, at the same time that expression of H-Ras(G12V) was induced by the addition of Dox. By itself, EHT 1864 had no significant effect on the baseline level of active Arf6 in cells that were not expressing active H-Ras (−Dox) (Fig. 7B). However the Rac inhibitor prevented the decrease in active Arf6 (Fig. 7B) and the accumulation of vacuoles (Fig. 7C) in the cells expressing H-Ras(G12V). These results strongly suggest that the decline in the amount of active Arf6 that occurs in cells expressing H-Ras(G12V) is caused by the increase in the amount of active Rac1.
Figure 7.
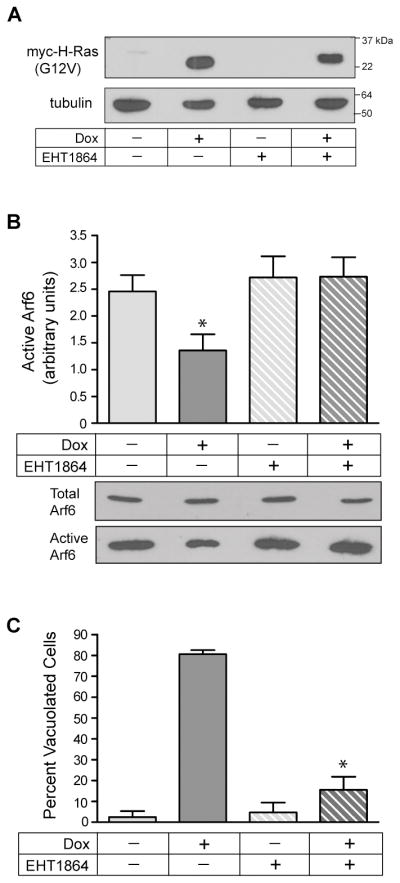
Expression of activated H-Ras causes a Rac1-dependent decrease in the amount of active Arf6, which is essential for vacuolization of glioblastoma cells. A) U251-C18 cells were incubated for 3d in the presence or absence of Dox (1 μg/ml), with or without the Rac1 inhibitor, EHT 1864 (25 μM). Inducible expression of myc-H-Ras(G12V) was verified by western blot analysis. B) Arf6 activation assays were performed on cells subjected to each condition, with active Arf6 normalized to total Arf6 as described in Materials and Methods. The results are the mean (± SD) of three independent determinations performed on separate cultures. The decrease in active Arf6 in the cells expressing H-Ras(G12V) (+Dox alone) (*) was significant at p < 0.01compared with the cells not expressing Ras (−Dox) or the cells incubated with the Rac inhibitor, EHT 1864. Representative immunoblots of the raw Arf-6 GTP pull-downs are shown below the bar graph. C) At the time that cultures were harvested for the Arf6 activation assays, parallel cultures were examined by phase contrast microscopy to determine the percentage of cells that were vacuolated. The results of three separate experiments (mean ± SD) are shown. The suppression of vacuolization caused by addition of the Rac inhibitor to cultures expressing H-Ras(G12V) (+Dox, +EHT 1864) (*) was significant at P < 0.0001 compared to the cells expressing H-Ras(G12V) without the Rac inhibitor (+Dox, −EHT 1864).
To rule out the possibility that the reciprocal changes in Rac1and Arf6 that occur in response to expression of H-Ras(G12V) might be a unique feature of the U251 glioblastoma cell line, we compared the effects of retrovirus-mediated H-Ras(G12V) expression in U251 cells side-by-side with U20S human osteosarcoma cells (Supplementary Data, Fig. S2). The results showed that the response of the osteosarcoma cell line was even more dramatic than the glioblastoma cell line. That is, Ras(G12V) expression caused a 10-fold increase in the amount of active Rac1 and a 60% decrease in the amount of active Arf6, coinciding with the accumulation of cytoplasmic vacuoles that incorporated the fluid-phase tracer, Lucifer yellow.
Rac1 Affects Arf6 via the Arf GAP, GIT1
We next addressed the question of how the activation of Rac1 might result in a shift in the nucleotide state of Arf6 toward the inactive GDP-bound form. Active Rac GTPases operate through a large number of downstream effectors to influence actin dynamics, cell migration, gene expression, cell survival and endocytosis (44). However, only a few Rac1 effectors offer potential connections with Arf6. Arfaptin-2/POR1 is a mediator of actin dynamics and membrane ruffling that can bind to both Rac1 and Arf6 (45;46). However, Arfaptin-2 binds preferentially to the inactive GDP form of Rac1 (47) and has no known effect on Arf6 GTP hydrolysis. A more likely candidate to link Rac1 to the observed decrease in GTP-bound Arf6 would be an Arf6 GAP (GTPase activating protein) that could interact with Rac1. Among the large family of Arf GAPs (48), GIT1 (also known as Cat-1 or p95-APP1) stands out because it can associate with both Rac1 and Rac3. Rac1-GDP is known to associate with GIT1 through its interaction with the PAK-interacting Rho exchange factor, βPIX. This in turn affects association of the GIT1/PIX complex with paxillin, with consequences for cell-matrix adhesion and endosomal trafficking (49–51). However, evidence from a recent study indicates that the GTP forms of Rac1 and especially Rac3 can also interact with GIT1 (52). In the case of Rac3, interaction of the GTP-bound form with GIT1 is direct, and it results in a reduction of Arf6 activity, presumably through stimulation of the GAP function of GIT1 (52). We hypothesized that if Rac1-GTP interaction with GIT1 has the same effect, GIT1 might be a key mediator of the Rac1-dependent decline in active Arf6.
To explore this possibility, we used the stable U251-Rac1(G12V)tet cell line in which constitutively active myc-Rac1(G12V) is conditionally expressed in the presence of Dox, resulting in the induction of methuosis (15). As shown in Fig. 8A, when myc-Rac1(G12V) was induced in these cells, immunoprecipitation of the active myc-Rac1(G12V) protein revealed that endogenous GIT1 was associated with it. If Dox was omitted, no detectable myc-Rac1(G12V) was expressed, and the immunoprecipitate did not contain GIT1, ruling out non-specific pull down of GIT1 with the myc antibody. Additionally, when the same experiment was done with U251 cells expressing myc-H-Ras(G12V) instead of myc-Rac1(G12V), no GIT1 was detected in the myc-H-Ras(G12V) immunoprecipitate (Fig. 8B). These observations indicate that the active form of Rac1 can associate with GIT1 in glioblastoma cells.
