

Abstract
Ferlins are known to regulate plasma membrane repair in muscle cells, and are linked to muscular dystrophy and cardiomyopathy. Recently, using proteomic analysis of caveolae/lipid rafts, we reported that endothelial cells (EC) express Myoferlin and that it regulates membrane expression of vascular endothelial growth factor receptor-2 (VEGFR-2).
Objective
To document the presence of other Ferlins in EC.
Results
We report that EC express another Ferlin, Dysferlin, and that in contrast to Myoferlin, it does not regulate VEGFR-2 expression levels or downstream signalling (nitric oxide and Erk1/2 phosphorylation). Instead, loss of Dysferlin in sub-confluent EC results in deficient adhesion followed by growth arrest, an effect not observed in confluent EC. In vivo, Dysferlin was also detected in intact and diseased blood vessels of rodent and human origin, and angiogenic challenge of Dysferlin-null mice results in impaired angiogenic response compared to control mice. Mechanistically, loss of Dysferlin in cultured EC causes poly-ubiquitination and proteasomal degradation of platelet endothelial cellular adhesion molecule-1 (PECAM-1/CD31), an adhesion molecule essential for angiogenesis. In addition, adenoviral-mediated gene transfer of PECAM-1 rescues the abnormal adhesion of EC caused by Dysferlin gene silencing.
Conclusion
Our data describe a novel pathway for PECAM-1 regulation and broaden the functional scope of Ferlins in angiogenesis and specialized Ferlin-selective protein cargo trafficking in vascular settings.
Introduction
Dysferlin and Myoferlin are members of the Ferlin family of proteins. The name Ferlin is derived from FER-1, a protein required for the correct fusion of specialized membranous organelles with the plasma membrane of sperms during spermiogenesis in Caenorhabditis elegans1. In mammalian cells, Dysferlin was the first Ferlin shown to regulate membrane fusion events at the plasma membrane of skeletal muscle cells2. Akin to the application of ‘patches’ at sites of damage, endovesicle fusion occurs at the sarcomere of skeletal muscle following physical injury3; the triggering effect of unregulated extracellular calcium (Ca2+) entry into cells is believed to activate the Ca2+-binding C2 domains of Ferlins and lead to specific interactions with membrane phospholipids and fusion of membrane vesicles4, 5. Interestingly, a growing number of peripheral functions were subsequently attributed to the presence of Ferlins in various settings; changes in Dysferlin activity or expression have been reported in preeclampsia6, cardiomyopathy7, Alzheimer’s disease8 and multiple sclerosis9, whereas Myoferlin, another Ferlin family member, plays a role in the pathogenesis of both muscular dystrophy and cardiomyopathy10, 11. Another salient example of growing functions for Ferlins is Otoferlin, which is linked to a recessive form of deafness in humans and mice 12.
Recently, we have documented the unexpected expression of Myoferlin in cultured vascular endothelial cells (EC) and intact blood vessels though proteomics identification of caveolae and/or lipid rafts resident proteins13. Loss of Myoferlin results in attenuation of proliferation, migration and nitric oxide (NO) release following vascular endothelial growth factor (VEGF) challenge and this coincides with a near complete loss of surface expression of VEGF receptor-2 (VEGFR-2) due to increased poly-ubiquitination and degradation13. This, combined with our recent characterization of Myoferlin in EC endocytosis14 raises the possibility that the involvement of Ferlins in non-muscle systems may reside in their yet poorly described ability to regulate both vesicle fusion and client protein trafficking as cargos of putative membrane-bound ‘patches’.
Herein, we report that cultured EC also express Dysferlin, and that in stark contrast to Myoferlin, Dysferlin does not participate in VEGFR-2 expression. Instead, we show that Dysferlin gene silencing causes near-complete inhibition of proliferation in sub-confluent EC due to EC detachment from their growth surface and most intriguingly, confluency or sufficient cell-cell contact can provide complete protection against such adhesion defect. Dysferlin protein expression can be detected in the aorta, mesenteric and coronary arteries of rodent or human origins and agonist-induced angiogenic challenge of Dysferlin-null mice results in deficient angiogenesis, supporting an active role for Dysferlin in endothelial homeostasis in vivo and in vitro. Mechanistically, we show that the loss of Dysferlin in sub-confluent cells causes mislocalisation followed by poly-ubiquitination and proteasomal degradation of platelet-endothelial cellular adhesion molecule (PECAM)-1, a transmembrane protein essential for angiogenesis. Furthermore, the adhesion defect caused by Dysferlin silencing can be rescued by adenoviral over-expression of PECAM-1. Together, these data identify a novel pathway for PECAM-1 regulation, and confirm that Dysferlin participates in vascular homeostasis. We also propose that Ferlins are profoundly heterogeneous in their capacity to regulate different membrane remodelling events, and this is likely attributable to the fusion of membrane vesicles containing unique protein cargo.
Materials and Methods
Cell culture
Native BAEC and HUVEC were isolated from bovine aorta and purchased from Lonza (Bazel, Switzerland) respectively. Cells were grown in high glucose Dulbecco’s modified Eagle’s (DMEM) or M199 mediums (Invitrogen) supplemented with FBS (Hyclone, South Logan, UT), L-glutamine, ECGS and penicillin-streptomycin (Sigma, St. Louis, MI). Full-length PECAM-1-coding adenovirus was amplified and used as previously described15. Transwells (0.4 μM pores) were from Corning.
RNA isolation, RT-PCR and Northern blot analysis
Total RNA was isolated from confluent cells with a RNA extraction kit (Qiagen). cDNA synthesis was performed using dT oligos (Superscript, Invitrogen). PCR was performed using the following degenerated bovine-human Dysferlin-specific PCR oligos: 5-GGAACAGGTTCCTGGGGGAAGC-3 and 5-TGGTGGAGGCCAGCGGCAG-3. Northern blot analysis was performed using a 269 bp human Dysferlin probe synthesized with the 5-GGAACAGGTTCCTGGGGGAAGC-3 and 5-GTCAGGCAGAGTCGGGGAGG-3 oligos with high homology to bovine and human Dysferlin cDNA13.
