

Abstract
Because carbonyl groups are ubiquitous in organic chemistry, the ability to synthesize functionalized carbonyl compounds, particularly enantioselectively, is an important objective. We have developed a straightforward and versatile method for catalytic asymmetric carbon–carbon bond formation at the γ-position of carbonyl compounds, specifically, phosphine-catalyzed additions of malonate esters to γ-substituted allenoates and allenamides. Mechanistic studies have provided insight into the reaction pathway.
Keywords: allene, catalysis
There are a variety of powerful methods for functionalizing carbonyl compounds in the α- and the β-positions, including classic approaches such as reacting enolates with electrophiles and adding nucleophiles to α,β-unsaturated carbonyl compounds (1, 2). In contrast, there are few general processes for incorporating new substituents in the γ-position of carbonyl compounds, particularly catalytic asymmetric reactions that form carbon–carbon bonds (3).
In pioneering early investigations, Trost and Lu demonstrated that phosphines can catalyze additions of certain carbon, nitrogen, and oxygen nucleophiles to the γ-position of 2-butynoates and 2,3-butadienoates (Eq. 1, EWG = ester), as well as related compounds (4–8); for these electrophiles, the γ-carbon of the product is not a stereocenter. In contrast, for additions to homologues of these electrophiles, there is the potential to simultaneously form a new bond and to control the stereochemistry of the γ-carbon (Eq. 2). Until recently, however, there were only isolated examples of intermolecular reactions of this type [≤ 30% yield (9–11); instead, another phosphine-catalyzed process, isomerization to the dienone (Eq. 2) (12, 13), often intervened], and there were no reports of an enantioselective variant. 

Since 2009, some progress has been described in achieving asymmetric γ additions of the type outlined in Eq. 2, specifically, chiral phosphine-catalyzed additions of nitromethane (14) and alkylthiols (15). Due to the central importance of carbon–carbon bond formation in organic chemistry, we are particularly interested in the use of carbon nucleophiles in this powerful functionalization process. Unfortunately, the only method that has been reported for catalytic enantioselective γ additions of carbon nucleophiles is limited to a single nucleophilic partner, nitromethane. Even nitroethane did not furnish satisfactory results (14).
1,3-Dicarbonyl compounds are an important family of carbon-based (pro)nucleophiles that have been widely exploited in asymmetric catalysis (16), including enantioselective γ-additions to 2-butynoates and 2,3-butadienoates (which cannot undergo undesired isomerization to a dienoate) to generate a δ-stereocenter (17). To the best of our knowledge, 1,3-dicarbonyl compounds have not been employed as nucleophiles in γ-additions of the type illustrated in Eq. 2, which would lead to the formation of a carbon–carbon bond between two tertiary centers. In this paper, we establish that such γ-additions of secondary carbon nucleophiles to γ-substituted electrophiles can indeed be achieved by a chiral phosphine catalyst in good yield and enantiomeric excess (ee) (Eq. 3). 
Thus, phosphepine 1, which was originally developed as a chiral ligand for transition metals (18), together with 2-methoxyphenol, efficiently catalyzes the γ-addition of diallyl malonate to a γ-substituted allenoate, furnishing the desired product in 98% yield and 95% ee (entry 1 of Table 1). In the absence of phosphepine 1, no γ-addition is observed (entry 2). If the protic additive (2-methoxyphenol) is omitted, or if the reaction is conducted at room temperature, then a lower yield and ee are obtained (entries 3 and 4). Chiral phosphines that have been employed in other catalytic asymmetric processes, including γ-additions, are not as effective as 1 (entries 5–8) (14, 15, 19–21).
Table 1.
Effect of reaction parameters on the catalytic asymmetric γ-addition of a malonate ester to a γ-substituted allenoate 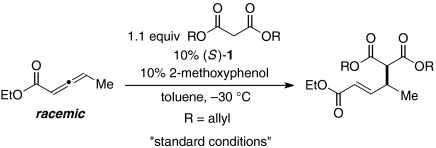
| Entry | Change from the “standard conditions” | Yield, %* | ee, % |
| 1 | None | 98 | 95 |
| 2 | No (S)-1 | < 5 | — |
| 3 | No 2-methoxyphenol | 24 | 71 |
| 4 | Room temperature instead of −30 °C | 91 | 90 |
| 5 | (S)-2 instead of (S)-1 | 80 | 95 |
| 6 | (S)-3 instead of (S)-1 | < 5 | — |
| 7 | (S)-4 instead of (S)-1 | 93 | 93 |
| 8 | (+)-TangPhos instead of (S)-1 | 50 | 17 |
All data are the average of two experiments.
*The yield was determined by GC analysis with n-decane as an internal standard. 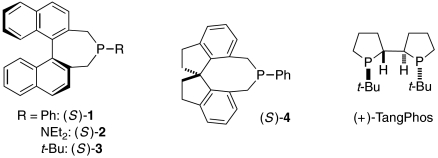
Under our optimized conditions, an array of catalytic enantioselective γ-additions of malonate esters to readily accessible γ-substituted allenoates proceed in good yield and ee (Table 2). Thus, formation of the desired carbon–carbon bond between two tertiary carbons occurs smoothly in the presence of functional groups such as alkynes, halides, ethers, acetals, esters, and alkenes.
Table 2.
Catalytic asymmetric γ-additions of malonate esters to γ-substituted allenoates 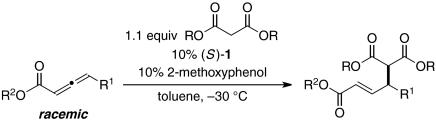
| Entry | R1 | R2 | R | Yield, %* | ee, % |
| 1 | Me | 94 | 94 | ||
| 2 | 88 | 92 | |||
| 3 | (CH2)3CI | 91 | 93 | ||
| 4 | (CH2)4OTIPS | 78 | 87 | ||
| 5 | (CH2)4OBn | Et | Allyl | 78 | 90 |
| 6 |  |
71 | 94 | ||
| 7 | (CH2)3CO2Me | 77 | 94 | ||
| 8 |
|
71 |
86 |
||
| 9 | n-Pr | 84 | 93 | ||
| 10 | Bn | Et | 65 | 90 | |
| 11 | CH2OMe | 74 | 93 | ||
| 12† | 71 | 85 |
All data are the average of two experiments.
*Yield of purified product. Only the E isomer is observed.
†15% (S)-1 was used.
Furthermore, in a preliminary study under our standard reaction conditions, we have determined that phosphepine 1 can catalyze the γ-addition of an α-substituted 1,3-dicarbonyl compound to an allenoate, thereby achieving asymmetric carbon–carbon bond formation between a tertiary and a quaternary stereocenter with good ee (and modest diastereoselectivity; Eq. 4). 
The conditions that we developed for γ-additions of malonate esters to allenoates can be applied, without modification, to reactions of allenamides (Table 3) (22). Good enantioselectivities are observed with a range of substrates.
Table 3.
Catalytic asymmetric γ-additions of malonate esters to γ-substituted Weinreb allenamides 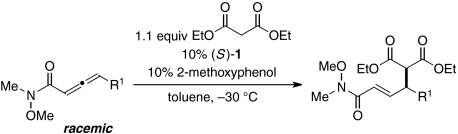
| Entry | R1 | Yield, %* | ee, % |
| 1 | Me | 71 | 95 |
| 2 | n-Pr | 66 | 93 |
| 3 | (CH2)2Ph | 66 | 94 |
| 4 | 70 | 94 |
All data are the average of two experiments.
*Yield of purified product. Only the E isomer is observed.
The enantioenriched products generated by phosphine-catalyzed γ-additions to activated allenes can be transformed into other useful compounds. For example, deallylation/decarboxylation can provide a carboxylic acid [Eq. 5; see C1–C6 of amphidinolide X: (23)], which can be cyclized to form a butyrolactone with good diastereoselectivity (Eq. 6; 24, 25). 
Several observations regarding this method for the addition of 1,3-dicarbonyl compounds to γ-substituted allenes are noteworthy. First, in contrast to other phosphine-catalyzed enantioselective γ-addition reactions (14, 15), kinetic resolution of the allene is observed (Eq. 7; the ee of the product is constant during the reaction). To the best of our knowledge, there are few examples of kinetic resolutions of allenes (26), and none that involve phosphine catalysis (8). 
In addition, we have determined that the resting state of the catalyst during the γ-addition process is phosphepine 1 itself (not the protonated catalyst or a catalyst-allene adduct). Furthermore, the rate law for the reaction is first order in catalyst and in allene, and zero order in malonate. Finally, the ee of the product correlates linearly with the ee of the catalyst. These observations are consistent with a pathway in which the first step of the reaction is turnover-limiting (Fig. 1).
Fig. 1.