Figure 8.
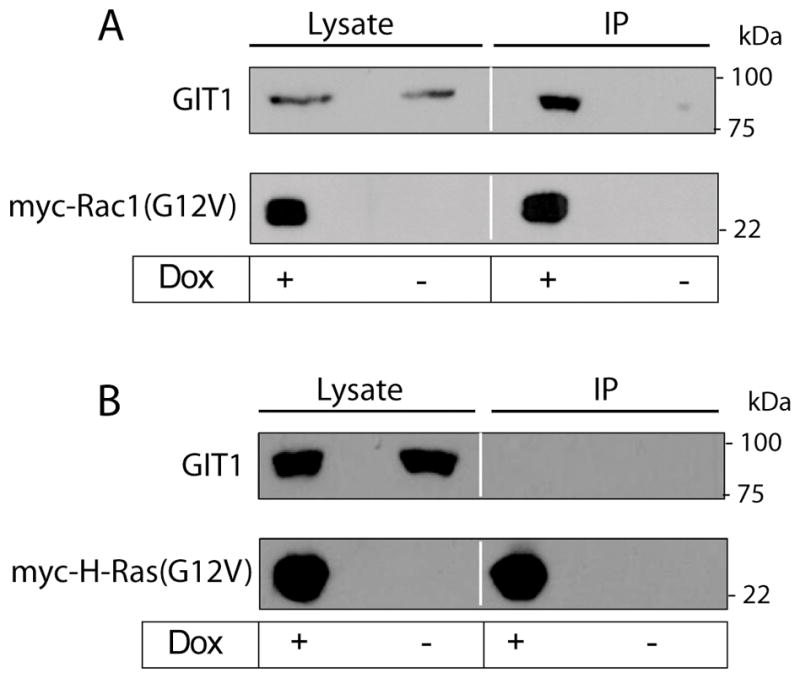
Active Rac1 interacts with the Arf6 GAP, GIT1. A) Three days after adding Dox to induce expression of myc-Rac1(G12V) in a stable U251 cell line, cells were lysed and the myc-tagged protein was collected on agarose beads conjugated with anti myc antibody. Samples of whole-cell lysate and proteins eluted from the myc-agarose beads were subjected to western blot analysis with monoclonal antibodies against GIT1 or myc. Immunoprecipitation of lysate from cells that did not receive Dox was performed to control for non-specific precipitation of GIT1. B) The same experiment was performed with stable U251 cells induced to express myc-H-Ras(G12V) to establish that GIT1 does not associate with activated H-Ras.
To assess the functional importance of GIT1 for the induction of methuosis, we generated two stable U251 cell lines in which expression of GIT1 was strongly suppressed by shRNAs targeting different sequences in the GIT1 mRNA (Fig. 9A). As a control we used a stable U251 cell line expressing shRNA against GFP. We have previously observed that myc-Rac1(G12V) can induce a methuosis phenotype that is essentially identical to that induced by myc-H-Ras(G12V) (15)(Fig. 1). Therefore, to determine if the interaction of active Rac1 with GIT1 is required for the decline in the pool of active Arf6 and the induction of vacuolization, we introduced myc-Rac1(G12V) into the control and GIT1 knockdown cell lines by retroviral transduction (Fig. 9). In contrast with published studies in which suppression of GIT1 caused cell rounding and detachment of neuronal cells (31), the glioblastoma cell lines lacking GIT1 exhibited cell morphologies and growth rates that were comparable to the control cells. When myc-Rac1(G12V) was expressed in these cell lines , the control cell line showed an approximate 50% reduction in the amount of active Arf6, compared with the same cells infected with empty retroviral vector (Fig. 9B). In contrast, expression of Rac1(G12V) in the two GIT1 KD cell lines caused a negligible reduction in the amount of active Arf6 (Fig. 9B). The preservation of active Arf6 in the GIT1 KD cells was associated with a reduced amount of cytoplasmic vacuolization and cell death in response to expression of Rac1(G21V) (Fig. 9C–E). These findings support the idea that the reduction in the amount of Arf6-GTP caused by activation of Rac1 could be mediated, at least in part, through stimulation of the GAP function of GIT1. They also reinforce the notion that the reduction in the pool of active Arf6 is an important contributing factor to the endosomal trafficking defects that underlie the methuosis phenotype.
Figure 9.

Suppression of GIT1 expression prevents the Rac1(G12V)-mediated decrease in active Arf6 and protects glioblastoma cells from methuosis. Stable cell lines were generated from U251 cells infected with lentivirus encoding shRNA against GFP (control) and two different shRNAs against GIT1 (GIT1 a & b). A) The control and GIT1 knockdown cell lines were infected with retrovirus encoding myc-Rac1(G12V), and three days after infection the cells were subjected to western blot analysis to check expression of myc-Rac1(G12V) and GIT1. B) On the fourth day after infection the control and GIT1 knockdown cell lines were assayed for the amount of active Arf6 as described in Materials and Methods. The results show the amount of active Arf6 in the cells expressing myc-Rac1(G12V), expressed as percent of the value obtained for the same cell line infected with empty retroviral vector (mean + SD for three experiments). The differences between each of the GIT1 knockdown cell lines and the control cell line were significant at p<0.004. C & D) On the fourth day after infection with empty or myc-Rac1(G12V) retroviral vectors, the control and GIT1 knockdown cell lines were examined by phase contrast microscopy to determine the percentage of cells that were vacuolated. The results of three separate experiments (mean ± SD) are shown in the bar graph. The suppression of Rac1-induced vacuolization in the GIT1 knockdown cell lines was significant at p < 0.0001 compared to the control cell line. E) On the fifth day after infection with empty or myc-Rac1(G12V) lentiviral vectors, the control and GIT1 knockdown cells were assayed for cell viability using MTT. Assays were performed on quadruplicate cultures in a 96-well plate, and the results for the cells infected with myc-Rac1(G12V) retro virus were expressed as percent of the same cells infected with empty vector. The increased viability observed in each of the GIT1 knockdown cell lines was significant at p<0.007 relative to the shRNA control cell line.
Discussion
The prognosis for patients with glioblastoma remains poor, despite efforts to improve surgical, radiological and chemotherapeutic treatment strategies. A major problem is that glioblastoma cells harbor genetic mutations that render them resistant to apoptosis (53;54). The discovery of cell death mechanisms that do not depend on activation of the classical mitochondrial and death receptor-mediated apoptotic pathways could present new opportunities to slow the progression of these aggressive tumors. In this report we have begun to define the signaling pathways underlying a non-conventional form of cell death termed methuosis, which can be triggered by ectopic expression of constitutively activated Ras in a broad spectrum of glioblastoma cell lines (15), as well as gastric carcinoma cells (14) and osteosarcoma cells lines (Supplementary Data, Fig. S2).