Plasmids, cell transfection and visualization
BAEC were transfected with 2 μg of a β-Gal, HA-ubiquitin (a kind gift from Dr. Alex Toker; Beth Israel Deaconess Medical Centre) or a previously characterized GFP-Dysferlin-fused plasmid (N-terminus tagged, kindly provided by Dr. Kate Bushby; Newcastle University) for 6 h with Opti-MEM I and lipofectamine 2000, and visualized by using a Leica AOBS confocal microscope and confocal z-stack images were acquired at 1-μm intervals for 15 μm in accordance with the Nyquist criterion (Volocity software).
Western Blot (WB) analysis, immunoprecipitation (IP), immunofluorescence (IF) and CEM/LR isolation
The antibodies used for WB are β-coatamer protein (β-COP, rabbit, ABR), phospho- and total ERK-1/2 (rabbit and mouse, Cell Signalling), VEGFR-2 and Cav-1 (rabbit, Santa Cruz Biotechnologies), HSP90 (mouse, BD), Dysferlin Hamlet 1 and 2 (mouse, Novocastra), PECAM-1 (mouse and rabbitt, Santa Cruz), PECAM-1 and VE-cadherin (Cell Signal), HA (rat, Roche). IP was performed with protein-G-coated beads (Sigma). For cell IF, PECAM-1 antibody was from Cell Signal and used on paraformaldehyde-fixed HUVEC as described13. CEM/LR preparation was performed as described 16.
siRNA treatment
Bovine Dysferlin cDNA target sequences (Dharmacon) were (5’-3’): NNGGAAGAAAUCGGUGAGUGA (Dysf1), NNCGGACAAGCCGCAGGACUU (Dysf2) and their respective scrambled non-silencing controls NNAAUGAGGACUGGGGAAACG (NS1) and AAGGCCGGGGCCCAAAUUC (NS2). Human Dysferlin DNA target sequences were synthesized with the 4-for-silencing program (Xeragon/Qiagen) and target the following DNA sequences: Dysf#1 CAGCGTGAACCCTGTATGGAA, #2 CACGATTTCCTGCATATTCGA, #3 CTGGGTGACATCCATGAGACA, #4 CTCCCTGTTTGCGGCCTTCTA 13.
Proliferation, viability, apoptosis assays, NO release measurement and phosphorylation assay
BAEC treatment for proliferation and survival assays, and NO chemiluminescence were performed as described 13.
Ear and in vitro angiogenesis assay
Animal experiments were performed in accordance with the UBC Animal Care Committee. Adenoviruses encoding murine VEGF-A164 (approximately 7 × 108 viral particles in 15 μL) were injected into the right ear of Dysferlin-null or age and sex-matched wild type mice (JAX, Bar Harbor, ME). The left ear of mice was injected with a β-galactosidase (β-Gal) virus. After 6 days, animals were euthanized, ears isolated, cut longitudinally in two and embedded in OCT medium to obtain full-length sections of the central ear area. For in vitro angiogenesis assays, rat tail collagen gel (BD) was prepared by mixing 7 volumes of Collagen I solution (final 2 mg/mL in 0.02N acetic acid) with 1 volume of 10X MEM and 2 volumes of NaHCO3 (1.17 mg/mL final). A 200 μl cell-free layer of collagen mixture was allowed to gel, followed by addition of a 400 μL top layer of collagen mixture containing 5,000 HUVEC treated with siRNA. Cells were then grown by adding a third layer of M199 Medium containing 20% FBS. Tube-like structures and cell numbers were visually counted. Non-embedded HUVEC time 0 controls treated with siRNA showed greater than 90% viability.
Immuno-histochemistry
Human coronary artery specimens were obtained from the James Hogg iCAPTURE Biobank tissue collection. Vascular tissues fixed and subjected to antigen retrieval (20 mins, 120°C, 30psi) in citrate (Dako, Mississauga, ON), quenched, blocked in 10% normal rabbit serum, incubated with anti-Dysferlin goat antibodies (Santa Cruz Biotechnologies) and processed using the Vectastain biotin-avidin detection kit (Vector Laboratories) and NovaRED HP substrate reagent (Vector Laboratories). Image digitization of positive and negative (IgG) conditions was done at the same time using an Aperio ScanScope digital slide scanner. For ears, frozen sections (5 μm thick) were immunostained with rat monoclonal anti-mouse PECAM-1 or VE-Cadherin primary antibodies (BD Biosciences) and Cy5 conjugated secondary antibodies. Positive structures away from the cartilage area of ears were counted. No background (IgG) was subtracted.
Statistical analysis
Data are presented as mean ± standard of the mean and considered at statistically different if P < 0.05 by Analysis of Variance followed by Dunnet’s t test.
Results
Dysferlin mRNA and protein are expressed in cultured EC
To show Dysferlin mRNA expression in EC, RT-PCR and Northern blot analysis were performed on BAEC and HUVEC mRNA. In both cell lines, RT-PCR resulted in the detection of the expected 1.5 Kbp Dysferlin amplicon (Fig. 1A), whereas incubation of mRNA templates with RNAse H before RT caused loss of amplification (lanes 3, 5) thereby confirming mRNA dependency. Using a Dysferlin cDNA probe and BAEC and HUVEC mRNA (Fig. 1B, left), Northern blot analysis revealed the presence of a 7.5-9.9 Kbp transcript (arrow, right). WB using BAEC or HUVEC protein lysates confirmed Dysferlin protein expression with both Dysferlin antisera (Fig. 1C, 250kDa marker). Skeletal muscle protein and β-COP were used as positive and loading controls, respectively.
Figure 1. Cultured EC express Dysferlin.

A) Detection of a 1.5Kbp Dysferlin-specific amplicon by RT-PCR with primary BAEC or HUVEC mRNA as templates. Amplification was sensitive to RNAse treatment before RT. B) Northern blot analysis using BAEC and HUVEC total mRNA (left) showed positive Dysferlin mRNA expression (arrow; right). C) WB analysis of Dysferlin protein expression in HUVEC, BAEC and mouse skeletal muscle lysates using two antisera (Dysf Ab #1 and 2). Loading control was β-COP. D) Confocal optical section (xy plane) and sectional views (xz plane of nuclei and Golgi) showing GFP-Dysferlin expression in live BAEC. Color gradient is shown to illustrate GFP signal intensity levels. Scale bar 10 μm. E) Enrichment of Dysferlin in CEM/LR (light fractions) from HUVEC and BAEC lysates. Blotting against HSP90 and β-COP was performed to show lack of bulk and Golgi apparatus proteins in CEM/LR.