The hallmark cytopathological feature of methuosis is the displacement of much of the cellular cytoplasmic space by fluid filled vacuoles derived from macropinosomes or clathrin-independent endosomes (CIE) (15). Macropinosomes are formed when membrane ruffles (lamellipodia) enclose pockets of extracellular fluid and are internalized (55;56). We previously showed that cells undergoing methuosis in response to expression of constitutively active H-Ras(G12V) exhibit increased macropinocytotic uptake of fluid phase tracers (15). However, unlike normal macropinosomes, which either recycle back to the plasma membrane or merge with lysosomal compartments (57;58), labeled vesicles internalized into cells undergoing methuosis do not appear to fuse with lysosomes or release their contents at the cell surface (15). Thus, large vacuoles accumulate and eventually cause a necrosis-like rupture of the cell that cannot be prevented with caspase inhibitors.
The present studies establish that the methuosis phenotype induced by over-expression of activated H-Ras depends on downstream activation of the Rac1 GTPase (Figs 2–5). Rac1 is widely recognized as an important regulator of actin dynamics, integrin-mediated cell adhesion, and cell migration (44;59), but there is also abundant evidence implicating Rac1 in the control of endocytic vesicle trafficking (60;61). Active Rac1 appears to affect endocytosis at multiple levels. On one hand, Rac1 interacts with synaptojanin-2 and downregulates clathrin-mediated receptor endocytosis (62;63). However, the active form of Rac1 has the opposite effect on clathrin-independent endocytosis, stimulating membrane ruffling and promoting macropinocytosis (22). Our results strongly suggest that in glioblastoma cells where over-expression of activated H-Ras drives the continuous activation of endogenous Rac1, the chronic stimulation of macropinocytosis by Rac1 is an important factor in the biogenesis of the large cytoplasmic vacuoles that eventually compromise the integrity of the cell.
Our previous studies have indicated that methuosis involves dysfunction in other steps of the endosomal trafficking pathway beyond the initial stimulation of macropinocytosis. During methuosis, fluid-filled endocytic vesicles tend to accumulate instead of being recycled or merged with lysosomes (15). Our current results show that a decline in the pool of active Arf6 is important for vacuolization induced by activation of Rac1 (Fig. 7 & Supplementary Data, Fig. S2). It is likely that this is due to loss of function of Arf6-GTP, rather than gain of function of Arf6-GDP, since over-expression of the GDP-locked Arf6(T27N) does not induce vacuolization in U251 cells (unpublished observation). We also show that the decline in active Arf6 requires the presence of GIT1 (Fig. 9), an Arf6 GAP that can interact with Rac1 (Figs.8). Since activation of Arf6 is important for recycling macropinosomes back to the cell surface (64), these findings suggest a model wherein chronic activation of Rac1 plays a dual role, stimulating the formation of incoming macropinosomes while simultaneously impeding macropinosome recycling by decreasing the pool of Arf6-GTP. The resulting overload of fluid-filled vesicles may promote their abnormal fusion to generate progressively larger vacuoles which, in the absence of effective lysosomal clearance, fill the cytoplasm and disrupt the cells (Fig. 10). The postulated block in trafficking between clathrin-independent late endosomes and lysosomes is supported by the observation that vacuoles labeled with fluorescent dextran in glioma cells expressing H-Ras(G12V) do not merge with LysoTracker-positive compartments (15). Furthermore, although the Ras-induced vacuoles are not sufficiently acidic to sequester LysoTracker or acridine orange, and they do not contain lysosomal enzymes like cathepsin B, some of them do acquire LAMP1 (15) and Rab 7 (Supplementary Data, Fig. S3). These features are consistent with arrest at a late endosomal stage. The molecular basis for the latter trafficking defect remains to be defined, but Rac1 modulation of Rab7, which functions in trafficking between late endosomes and lysosomes, seems to be a likely possibility. In support of this concept, Sun et al. (65) have reported a direct interaction between Rac1 and Rab7 in osteoclasts. Very recently, Frasa et al. (66) described a novel protein named Armus, which associates with the GTP-bound form of Rac1 and inactivates Rab7 through its C-terminal GAP domain. Based on this observation, we predict that chronic activation of Rac1 might interfere with Rab7 mediated trafficking events at the interface between late endosomes and lysosomes (Fig. 10). Future investigation of the role of signaling between Rac1 and Rab7 promises to provide additional insights into the molecular basis for endosomal vacuolization in methuosis.
Figure 10.
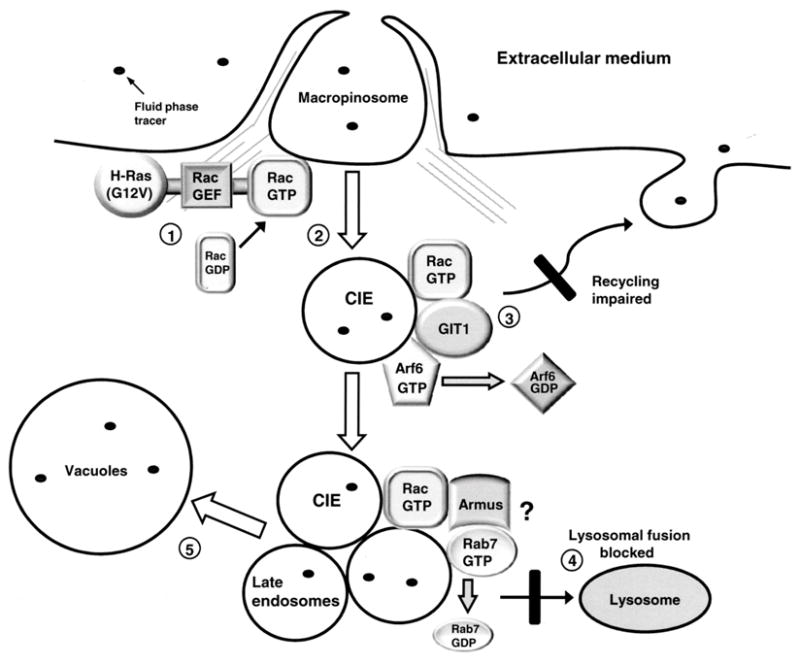
A hypothetical model for Ras-induced methuosis suggested by the present findings: 1. Expression of constitutively activated H-Ras reaches a threshold sufficient to stimulate the activity of an unidentified Rac1 guanine nucleotide exchange factor (GEF). 2. The resulting increase in the pool of active Rac1 enhances macropinocytosis. 3. As the pool of active Rac1 increases beyond normal physiological levels, Rac1-GTP associates with GIT1 and stimulates Arf6 GAP activity. The consequent decrease in the pool of active Arf6 impairs recycling of clathrin-independent endosomes (CIE). 4. CIEs derived from macropinosomes acquire some characteristics of late endosomes (Rab7, LAMP1), but fail to merge with lysosomes, due to trafficking defects at the late endosome/lysosome boundary. One possible mechanism for a block in lysosomal fusion could be Rac1 stimulation of the Rab7 GAP, Armus, resulting in inactivation of Rab7, as recently described by Frasa et al. (66). 5. The accumulated CIE and late endosomal vesicles coalesce to form progressively larger vacuolar structures, which ultimately displace most of the cytoplasm and lead to cell detachment from the substratum and disruption of cell membrane integrity.
At first glance our finding that inactivation of Arf6 contributes to vacuolization of cells expressing active Ras might seem contradictory to the observations of Porat-Shilom et al. (43), who found that active Arf6(Q67L) induces the formation of vacuoles in cells expressing H-Ras(G12V). However, the vacuoles induced by active Arf6 originate at a different stage of the endocytic pathway than those induced by inactivation of Arf6. Specifically, in cells co-expressing H-Ras(G12V) with Arf6(Q67L), vacuoles arise because nascent macropinosomes are trapped at an early stage after internalization, resulting in vesicles that contain PI (4,5)P2 and PI(3,4,5)P3, but have not yet acquired Rab5 or Rab7, which mark early and late endosomes, respectively (43). In contrast, vacuoles that form when H-Ras(G12V) activates endogenous Rac1, with concordant inactivation of Arf6, have characteristics of a late endosomal compartment that has acquired LAMP1 (15) and Rab7 (Supplementary Data, Fig. S3). This would be consistent with our model (Fig. 10) wherein clathrin-independent endosomes fail to recycle and arrest at a stage before lysosomal fusion, but after the Rab5/Rab7 conversion (67).
The ability of activated Ras to stimulate macropinocytosis has been recognized for a number of years (68;69), but the connection between dysfunctional Ras-induced macropinocytosis and cell death in specific types of cancer cells has been appreciated only recently. The present study implicates the Rac1 and Arf6 GTPases as important downstream components in this process. A major unanswered question is why expression of constitutively activated Ras mutants can promote cell proliferation and survival through classical MAPK and PI3K signaling pathways in some types of cells, but can also cause disruptions of endocytic trafficking and cell death through alterations of Rac1 and Arf6 in others. The answer might lie, at least in part, in cell type-specific variations in the expression of nucleotide exchange factors that can link activation of Ras to Rac1. In this regard our finding that neither Tiam1 nor Eps8 was essential for Ras to induce methuosis raises the possibility that other poorly characterized nucleotide exchange factors (e.g., Tiam2/Stef) or novel signaling pathways may play such a role in glioblastoma cells.
Another unsolved puzzle is why Rac1 activation can promote cell motility and invasiveness (59;70;71), yet can also trigger cytopathological changes in vesicular trafficking that lead to cell death. Our results suggest that a key to understanding the paradoxical effects of Rac1 may lie in the balance between active Arf6 and Rac1. Under normal physiological conditions Arf6 cycles between the inactive GDP state and the active GTP state. Arf6-GTP promotes cell adhesion and migration by facilitating translocation of Rac1 to the leading edge of the cell membrane and stimulating activation of Rac1 via nucleotide exchange factors like the DOCK180/Elmo complex (72) or Kalirin (73). The multifunctional scaffold protein, GIT1, also appears to play an important role in Rac1 trafficking to points of cell adhesion (49;74). GIT1 can associate with both the GDP and GTP forms of Rac1, but the indirect interaction with Rac1-GDP through βPIX appears to predominate (52). However, based on the co-immunoprecipitation of Rac1(G12V) with GIT1 (Fig. 8), we speculate that in cells where overexpression of H-Ras(G12V) drives constitutive activation of endogenous Rac1, or in cells where active Rac1(G12V) is highly expressed, a direct interaction between the GTP form of Rac1 and GIT1 may be favored over the ‘normal’ interaction with Rac1-GDP. Direct interaction between Rac3-GTP and GIT1 has been shown to reduce Arf6 activity (52). Our observation that the Rac1(G12V)-induced decrease in active Arf6 was prevented by knockdown of GIT1 (Fig. 9) suggests that Rac1-GTP may have a similar ability to reduce the pool of active Arf6 by stimulating the GAP activity of GIT1.
It is important to note that while our model (Fig. 10) may apply to glioblastoma cells and other types of cancer cells (osteosarcoma, gastric carcinoma), it remains unclear if all cell types have the potential to respond to expression of activated Ras or Rac1 by undergoing vacuolization and cell death. We previously reported that in some types of cells (HEK293, human skin fibroblasts) expression of H-Ras(G12V) did not elicit the methuosis phenotype (15). However, the results obtained in the present study with graded H-Ras(G12V) expression (Fig. 3) raised the possibility that the lack of response previously seen in some cell lines could have been due to the level of Ras expression remaining below the threshold needed for Rac1 activation. We have since revisited this issue using a retroviral vector to obtain nearly identical levels of H-Ras(G12V) over-expression in U251, U20S and HEK293T cells, and found that under these conditions the HEK 293 cells can in fact be induced to undergo morphological changes typical of methuosis (Supplementary data, Fig. S4). Other factors that might influence the responses of particular cell types might include the expression level of GIT1 relative to other Arf6 GAPs or the expression level of Arf6 itself. The question of tissue-specific variations in these proteins in different types of normal and transformed cells has not received much attention. However, the recent finding that high grade human gliomas and glioma cell lines have elevated levels of Arf6 compared with low grade tumors or normal brain tissue (75) suggests that significant variations in Arf6 signaling mechanisms may occur in some types of cancer cells.