To characterize Dysferlin subcellular localization, we used a GFP-tagged version of Dysferlin shown to behave similarly to endogenous Dysferlin 17. GFP-positive signal was detected throughout live (unfixed) BAEC transiently expressing GFP-Dysferlin (Fig 1D; gradient color xy plane image). Immuno-fluorescence allowed the identification of nuclear and Golgi structures (DAPI and GM130, respectively, Fig. S1A), and high GFP-Dysferlin expression was found in xz-plane sectional views of the nucleus and Golgi apparatus (Fig. 1D, xz planes).
Finally, enrichment of Dysferlin in specialized cholesterol-enriched microdomains/lipid rafts (CEM/LR) of the plasma membrane was determined by sucrose fractionation as previously described for Myoferlin13. Dysferlin was found to be enriched in BAEC and HUVEC light, cholesterol-rich CEM/LR fractions (Fig. 1E); these fractions were also enriched in caveolae protein Cav-1 and lacked Golgi/post-Golgi contaminants (β-COP, heavy fractions) and bulk plasma membrane markers such as HSP90 13. Similarly to Myoferlin, cholesterol disruption using methyl-β-cyclodextrin increased Dysferlin solubility in Triton X-100-based buffers (data not shown) 13, an additional sign that Dysferlin is a CEM/LR resident protein. Collectively, these data identify Dysferlin as an EC protein enriched in cholesterol-rich microdomains.
Loss of Dysferlin does not impair VEGFR-2 expression or signalling in EC
Since Myoferlin was shown to regulate VEGFR-213 we hypothesized that Dysferlin also participates in VEGFR-2 trafficking, and gene silencing techniques were used to perform loss of function studies. Transfection of near confluent BAEC or HUVEC cultures with bovine (Dysf1, Dysf2) or human Dysferlin siRNA (Dysf1-Dysf4) (75 nM; Dysf1-2 shown) caused up to 52, 68 and 81% (BAEC) and 62, 82 and 94% (HUVEC) decrease in Dysferlin protein expression at 24, 48 and 72h respectively compared with two scrambled non-silencing siRNA (NS1, NS2) (Fig. 2A,B; 72h shown). Surprisingly, IP and WB analysis revealed that Dysf1-2 (72 h) did not cause down-regulation of VEGFR-2 and Tie-2 (another angiogenic tyrosine kinase receptor) (Fig. 2A,B) or VEGF/VEGFR-2-induced NO release and Erk1/2 phosphorylation (Fig. S1B,C)13. Together, these data indicate that Dysferlin gene silencing does not decrease VEGFR-2 expression or downstream signaling and document specific ‘cargo’ proteins for Dysferlin vs Myoferlin-dependent trafficking events.
Figure 2. Dysferlin gene silencing causes loss of proliferation through impaired adhesion in sub-confluent but not confluent EC.

A,B) Down-regulation of Dysferlin in near-confluent BAEC or HUVEC did not decrease VEGFR-2 or Tie-2 expression. Cells were treated with bovine or human-specific Dysferlin (Dysf1-2) or scrambled non-silencing siRNA (NS1-2). WB analysis against Dysferlin (top) or VEGFR-2 or Tie-2 following IP (IP) (bottom blots) was performed. C) Dysferlin gene silencing decreases BAEC proliferation. BAEC were seeded (approx. 5% confluency), transfected with siRNA sequences and starved in 0.1% FBS. Cells were then stimulated in 10% FBS or starved further and counted at 24, 48, or 72h. N = 8 in triplicate per condition. D) Loss of Dysferlin caused rapid defect in BAEC adhesion. Cells were treated as described in C and the adherent vs total cell ratio was determined by collecting unadhered cells and trypsinizing adhered cells for quantification. N = 6 per group in duplicate. E) Decreased angiogenesis following Dysferlin knock-down in vitro. Following siRNA treatment, confluent HUVEC were embedded in collagen gel with 10% FBS, and average length of tube-like structures per area (length mm/mm2) and proliferation (HUVEC/mm2) were quantified at 24 and 48 post-21 embedding. Typical data are shown, presented as mean +/- S.E.M. * P<0.001 compared to their respective non-silencing siRNA controls.
Dysferlin regulates proliferation and adhesion of sub-confluent EC
To determine if Dysferlin regulates basic EC functions such as proliferation, sub-confluent BAEC (covering less than 5% of growth surface) were treated with siRNA sequences (6h; Day -1), starved (0.1% FBS) to induce G0 synchronization, and incubated in 10% FBS containing medium, inducing a time-dependent increase in proliferation of control BAEC (Fig. 2C, black bars, left; 24, 48 and 72 h) whereas Dysferlin siRNA-treated BAEC (white bars) showed near complete (>85%; 72 h) inhibition of proliferation (P < 0.001). Incubation of BAEC in starvation medium caused minimal proliferation of control cells (Fig. 2C, black bars, right) but Dysferlin siRNA-treated cells (white bars) showed lower counts than Day 0. This suggested that decreased proliferation is not the primary outcome of Dysferlin knock-down but rather a secondary consequence of another cellular defect.
To test if loss of Dysferlin causes necrosis or apoptosis, Trypan blue exclusion and Caspase-8 activity were determined in total BAEC (adherent and non-adherent) grown under high FBS conditions (similarly to Fig. 2C). Forty-eight (48) h after siRNA treatment, no difference was observed between the Dysferlin and non-silencing siRNA-treated groups (Fig. S2A, B), suggesting that loss of Dysferlin did not promote necrosis or apoptosis.