Our initial characterization of methuosis has focused on cells where this form of death can be triggered by ectopic expression of activated Ras or Rac1. However, we believe that methuosis may represent just one example of a larger group of cytopathologies characterized by dysfunctional trafficking and vacuolization of early or late endosomal compartments. For instance, in cells exposed to the Helicobacter pylori VacA toxin (76) or the PI3K inhibitor, wortmannin (77), cells are disrupted by accumulation of large vacuoles that arise from reorganization of late endosomes and lysosomes. Interestingly, the induction of vacuoles by VacA appears to depend on Rac1 signaling, since it is potentiated by expression of activated Rac1 and inhibited by dominant-negative Rac1 (18). In another case, cultured cells over-expressing a peripheral nerve myelin protein, Gas3/PMP22 (growth arrest specific 3/peripheral myelin protein 22), have been found to accumulate vacuoles derived from an Arf6-dependent non-clathrin endosome recycling compartment (78). These observations suggest that perturbation of Rac1 and Arf6 dependent trafficking pathways may be a common mechanism in various cytopathologies associated with cytoplasmic vacuolization and necrosis-like cell death. Ultimately, as more is learned about the relevant signaling pathways, the identification of drug-like compounds that can trigger methuosis in tumor cells may provide new opportunities for treatment of cancers that do not respond well to conventional agents that work by inducing apoptosis.
Supplementary Data
Figure S1. Expression of activated Rac1 induces caspase-independent death in a stable U251 glioblastoma cell line. A) U251-Rac1(G12V)tet cells were switched to medium with or without Dox, and one day later 50 μM z-VAD-fmk was added to half of the cultures. On day-6, A) live cells were examined by phase contrast microscopy, B) western blot analysis was performed to verify expression of myc-Rac1(G12V) and C) attached and detached cells were harvested for analysis of PARP cleavage. There were no detached cells in the −Dox controls. D) Cells expressing Rac1(G12V) were subjected to MTT assays over a period of 7 days following induction of Rac1(G12V) to compare their viability with and without zVAD (the inhibitor was replenished every day). The results shown in the bar graph are from three independent experiments (mean ± SD). The decreases in viable cells on day 3 and day 7 (+/− zVAD) were significant at p≤0.04.
Figure S2. : Expression of active H-Ras in osteosarcoma cells causes reciprocal changes in active Rac1 and Arf6 comparable to those observed in glioblastoma cells. U251 and O2OS cells were infected with retrovirus encoding myc-H-Ras(G12V) or retrovirus lacking a cDNA insert (empty vector, EV). Three days later the cells were subcultured into 100 mm dishes for Arf6 and Rac1 activation assays or 35 mm dishes for Lucifer yellow labeling. A) Western blots showing comparable expression of myc-Ras(G12V) in both cell lines. B) On day-4 after infection, cells were harvested and assayed for endogenous active Rac1 and Arf6, as described in Materials and Methods. The results are from three independent experiments (mean ± SD). The increases in the amount of active Rac1 upon H-Ras(G12V) expression are significant at p≤0.005 for both the cell lines. The decreases in the amount of active Arf6 in the U251 and U2OS cells expressing H-Ras(G12V) were significant at p<0.003. C) On day- 4, live cells were photographed and at least 100 cells were scored to determine the percentage of vacuolated cells in each culture. The results are from three independent experiments (mean ± SD). D) The phase lucent vacuoles induced by the expression of myc-H-Ras(G12V) in U251 and U2OS cells incorporate the extracellular fluid-phase tracer, Lucifer yellow after a 15 min incubation.
Figure S3. Vacuoles in cells undergoing methuosis contain Rab7. A stable U251 glioblastoma cell line that expresses myc-Rac1(G12V) under the control of a tetracycline responsive promoter (15) was grown in the presence of 1 μg/ml Dox for 3 days to induce Rac1(G12V) expression and methuosis. The cells were then fixed and subjected to phase contrast and immunofluorescence microscopy. Localization of endogenous Rab7 was established with a rabbit polyclonal antibody directed against Rab7 (Cell Signaling Technology, Danvers, MA), followed by goat anti-rabbit IgG labeled with Alexa Fluor 488, using immunofluorescence procedures described previously (15). The same cell is shown in the phase contrast and immunofluorescence pictures. The selected cell is typical of most of the vacuolated cells that were examined, with some vacuoles clearly rimmed by Rab7 and others negative for Rab7.
Figure S4. Activated H-Ras can induce vacuolization in multiple cell lines when expressed at comparable levels. The indicated cell lines were infected with retrovirus encoding myc-H-Ras(G12V) or retrovirus with no cDNA insert (empty vector, EV). A) On the fourth day after infection the cells were harvested for western blot analysis with a monoclonal antibody specific for H-Ras (Santa Cruz Biotechnology, Santa Cruz, CA). The results show that the level of myc-H-Ras(G12V) expression, relative to endogenous H-Ras, was similar in all three cell lines. B) Phase contrast microscopy demonstrates that all three cell lines had accumulated numerous cytoplasmic vacuoles by the third day after infection.
Acknowledgments
We thank Dr. Laurent Désiré and ExonHit Therapeutics for providing EHT 1864, and Dr. Amy Wilson-Delfosse for the Rac1(G12V) construct.
Grant Support: The work was supported by grant R01 CA115495 (W. Maltese) from the National Institutes of Health.