To determine if the decreased proliferation observed in Fig. 2C was a result of aberrant adhesion, the ratio of adhered vs total cells was established in BAEC grown under similar conditions. Twenty-four (24), 48 and 72 h following Dysferlin siRNA treatment, up to 28, 70 and 81% of BAEC and 25, 34 and 42% of HUVEC showed deficient adhesion (Fig. 2D and S2C,D; P < 0.001 compared to non-silencing siRNA). High FBS (10%) concentrations, which caused active BAEC growth, did not prevent the loss of adhesion (Fig. 2D). In HUVEC, loss of adhesion peaked at 120h post-siRNA treatment (68% deficiency; data not shown). Incubation of Dysferlin siRNA-treated BAEC in normal BAEC-conditioned medium or co-cultured with untreated confluent BAEC grown on Transwells, which allowed the release of soluble factors, did not rescue the adhesion defect (data not shown), arguing against aberrant soluble cell signaling as a result of Dysferlin silencing. Moreover, coating of plates with gelatin or poly-arginine to compensate for changes in growth matrix requirements did not improve adhesion either (data not shown). In contrast, BAEC seeded at higher or near-confluency levels improved or completely rescued adhesion after Dysferlin siRNA treatment, respectively (Fig. S2E) indicating that increased cell-cell contact likely rescues adhesion to the growth surface. Re-adhesion assays provided evidence that confluent (protected) cells transfected with Dysferlin siRNA exhibited impaired adhesion when trypsinized and re-seeded 24h and 48h post-transfection at low confluency levels (Fig. S2F), which suggests that the protective effect of cell-cell contact is only transient. As expected, proliferation assays performed with cells seeded at approximately 50% confluency, which allows significant cell-to-cell contact, following Dysferlin siRNA treatment induced similar proliferation rates as the control siRNA-treated group (10% FBS; Fig. S3A). Hence, these data depict a role for Dysferlin in regulating the adhesion machinery and mitogenesis in sub-confluent EC.
Angiogenesis is known to rely on the partial detachment of EC from their basal membrane followed by migration and proliferation to form new vessels. To assess the effect of Dysferlin silencing on angiogenesis in vitro, HUVEC were treated with Dysferlin siRNA sequences and imbedded in collagen I gel. As depicted in Fig. 2E, loss of Dysferlin caused a decrease in tube-like structure formation compared to control siRNA treatment (top panels, 24 and 48h) as well as shorter tube-like structures and decreased proliferation (bottom left and right) under high FBS conditions, an indication of blunted angiogenesis in vitro following Dysferlin gene silencing.
Dysferlin is expressed in human and rodent blood vessels
Since Dysferlin is stably expressed in cultured EC, immunohistochemistry was performed to characterize its expression in vascular tissues by using two different Dysferlin antisera (C19 and E20 goat; C19 shown; Fig. 3A-D). Antibody specificity was confirmed using paraffin-embedded sections of wild type (WT) and Dysferlin-null mice aortas. Dysferlin (brown staining) was highly detected in the endothelial layer (intima I, arrows), adventitia (A) including the extracellular material, and to a lesser extent the medial (M) smooth muscle cells of WT vessels, with little staining in Dysferlin-null aorta (Fig. 3A). Dysferlin was also detected in human coronary arteries with age-related hyperplasia (Fig. 3B, neointima, NI), intact rat aorta (Fig. 3C) and smaller vessels such as the mouse superficial femoral artery (Fig. 3D). Cell nuclei were stained in blue. Staining of adjacent section with isotype-matched IgG antisera (right) produced little staining, supporting Dysferlin expression in vivo.
Figure 3. Dysferlin is expressed in blood vessels.

A) Specificity of Dysferlin staining was confirmed by positive and negative Dysferlin detection in WT and Dysferlin-null mouse aorta paraffin sections, respectively. Scale bar 20 μm. B-D) Shown are 5 μm-thick adjacent sections of a human coronary artery with age-related hyperplasia (B; scale bar 100 μm), rat aorta (C; scale bar 25 μm) and mouse superficial femoral artery (D; scale bar 20 μm) stained with one of two Dysferlin antiserum (left image, brown color) or a non-immune IgG (right). Adventia (A), media (M) and neointima (NI). Counter-stain was performed with hematoxylin (blue). All immunostaining and image processing were performed in parallel, allowing direct comparison. Arrows indicate endothelial staining.
Dysferlin-null mice show impaired agonist-induced angiogenesis
The high expression of Dysferlin in blood vessels and its role in EC biology suggest that Dysferlin might influence vascular homeostasis in vivo, such as angiogenesis. To directly test this hypothesis, adenoviruses coding for murine VEGF164 (AdVEGF), an endothelial-specific angiogenic agonist, or β-galactosidase (Adβ-Gal) were intradermally injected into the ears of Dysferlin-deficient mice (Dysf -/-) and wild-type controls. Six days post-injection, visualization and quantification (Fig. 4A) of EC-positive structures by anti-VE-cadherin or PECAM-1 immunochemistry (red channel, PECAM-1 shown) documented a 2.4-fold increase (*P < 0.05, **P < 0.01) in total blood vessel count induced by AdVEGF in WT mice (n = 7), whereas in Dysf -/- mice the increase was only 0.8-fold and statistically different from WT mice (+ < 0.05) (Fig. 4A), indicating decreased angiogenesis in Dysferlin-null mice. However, VEGF- and β-Gal-induced edema formation, quantified by measuring the thickness of H&E stained ears sections as an indication of VEGF/VEGFR-2 activity18, was found to be similar between WT and Dysf-/- mice (Fig. 4B). Moreover, quantification of VEGFR-2 expression in EC-rich lung extracts (Fig. C) was also similar. These data indicate that genetic loss of Dysferlin leads to blunted neo-vascularization without causing autonomous defects in EC or decreased VEGF/VEGFR-2 signaling.
Figure 4. Dysferlin regulates neovascularisation.
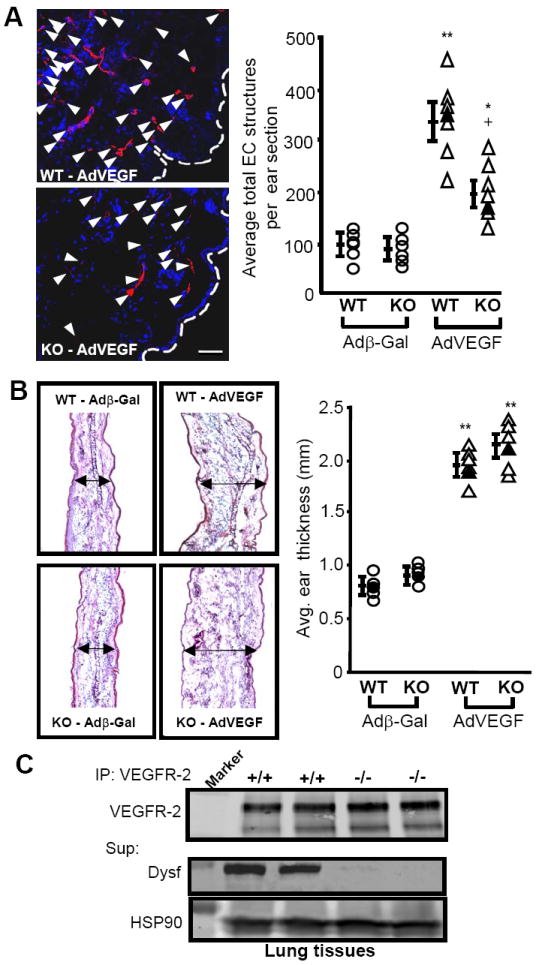
A) Genetic loss of Dysferlin caused abrogated angiogenic response to VEGF. Representative confocal optical sections (left) showing EC-positive structures (red channel) in ears of Dysferlin-null mice following adenoviral delivery of VEGF compared with age and sex-matched WT littermates. Control adenovirus encoded for or β-Gal. Blue channel represents DAPI (nucleus) staining. Scale bar 50 μm. Right, individual quantification of average total EC structures per ear section (n = 7 per group). *P < 0.05, **P < 0.01 compared with control virus, +P < 0.05 compared with WT. Black markers indicate ears shown on left panel. B) Adenoviral delivery of VEGF causes similar ear oedema in both WT and Dysferlin-null mice. Left, a section of the ears described in A was stained with H&E and midsection thickness was determined using a microscope. Black arrows describe thickness. Right, individual quantification of ear thickness. **P < 0.001 compared with control virus. Black markers indicate specimen depicted on left. C) Similar lung VEGFR-2 expression levels (240 and 210 kDa isoforms) in WT and Dysferlin-null mice, determined by IP and WB against VEGFR-2, or Dysferlin and HSP90 using the supernatant. Marker is 250 or 100 kDa.
Dysferlin deficiency causes PECAM-1 down-regulation in sub-confluent cells
VE-Cadherin and PECAM-1 are the two adhesion molecules that undergo one of the most significant relocalization in sub-confluent vs confluent EC19 and we hypothesized that they could be linked to the adhesion defect we observed in sub-confluent EC. Dysferlin knock-down (24h) had no effect on VE-Cadherin (110-130 kDa) levels (Fig. 5A) whereas PECAM-1 expression (110-125 kDa) was decreased by up to 92% in sub-confluent BAEC (25% confluence) but only by up to 19% in confluent BAEC. In sub-confluent HUVEC (25-50% confluent), PECAM-1 levels were reduced by up to 60%(140 kDa, marker 150 kDa) following Dysferlin siRNA treatment (Fig. 5B). PECAM-1 staining in HUVEC was characterized as numerous punctas throughout the cytoplasm, with a higher density around the peri-nuclear region (Fig. 5B, red channel, white arrows) whereas PECAM-1 detection was weaker and less diffused following a 24h Dysferlin siRNA treatment in the remaining adhered cells (right). In approximately 60% confluent (mostly protected) cells, the majority of PECAM-1 localized at cell junctions, covering 82% of cell-cell contact zones with ‘thick’ PECAM-1 positive structures (Fig. 5B bottom left and inset), whereas Dysferlin silencing caused a slight decreased in PECAM-1 staining at cell junctions, with 73% of cell-cell contact areas stained positive for slightly ‘thinner’ PECAM-1-positive structures (bottom right). Confluent HUVEC treated with Dysferlin or non-silencing siRNA showed similar PECAM-1 staining almost exclusively at cell-cell junctions (data not shown). Hence, loss of PECAM-1 is a likely candidate to rationalize the adhesion deficiency observed following Dysferlin knock-down in sub-confluent BAEC.
Figure 5. Dysferlin gene silencing causes adhesion defect though PECAM-1 poly-ubiquitination and degradation.


A) Decreased PECAM-1 expression following Dysferlin gene silencing in sub-confluent (left) but not confluent (right) BAEC. WB against Dysferlin, VE-Cadherin, PECAM-1 (marker 250 or 100 kDa) and HSP90 (loading control) was performed. B) Decreased PECAM-1 expression in sub-confluent HUVEC treated with human Dysferlin siRNA (24h; left). Dysferlin, PECAM-1 and β-COP expression (loading control) was analyzed by WB. Right, confocal immunofluorescence imaging (single optical sections) in 5 or 60% confluent HUVEC showing PECAM-1 (red) and nuclei (blue). Arrows indicate areas of high PECAM-1 staining and insets show magnified view of cell-cell junction. Scale bars 10 μm. C-D) PECAM-1 over-expression through adenovirus infection had no effect on adhesion of non-silencing siRNA-treated cells (solid markers) but rescued deficient adhesion by 59 and 68% in Dysferlin-silenced BAEC and HUVEC, respectively (approx 5% confluency time 0). * P<0.05, **P<0.01 compared with respective Adβ-Gal-treated control. E) Dysferlin silencing increased PECAM-1 poly-ubiquitination. Sub-confluent (25%) BAEC (left) and HUVEC (right) were transfected with a HA-ubiquitin plasmid and treated 24h later with control and Dysferlin siRNA sequences for 16h, proteins were collected and immuno-precipitated against PECAM-1 and blotted against HA. Marker (Mrk) is 100 (left) or 150 kDa (right). F) Inhibition of proteasome degradation with MG132 (6 h, 2×10-6 M) partly rescued loss of PECAM-1 in sub-confluent cells (25%) treated with Dysferlin siRNA (24h). G) Dysferlin IP in HUVEC (25% confluent, top panel) resulted in PECAM-1 co-IP (bottom blot). IP with non-immune IgG control did not result in Dysferlin or PECAM recovery. Supernatant HSP90 was used as a loading control. H) Single confocal optical section showing GFP-Dysferlin (green) co-localization (yellow, arrows) with PECAM-1 (red) around the peri-nuclear regions and multiple cellular punctas (inset). Nucleus is shown in blue (DAPI). Scale bar 20 μm.