References
- 1.Bursch W, Ellinger A, Gerner C, Frohwein U, Schulte-Hermann R. Programmed cell death (PCD). Apoptosis, autophagic PCD, or others? Ann N Y Acad Sci. 2000;926:1–12. doi: 10.1111/j.1749-6632.2000.tb05594.x. [DOI] [PubMed] [Google Scholar]
- 2.Lockshin RA, Zakeri Z. Apoptosis, autophagy, and more. Int J Biochem Cell Biol. 2004;36:2405–2419. doi: 10.1016/j.biocel.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 3.Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. 2004;23:2891–2906. doi: 10.1038/sj.onc.1207521. [DOI] [PubMed] [Google Scholar]
- 4.Sperandio S, de Belle I, Bredesen DE. An alternative, nonapoptotic form of programmed cell death. Proc Natl Acad Sci U S A. 2000;97:14376–14381. doi: 10.1073/pnas.97.26.14376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang Y, Li X, Wang L, Ding P, Zhang Y, Han W, Ma D. An alternative form of paraptosis-like cell death, triggered by TAJ/TROY and enhanced by PDCD5 overexpression. J Cell Sci. 2004;117:1525–1532. doi: 10.1242/jcs.00994. [DOI] [PubMed] [Google Scholar]
- 6.Majno G, Joris I. Apoptosis, oncosis, and necrosis. An overview of cell death. Am J Pathol. 1995;146:3–15. [PMC free article] [PubMed] [Google Scholar]
- 7.Trump BF, Berezesky IK, Chang SH, Phelps PC. The pathways of cell death: oncosis, apoptosis, and necrosis. Toxicol Pathol. 1997;25:82–88. doi: 10.1177/019262339702500116. [DOI] [PubMed] [Google Scholar]
- 8.Suarez Y, Gonzalez L, Cuadrado A, Berciano M, Lafarga M, Munoz A. Kahalalide F, a new marine-derived compound, induces oncosis in human prostate and breast cancer cells. Mol Cancer Ther. 2003;2:863–872. [PubMed] [Google Scholar]
- 9.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 10.Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, Hu X. Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007;6:1641–1649. doi: 10.1158/1535-7163.MCT-06-0511. [DOI] [PubMed] [Google Scholar]
- 11.Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, Cibas ES, Brugge JS. A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell. 2007;131:966–979. doi: 10.1016/j.cell.2007.10.040. [DOI] [PubMed] [Google Scholar]
- 12.Syntichaki P, Tavernarakis N. Death by necrosis. Uncontrollable catastrophe, or is there order behind the chaos? EMBO Rep. 2002;3:604–609. doi: 10.1093/embo-reports/kvf138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Chi S, Kitanaka C, Noguchi K, Mochizuki T, Nagashima Y, Shirouzu M, Fujita H, Yoshida M, Chen W, Asai A, Himeno M, Yokoyama S, Kuchino Y. Oncogenic Ras triggers cell suicide through the activation of a caspase-independent cell death program in human cancer cells. Oncogene. 1999;18:2281–2290. doi: 10.1038/sj.onc.1202538. [DOI] [PubMed] [Google Scholar]
- 15.Overmeyer JH, Kaul A, Johnson EE, Maltese WA. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol Cancer Res. 2008;6:965–977. doi: 10.1158/1541-7786.MCR-07-2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clarke PH. Developmental cell death:morphological diversity and multiple mechanisms. Anat Embryol. 1990;181:195–213. doi: 10.1007/BF00174615. [DOI] [PubMed] [Google Scholar]
- 17.Schweichel J-U, Merker HJ. The morphology of various types of cell death in prenatal tissues. Teratology. 1973;7:253–266. doi: 10.1002/tera.1420070306. [DOI] [PubMed] [Google Scholar]
- 18.Hotchin NA, Cover TL, Akhtar N. Cell vacuolation induced by the VacA cytotoxin of Helicobacter pylori is regulated by the Rac1 GTPase. J Biol Chem. 2000;275:14009–14012. doi: 10.1074/jbc.c000153200. [DOI] [PubMed] [Google Scholar]
- 19.Suzuki J, Ohnishi H, Wada A, Hirayama T, Ohno H, Ueda N, Yasuda H, Iiri T, Wada Y, Futai M, Mashima H. Involvement of syntaxin 7 in human gastric epithelial cell vacuolation induced by the Helicobacter pylori-produced cytotoxin VacA. J Biol Chem. 2003;278:25585–25590. doi: 10.1074/jbc.M212445200. [DOI] [PubMed] [Google Scholar]
- 20.Walker RM, McElligott TF. Furosemide induced hepatotoxicity. J Pathol. 1981;135:301–314. doi: 10.1002/path.1711350407. [DOI] [PubMed] [Google Scholar]
- 21.Kaul A, Overmeyer JH, Maltese WA. Activated Ras induces cytoplasmic vacuolation and non-apoptotic death in glioblastoma cells via novel effector pathways. Cell Signal. 2007;19:1034–1043. doi: 10.1016/j.cellsig.2006.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, Hall A. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–410. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 23.Castellano F, Montcourrier P, Chavrier P. Membrane recruitment of Rac1 triggers phagocytosis. J Cell Sci. 2000;113:2955–2961. doi: 10.1242/jcs.113.17.2955. [DOI] [PubMed] [Google Scholar]
- 24.Lambert JM, Lambert QT, Reuther GW, Malliri A, Siderovski DP, Sondek J, Collard JG, Der CJ. Tiam1 mediates Ras activation of Rac by a PI(3)K-independent mechanism. Nat Cell Biol. 2002;4:621–625. doi: 10.1038/ncb833. [DOI] [PubMed] [Google Scholar]
- 25.Miura K, Nam JM, Kojima C, Mochizuki N, Sabe H. EphA2 engages Git1 to suppress Arf6 activity modulating epithelial cell-cell contacts. Mol Biol Cell. 2009;20:1949–1959. doi: 10.1091/mbc.E08-06-0549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kaul A, Maltese WA. Killing of cancer cells by the photoactivatable protein kinase C inhibitor, calphostin C, involves induction of endoplasmic reticulum stress. Neoplasia. 2009;11:823–834. doi: 10.1593/neo.09388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zeng X, Overmeyer JH, Maltese WA. Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci. 2006;119:259–270. doi: 10.1242/jcs.02735. [DOI] [PubMed] [Google Scholar]
- 28.Johnson EE, Overmeyer JH, Gunning WT, Maltese WA. Gene silencing reveals a specific function of hVps34 phosphatidylinositol 3-kinase in late versus early endosomes. J Cell Sci. 2006;119:1219–1232. doi: 10.1242/jcs.02833. [DOI] [PubMed] [Google Scholar]