To confirm that loss of PECAM-1 causes the adhesion defect in sub-confluent EC following Dysferlin knock-down, PECAM-1 was over-expressed using an adenovirus encoding for mouse PECAM-1 before Dysferlin gene silencing. Infection with AdPECAM-1 (50 MOI;) caused a 3-fold increase in total PECAM-1 expression compared to AdβGal-treated cells (Fig. S3B) and rescued by 59 and 68% the adhesion defect in BAEC and HUVEC, respectively (Fig. 5C-D, white circle vs white square) thereby confirming PECAM-1 as the main, but likely not exclusive, deficient gene product causing cell detachment following Dysferlin gene silencing.
Dysferlin forms a complex with PECAM-1, which prevents its poly-ubiquitination and degradation
In an attempt to show that Dysferlin down-regulation causes PECAM-1 protein degradation, EC were transfected with a HA-ubiquitin plasmid, treated with Dysferlin siRNA and PECAM-1 poly-ubiquitination was visualized by PECAM-1 IP and anti-HA blotting. An acute (16h) treatment with Dysferlin siRNA (Dysf2) in 25% confluent BAEC and HUVEC caused a 49% and 55% decrease in PECAM-1 expression compared to matching control siRNA (NS2) (Fig. 5E) and induced a drastic increase in PECAM-1 HA-ubiquitination. Inhibition of protein degradation with proteasome inhibitor MG132 (6h pretreatment) partially rescued (68%) loss of PECAM-1 expression following a 24h acute treatment with bovine Dysferlin siRNA (Fig. 5F). Control HUVEC showed quick signs of toxicity to MG132 (2h) and were not tested for PECAM-1 degradation. Dysferlin IP from similarly treated HUVEC cells resulted in a drastically greater co-IP of PECAM-1 than a control IgG IP (Fig. 5G). Moreover, robust co-localization was observed between GFP-Dysferlin (green) and PECAM-1 (red) around the Golgi apparatus and numerous cytoplasmic punctas in fixed non-confluent HUVEC (Fig. 5H, inset). Together, these data lend credence to the hypothesis that Dysferlin forms a complex with PECAM-1, which could participate in preventing PECAM-1 poly-ubiquitination and proteasome-dependent degradation.
Discussion
The current identification of Dysferlin in multiple EC lines and vascular tissues confirms the growing occurrence of Ferlins in non-muscle cells20. Initially believed to repair the sarcolemma of skeletal muscle cells2, the presence of Ferlins have been documented to regulate biological activities in other tissues, and as such are now believed to play other roles besides relatively simple membrane ‘patching’ events. Since we document its presence in multiple cellular compartments, such as CEM/LR, and in the media and adventitia of vessels, this supports the concept that Dysferlin may participate in complex signalling events not only in EC but also SMC and fibroblasts. The Dysferlin protein sequence contains many predicted protein binding domains as well as a nuclear localisation signal which further support a role in cellular signalisation21.
The positive role of Dysferlin in EC-driven new blood vessel formation was directly assessed in Dysferlin-null mice with a relatively short (6 days) angiogenesis assay performed in conjunction with an EC-specific agonist (VEGF). VEGF is well-known to elicit EC-derived capillary growth as these spouting and newly-formed vessels lack stabilizing pericytes/SMC, are highly unstable and undergo vaso-obliteration 2 weeks post-injection (data not shown), thereby limiting interferences of our observations with other cell types (SMC, supportive tissues) and further stressing the importance of Dysferlin to EC activity. This model produces no immune inflammation compared to other angiogenesis assays, such as matrigel plugs, which eliminates potential interferences from inflammatory cells in the EC phenotype of Dysferlin-null mice.
Dysferlin-dependent regulation of PECAM-1 expression and adhesion
PECAM-1 is highly expressed in the vasculature, with approximately one million copies reported on the surface of EC22, 23. Maintenance of the lateral localization of PECAM-1 at EC junctions requires significant cell-cell contact in order to allow encounter with another homophilic PECAM-1 molecule 19. Hence, PECAM-1’s unique spatial localization in confluent vs non-confluent cells makes it a prime candidate to rationalize the adhesion defect observed in EC with little to no cell-cell contact following Dysferlin gene silencing. Interestingly, it has been postulated that PECAM-1 might exert its effect at EC membrane by regulating intracellular Ca2+ levels24, which is in line with the Ca2+-sensing nature of Ferlins. A membrane network linked at intervals to the junctional surface is believed to exist just below the plasmalemma, and intracellular vesicle-like PECAM-1 stores are found in this compartment and constitutively recycles along EC borders25, which supports the concept that such PECAM-1-containing vesicles trafficking is Dysferlin- and Ca2+-regulated. This, combined with evidence showing that PECAM-1 interacts with other adhesion molecules such as αvβ3 26, directly modulates EC adhesion and motility27, and plays an important role in ‘inside-out’ and ‘outside-in’ signalling events in EC28, which is reminiscent of Ferlin activity, lends credence to our observations linking low PECAM-1 expression to Dysferlin silencing-induced adhesion deficiency.
Dysferlin, PECAM-1 and angiogenesis
Unchallenged Dysferlin or PECAM-1-null mice do not show obvious signs of vascular defects, although angiogenic response is blunted in PECAM-1-null mice29, 30. PECAM-1 is known to serve as a scaffold for various signalling molecules such as SHIP, PLC and SHP-2, and appropriate PECAM-1 localization is essential to proper assembly and regulation of these signalling complexes31. When the proven importance of PECAM-1 to angiogenesis is taken into the context of our data showing deficient adhesion, proliferation and tube formation following Dysferlin knock-down, this not only supports our findings describing deficient angiogenesis in mice lacking Dysferlin but also suggests that more vascular defects are likely to be found in Dysferlin-null mice. Angiogenesis requires EC proliferation, migration and three-dimensional assembly into tubular structures, and our data strongly suggest that these steps require Dysferlin. It is of interest to note that signs of deficient inflammation in skeletal muscle of Dysferlin-null mice have recently been reported32. However, the pluripotency of PECAM-1 signalling activities along with the unknown intracellular binding partners of Dysferlin complicate our understanding of the different signalling events that lead to aberrant vs normal adhesion. To add to the complexity of our data, most reports on PECAM-1 study confluent EC, i.e. with junctional PECAM-1 localization, rather than sub-confluent EC.
Ferlins in protein cargo trafficking of EC
The concept that Ferlin membrane patches fusing at the plasma membrane contain ‘cargo’ was initially proposed by data showing deficient neurotransmitter exocytosis in Otoferlin-null mice12. Synaptotagmins, which share structural similarities with Dysferlin 3, 33, are well described trafficking proteins and their C2 domains allow them to act as calcium sensors involved in neurotransmitter and hormone secretion in various cell type34. Hence the documented function of C2 domain-containing proteins in the regulation of fusion events and cargo trafficking further support a role for Dysferlin in cargo trafficking processes, and it is likely that PECAM-1 is a key but not exclusive protein cargo of Dysferlin-regulated membrane ‘patches’. On the other hand, data supporting profound differences in protein trafficking between Dysferlin and Myoferlin-containing membrane vesicles in EC are normal VEGFR-2 expression and VEGFR-2 dependent edema in Dysferlin-null mice as well as intact VEGFR-2 expression and downstream signalling following Dysferlin siRNA treatment in vitro. Such theory that Ferlin mediate unique protein cargo-specific membrane fusion events rationalize the observation that loss of either Myoferlin or Dysferlin causes Limb-girdle muscular dystrophy that cannot be compensated by other Ferlins since specific cargo is involved. Further investigation of the protein cargo of these membrane patches is therefore clearly warranted.
Supplementary Material
Acknowledgments
This work is supported by grants from the Canadian Institutes for Health Research (CIHR), Michael Smith Foundation for Health Research (MSFHR), Canadian Foundation for Innovation, British Columbia Knowledge Development Fund, British Columbia Proteomics Network (P.B.) and National Institutes of Health EY016695 (N.S.). C.Y, A.S. and P.B. are supported by salary awards from CIHR and MSFHR.
Footnotes
The authors have no disclosure to report.
References
- 1.Washington NL, Ward S. FER-1 regulates Ca2+ -mediated membrane fusion during C. elegans spermatogenesis. J Cell Sci. 2006;119:2552–2562. doi: 10.1242/jcs.02980. [DOI] [PubMed] [Google Scholar]
- 2.Bansal D, Miyake K, Vogel SS, Groh S, Chen CC, Williamson R, McNeil PL, Campbell KP. Defective membrane repair in dysferlin-deficient muscular dystrophy. Nature. 2003;423:168–172. doi: 10.1038/nature01573. [DOI] [PubMed] [Google Scholar]
- 3.Bansal D, Campbell KP. Dysferlin and the plasma membrane repair in muscular dystrophy. Trends Cell Biol. 2004;14:206–213. doi: 10.1016/j.tcb.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Davis DB, Doherty KR, Delmonte AJ, McNally EM. Calcium-sensitive phospholipid binding properties of normal and mutant ferlin C2 domains. J Biol Chem. 2002;277:22883–22888. doi: 10.1074/jbc.M201858200. [DOI] [PubMed] [Google Scholar]
- 5.Lennon NJ, Kho A, Bacskai BJ, Perlmutter SL, Hyman BT, Brown RH., Jr Dysferlin interacts with annexins A1 and A2 and mediates sarcolemmal wound-healing. J Biol Chem. 2003;278:50466–50473. doi: 10.1074/jbc.M307247200. [DOI] [PubMed] [Google Scholar]
- 6.Lang CT, Markham KB, Behrendt NJ, Suarez AA, Samuels P, Vandre DD, Robinson JM, Ackerman WEt. Placental dysferlin expression is reduced in severe preeclampsia. Placenta. 2009;30:711–718. doi: 10.1016/j.placenta.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wenzel K, Geier C, Qadri F, Hubner N, Schulz H, Erdmann B, Gross V, Bauer D, Dechend R, Dietz R, Osterziel KJ, Spuler S, Ozcelik C. Dysfunction of dysferlin-deficient hearts. J Mol Med. 2007;85:1203–1214. doi: 10.1007/s00109-007-0253-7. [DOI] [PubMed] [Google Scholar]
- 8.Galvin JE, Palamand D, Strider J, Milone M, Pestronk A. The muscle protein dysferlin accumulates in the Alzheimer brain. Acta Neuropathol. 2006;112:665–671. doi: 10.1007/s00401-006-0147-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hochmeister S, Grundtner R, Bauer J, Engelhardt B, Lyck R, Gordon G, Korosec T, Kutzelnigg A, Berger JJ, Bradl M, Bittner RE, Lassmann H. Dysferlin is a new marker for leaky brain blood vessels in multiple sclerosis. J Neuropathol Exp Neurol. 2006;65:855–865. doi: 10.1097/01.jnen.0000235119.52311.16. [DOI] [PubMed] [Google Scholar]
- 10.Davis DB, Delmonte AJ, Ly CT, McNally EM. Myoferlin, a candidate gene and potential modifier of muscular dystrophy. Hum Mol Genet. 2000;9:217–226. doi: 10.1093/hmg/9.2.217. [DOI] [PubMed] [Google Scholar]
- 11.Doherty KR, Cave A, Davis DB, Delmonte AJ, Posey A, Earley JU, Hadhazy M, McNally EM. Normal myoblast fusion requires myoferlin. Development. 2005;132:5565–5575. doi: 10.1242/dev.02155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roux I, Safieddine S, Nouvian R, Grati M, Simmler MC, Bahloul A, Perfettini I, Le Gall M, Rostaing P, Hamard G, Triller A, Avan P, Moser T, Petit C. Otoferlin, defective in a human deafness form, is essential for exocytosis at the auditory ribbon synapse. Cell. 2006;127:277–289. doi: 10.1016/j.cell.2006.08.040. [DOI] [PubMed] [Google Scholar]