- 29.Ladd PD, Butler JS, Skalnik DG. Identification of a genomic fragment that directs hematopoietic-specific expression of Rac2 and analysis of the DNA methylation profile of the gene locus. Gene. 2004;341:323–333. doi: 10.1016/j.gene.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 30.Corbetta S, Gualdoni S, Albertinazzi C, Paris S, Croci L, Consalez GG, de CI. Generation and characterization of Rac3 knockout mice. Mol Cell Biol. 2005;25:5763–5776. doi: 10.1128/MCB.25.13.5763-5776.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hajdo-Milasinovic A, Ellenbroek SI, van Es S, van d V, Collard JG. Rac1 and Rac3 have opposing functions in cell adhesion and differentiation of neuronal cells. J Cell Sci. 2007;120:555–566. doi: 10.1242/jcs.03364. [DOI] [PubMed] [Google Scholar]
- 32.Bolis A, Corbetta S, Cioce A, de CI. Differential distribution of Rac1 and Rac3 GTPases in the developing mouse brain: implications for a role of Rac3 in Purkinje cell differentiation. Eur J Neurosci. 2003;18:2417–2424. doi: 10.1046/j.1460-9568.2003.02938.x. [DOI] [PubMed] [Google Scholar]
- 33.Campbell SL, Khosravi-Far R, Rossman KL, Clark GJ, Der CJ. Increasing complexity of Ras signaling. Oncogene. 1998;17:1395–1413. doi: 10.1038/sj.onc.1202174. [DOI] [PubMed] [Google Scholar]
- 34.Giehl K. Oncogenic Ras in tumour progression and metastasis. Biol Chem. 2005;386:193–205. doi: 10.1515/BC.2005.025. [DOI] [PubMed] [Google Scholar]
- 35.Shutes A, Onesto C, Picard V, Leblond B, Schweighoffer F, Der CJ. Specificity and mechanism of action of EHT 1864, a novel small molecule inhibitor of Rac family small GTPases. J Biol Chem. 2007;282:35666–35678. doi: 10.1074/jbc.M703571200. [DOI] [PubMed] [Google Scholar]
- 36.Radhakrishna H, Al Awar O, Khachikian Z, Donaldson JG. ARF6 requirement for Rac ruffling suggests a role for membrane trafficking in cortical actin rearrangements. J Cell Sci. 1999;112:855–866. doi: 10.1242/jcs.112.6.855. [DOI] [PubMed] [Google Scholar]
- 37.Welch HC, Coadwell WJ, Stephens LR, Hawkins PT. Phosphoinositide 3-kinase- dependent activation of Rac. FEBS Lett. 2003;546:93–97. doi: 10.1016/s0014-5793(03)00454-x. [DOI] [PubMed] [Google Scholar]
- 38.Li N, Batzer A, Daly R, Yajnik V, Skolnik E, Chardin P, Bar-Sagi D, Margolis B, Schlessinger J. Guanine-nucleotide-releasing factor hSos1 binds to Grb2 and links receptor tyrosine kinases to Ras signalling. Nature. 1993;363:85–88. doi: 10.1038/363085a0. [DOI] [PubMed] [Google Scholar]
- 39.Innocenti M, Zippel R, Brambilla R, Sturani E. CDC25(Mm)/Ras-GRF1 regulates both Ras and Rac signaling pathways. FEBS Lett. 1999;460:357–362. doi: 10.1016/s0014-5793(99)01374-5. [DOI] [PubMed] [Google Scholar]
- 40.Innocenti M, Tenca P, Frittoli E, Faretta M, Tocchetti A, Di Fiore PP, Scita G. Mechanisms through which Sos-1 coordinates the activation of Ras and Rac. J Cell Biol. 2002;156:125–136. doi: 10.1083/jcb.200108035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peters PJ, Hsu VW, Ooi CE, Finazzi D, Teal SB, Oorschot V, Donaldson JG, Klausner RD. Overexpression of wild-type and mutant ARF1 and ARF6: distinct perturbations of nonoverlapping membrane compartments. J Cell Biol. 1995;128:1003–1017. doi: 10.1083/jcb.128.6.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Radhakrishna H, Donaldson JG. ADP-ribosylation factor 6 regulates a novel plasma membrane recycling pathway. J Cell Biol. 1997;139:49–61. doi: 10.1083/jcb.139.1.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Porat-Shliom N, Kloog Y, Donaldson JG. A unique platform for H-Ras signaling involving clathrin-independent endocytosis. Mol Biol Cell. 2008;19:765–775. doi: 10.1091/mbc.E07-08-0841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 45.Van Aelst L, Joneson T, Bar-Sagi D. Identification of a novel Rac1-interacting protein involved in membrane ruffling. EMBO J. 1996;15:3778–3786. [PMC free article] [PubMed] [Google Scholar]
- 46.D’Souza-Schorey C, Boshans RL, McDonough M, Stahl PD, Van Aelst L. A role for POR1, a Rac1-interacting protein, in ARF6-mediated cytoskeletal rearrangements. EMBO J. 1997;16:5445–5454. doi: 10.1093/emboj/16.17.5445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Shin OH, Exton JH. Differential binding of arfaptin 2/POR1 to ADP-ribosylation factors and Rac1. Biochem Biophys Res Commun. 2001;285:1267–1273. doi: 10.1006/bbrc.2001.5330. [DOI] [PubMed] [Google Scholar]
- 48.Inoue H, Randazzo PA. Arf GAPs and their interacting proteins. Traffic. 2007;8:1465–1475. doi: 10.1111/j.1600-0854.2007.00624.x. [DOI] [PubMed] [Google Scholar]
- 49.Di Cesare A, Paris S, Albertinazzi C, Dariozzi S, Andersen J, Mann M, Longhi R, de CI. p95-APP1 links membrane transport to Rac-mediated reorganization of actin. Nat Cell Biol. 2000;2:521–530. doi: 10.1038/35019561. [DOI] [PubMed] [Google Scholar]
- 50.Turner CE, Brown MC, Perrotta JA, Riedy MC, Nikolopoulos SN, McDonald AR, Bagrodia S, Thomas S, Leventhal PS. Paxillin LD4 motif binds PAK and PIX through a novel 95-kD ankyrin repeat, ARF-GAP protein: A role in cytoskeletal remodeling. J Cell Biol. 1999;145:851–863. doi: 10.1083/jcb.145.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paris S, Longhi R, Santambrogio P, de CI. Leucine-zipper-mediated homo- and hetero-dimerization of GIT family p95-ARF GTPase-activating protein, PIX-, paxillin-interacting proteins 1 and 2. Biochem J. 2003;372:391–398. doi: 10.1042/BJ20030047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hajdo-Milasinovic A, van der Kammen RA, Moneva Z, Collard JG. Rac3 inhibits adhesion and differentiation of neuronal cells by modifying GIT1 downstream signaling. J Cell Sci. 2009;122:2127–2136. doi: 10.1242/jcs.039958. [DOI] [PubMed] [Google Scholar]