- 13.Bernatchez PN, Acevedo L, Fernandez-Hernando C, Murata T, Chalouni C, Kim J, Erdjument-Bromage H, Shah V, Gratton JP, McNally EM, Tempst P, Sessa WC. Myoferlin regulates vascular endothelial growth factor receptor-2 stability and function. J Biol Chem. 2007;282:30745–30753. doi: 10.1074/jbc.M704798200. [DOI] [PubMed] [Google Scholar]
- 14.Bernatchez PN, Sharma A, Kodaman P, Sessa WC. Myoferlin is critical for endocytosis in endothelial cells. Am J Physiol Cell Physiol. 2009;297:C484–492. doi: 10.1152/ajpcell.00498.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kondo S, Scheef EA, Sheibani N, Sorenson CM. PECAM-1 isoform-specific regulation of kidney endothelial cell migration and capillary morphogenesis. Am J Physiol Cell Physiol. 2007;292:C2070–2083. doi: 10.1152/ajpcell.00489.2006. [DOI] [PubMed] [Google Scholar]
- 16.Acevedo L, Yu J, Erdjument-Bromage H, Miao RQ, Kim JE, Fulton D, Tempst P, Strittmatter SM, Sessa WC. A new role for Nogo as a regulator of vascular remodeling. Nat Med. 2004;10:382–388. doi: 10.1038/nm1020. [DOI] [PubMed] [Google Scholar]
- 17.Hernandez-Deviez DJ, Howes MT, Laval SH, Bushby K, Hancock JF, Parton RG. Caveolin regulates endocytosis of the muscle repair protein, dysferlin. J Biol Chem. 2008;283:6476–6488. doi: 10.1074/jbc.M708776200. [DOI] [PubMed] [Google Scholar]
- 18.Chang SH, Feng D, Nagy JA, Sciuto TE, Dvorak AM, Dvorak HF. Vascular permeability and pathological angiogenesis in caveolin-1-null mice. Am J Pathol. 2009;175:1768–1776. doi: 10.2353/ajpath.2009.090171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Albelda SM, Oliver PD, Romer LH, Buck CA. EndoCAM: a novel endothelial cell-cell adhesion molecule. J Cell Biol. 1990;110:1227–1237. doi: 10.1083/jcb.110.4.1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Karsan A, Blonder J, Law J, Yaquian E, Lucas DA, Conrads TP, Veenstra T. Proteomic analysis of lipid microdomains from lipopolysaccharide-activated human endothelial cells. J Proteome Res. 2005;4:349–357. doi: 10.1021/pr049824w. [DOI] [PubMed] [Google Scholar]
- 21.Glover L, Brown RH., Jr Dysferlin in membrane trafficking and patch repair. Traffic. 2007;8:785–794. doi: 10.1111/j.1600-0854.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 22.Newman PJ. The role of PECAM-1 in vascular cell biology. Ann N Y Acad Sci. 1994;714:165–174. doi: 10.1111/j.1749-6632.1994.tb12041.x. [DOI] [PubMed] [Google Scholar]
- 23.Jackson DE. The unfolding tale of PECAM-1. FEBS Lett. 2003;540:7–14. doi: 10.1016/s0014-5793(03)00224-2. [DOI] [PubMed] [Google Scholar]
- 24.Gurubhagavatula I, Amrani Y, Pratico D, Ruberg FL, Albelda SM, Panettieri RA., Jr Engagement of human PECAM-1 (CD31) on human endothelial cells increases intracellular calcium ion concentration and stimulates prostacyclin release. J Clin Invest. 1998;101:212–222. doi: 10.1172/JCI269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mamdouh Z, Chen X, Pierini LM, Maxfield FR, Muller WA. Targeted recycling of PECAM from endothelial surface-connected compartments during diapedesis. Nature. 2003;421:748–753. doi: 10.1038/nature01300. [DOI] [PubMed] [Google Scholar]
- 26.Piali L, Hammel P, Uherek C, Bachmann F, Gisler RH, Dunon D, Imhof BA. CD31/PECAM-1 is a ligand for alpha v beta 3 integrin involved in adhesion of leukocytes to endothelium. J Cell Biol. 1995;130:451–460. doi: 10.1083/jcb.130.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J, Sheibani N. Modulation of VE-cadherin and PECAM-1 mediated cell-cell adhesions by mitogen-activated protein kinases. J Cell Biochem. 2003;90:121–137. doi: 10.1002/jcb.10600. [DOI] [PubMed] [Google Scholar]
- 28.Kim CS, Wang T, Madri JA. Platelet endothelial cell adhesion molecule-1 expression modulates endothelial cell migration in vitro. Lab Invest. 1998;78:583–590. [PubMed] [Google Scholar]
- 29.Wang S, Sorenson CM, Sheibani N. Attenuation of retinal vascular development and neovascularization during oxygen-induced ischemic retinopathy in Bcl-2-/- mice. Dev Biol. 2005;279:205–219. doi: 10.1016/j.ydbio.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 30.Wang XQ, Sheibani N, Watson JC. Modulation of tumor endothelial cell marker 7 expression during endothelial cell capillary morphogenesis. Microvasc Res. 2005;70:189–197. doi: 10.1016/j.mvr.2005.08.004. [DOI] [PubMed] [Google Scholar]
- 31.Ilan N, Madri JA. PECAM-1: old friend, new partners. Curr Opin Cell Biol. 2003;15:515–524. doi: 10.1016/s0955-0674(03)00100-5. [DOI] [PubMed] [Google Scholar]
- 32.Roche JA, Lovering RM, Roche R, Ru LW, Reed PW, Bloch RJ. Extensive mononuclear infiltration and myogenesis characterize the recovery of dysferlin-null skeletal muscle from contraction-induced injuries. Am J Physiol Cell Physiol. 2009 doi: 10.1152/ajpcell.00122.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Britton S, Freeman T, Vafiadaki E, Keers S, Harrison R, Bushby K, Bashir R. The third human FER-1-like protein is highly similar to dysferlin. Genomics. 2000;68:313–321. doi: 10.1006/geno.2000.6290. [DOI] [PubMed] [Google Scholar]
- 34.Nalefski EA, Falke JJ. The C2 domain calcium-binding motif: structural and functional diversity. Protein Sci. 1996;5:2375–2390. doi: 10.1002/pro.5560051201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.