- 53.Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens A-C, Van Meir EG. Frequent co-alterations of TP53, p16/CDKN2A, p14arf, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9:469–479. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Furnari FB, Fenton T, Bachoo RM, Mukasa A, Stommel JM, Stegh A, Hahn WC, Ligon KL, Louis DN, Brennan C, Chin L, DePinho RA, Cavenee WK. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21:2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 55.Swanson JA, Watts C. Macropinocytosis. Trends Cell Biol. 1995;5:424–428. doi: 10.1016/s0962-8924(00)89101-1. [DOI] [PubMed] [Google Scholar]
- 56.Donaldson JG, Porat-Shliom N, Cohen LA. Clathrin-independent endocytosis: A unique platform for cell signaling and PM remodeling. Cell Signal. 2009;21:1–6. doi: 10.1016/j.cellsig.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Racoosin EL, Swanson JA. Macropinosome maturation and fusion with tubular lysosomes in macrophages. J Cell Biol. 1993;121:1011–1020. doi: 10.1083/jcb.121.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chan AY, Coniglio SJ, Chuang YY, Michaelson D, Knaus UG, Philips MR, Symons M. Roles of the Rac1 and Rac3 GTPases in human tumor cell invasion. Oncogene. 2005;24:7821–7829. doi: 10.1038/sj.onc.1208909. [DOI] [PubMed] [Google Scholar]
- 60.Ridley AJ. Rho proteins: linking signaling with membrane trafficking. Traffic. 2001;2:303–310. doi: 10.1034/j.1600-0854.2001.002005303.x. [DOI] [PubMed] [Google Scholar]
- 61.Symons M, Rusk N. Control of vesicular trafficking by Rho GTPases. Curr Biol. 2003;13:R409–R418. doi: 10.1016/s0960-9822(03)00324-5. [DOI] [PubMed] [Google Scholar]
- 62.Lamaze C, Chuang TH, Terlecky LJ, Bokoch GM, Schmid SL. Regulation of receptor- mediated endocytosis by Rho and Rac. Nature. 1996;382:177–179. doi: 10.1038/382177a0. [DOI] [PubMed] [Google Scholar]
- 63.Malecz N, McCabe PC, Spaargaren C, Qiu R, Chuang Y, Symons M. Synaptojanin 2, a novel Rac1 effector that regulates clathrin-mediated endocytosis. Curr Biol. 2000;10:1383–1386. doi: 10.1016/s0960-9822(00)00778-8. [DOI] [PubMed] [Google Scholar]
- 64.Grant BD, Donaldson JG. Pathways and mechanisms of endocytic recycling. Nat Rev Mol Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sun Y, Buki KG, Ettala O, Vaaraniemi JP, Vaananen HK. Possible role of direct Rac1-Rab7 interaction in ruffled border formation of osteoclasts. J Biol Chem. 2005;280:32356–32361. doi: 10.1074/jbc.M414213200. [DOI] [PubMed] [Google Scholar]
- 66.Frasa MA, Maximiano FC, Smolarczyk K, Francis RE, Betson ME, Lozano E, Goldenring J, Seabra MC, Rak A, Ahmadian MR, Braga VM. Armus is a Rac1 effector that inactivates Rab7 and regulates E-cadherin degradation. Curr Biol. 2010;20:198–208. doi: 10.1016/j.cub.2009.12.053. [DOI] [PubMed] [Google Scholar]
- 67.Rink J, Ghigo E, Kalaidzidis Y, Zerial M. Rab conversion as a mechanism of progression from early to late endosomes. Cell. 2005;122:735–749. doi: 10.1016/j.cell.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 68.Bar-Sagi D, Feramisco JR. Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science. 1986;233:1061–1068. doi: 10.1126/science.3090687. [DOI] [PubMed] [Google Scholar]
- 69.Walsh AB, Bar-Sagi D. Differential activation of the Rac pathway by Ha-Ras and K-Ras. J Biol Chem. 2001;276:15609–15615. doi: 10.1074/jbc.M010573200. [DOI] [PubMed] [Google Scholar]
- 70.Keely PJ, Westwick JK, Whitehead IP, Der CJ, Parise LV. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997;390:632–636. doi: 10.1038/37656. [DOI] [PubMed] [Google Scholar]
- 71.Edwards DC, Sanders LC, Bokoch GM, Gill GN. Activation of LIM-kinase by Pak1 couples Rac/Cdc42 GTPase signalling to actin cytoskeletal dynamics. Nat Cell Biol. 1999;1:253–259. doi: 10.1038/12963. [DOI] [PubMed] [Google Scholar]
- 72.Santy LC, Ravichandran KS, Casanova JE. The DOCK180/Elmo complex couples ARNO-mediated Arf6 activation to the downstream activation of Rac1. Curr Biol. 2005;15:1749–1754. doi: 10.1016/j.cub.2005.08.052. [DOI] [PubMed] [Google Scholar]
- 73.Koo TH, Eipper BA, Donaldson JG. Arf6 recruits the Rac GEF Kalirin to the plasma membrane facilitating Rac activation. BMC Cell Biol. 2007;8:29. doi: 10.1186/1471-2121-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Premont RT, Perry SJ, Schmalzigaug R, Roseman JT, Xing Y, Claing A. The GIT/PIX complex: an oligomeric assembly of GIT family ARF GTPase-activating proteins and PIX family Rac1/Cdc42 guanine nucleotide exchange factors. Cell Signal. 2004;16:1001–1011. doi: 10.1016/j.cellsig.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 75.Li M, Wang J, Ng SS, Chan CY, He ML, Yu F, Lai L, Shi C, Chen Y, Yew DT, Kung HF, Lin MC. Adenosine diphosphate-ribosylation factor 6 is required for epidermal growth factor-induced glioblastoma cell proliferation. Cancer. 2009;115:4959–4972. doi: 10.1002/cncr.24550. [DOI] [PubMed] [Google Scholar]
- 76.Molinari M, Galli C, Norais N, Telford JL, Rappuoli R, Luzio JP, Montecucco C. Vacuoles induced by Helicobacter pylori toxin contain both late endosomal and lysosomal markers. J Biol Chem. 1997;272:25339–25344. doi: 10.1074/jbc.272.40.25339. [DOI] [PubMed] [Google Scholar]
- 77.Reaves BJ, Bright NA, Mullock BM, Luzio JP. The effect of wortmannin on the localisation of lysosomal type I integral membrane glycoproteins suggests a role for phosphoinositide 3-kinase activity in regulating membrane traffic late in the endocytic pathway. J Cell Sci. 1996;109:749–762. doi: 10.1242/jcs.109.4.749. [DOI] [PubMed] [Google Scholar]
- 78.Chies R, Nobbio L, Edomi P, Schenone A, Schneider C, Brancolini C. Alterations in the Arf6-regulated plasma membrane endosomal recycling pathway in cells overexpressing the tetraspan protein Gas3/PMP22. J Cell Sci. 2003;116:987–999. doi: 10.1242/jcs.00326. [DOI] [PubMed] [Google Scholar]