Possible mechanism for the phosphine-catalyzed asymmetric γ-addition of a malonate ester to an activated allene (for the sake of simplicity, only one E/Z isomer of the intermediates is illustrated and all of the elementary steps are drawn as irreversible).
In summary, we have developed phosphine-catalyzed γ-additions of secondary nucleophiles to γ-substituted allenes, specifically, a versatile method for asymmetric carbon–carbon bond formation between malonate esters and 2,3-allenoates and 2,3-allenamides. Interestingly, a preliminary investigation suggests that the enantioselective synthesis of adjacent tertiary and quaternary stereocenters may even be possible through this approach. Mechanistic studies have furnished insight into the reaction pathway, including an example of a kinetic resolution of an allene. Because this straightforward method for the catalytic asymmetric γ-functionalization of carbonyl compounds complements existing methods for α- and β-functionalizations, additional efforts to exploit this powerful mode of reactivity are under way.
Supplementary Material
Acknowledgments.
We thank Dr. Sean W. Smith for preliminary studies and Dr. Meiliana Tjandra and Shaun D. Fontaine for experimental assistance. Support has been provided by the National Institutes of Health (National Institute of General Medical Sciences, Grant R01-GM57034), the University of Bologna (Marco Polo fellowship for R.S.), Merck Research Laboratories, and Novartis.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission. D.W.M. is a guest editor invited by the Editorial Board.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1003597107/-/DCSupplemental.
References
- 1.Carreira EM, Fettes A, Marti C. Catalytic enantioselective aldol reactions. Org Reactions. 2006;67:1–216. [Google Scholar]
- 2.Christoffers J, Koripelly G, Rosiak A, Roessle M. Recent advances in metal-catalyzed asymmetric conjugate additions. Synthesis. 2007:1279–1300. [Google Scholar]
- 3.Denmark SE, Heemstra JR, Beutner GL. Catalytic, enantioselective, vinylogous aldol reactions. Angew Chem Int Edit. 2005;44:4682–4698. doi: 10.1002/anie.200462338. [DOI] [PubMed] [Google Scholar]
- 4.Trost BM, Li C-J. Novel “umpolung” in C–C bond formation catalyzed by triphenylphosphine. J Am Chem Soc. 1994;116:3167–3168. [Google Scholar]
- 5.Trost BM, Li C-J. Phosphine-catalyzed isomerization-addition of oxygen nucleophiles to 2-alkynoates. J Am Chem Soc. 1994;116:10819–10820. [Google Scholar]
- 6.Zhang C, Lu X. Umpolung addition reaction of nucleophiles to 2,3-butadienoates catalyzed by a phosphine. Synlett. 1995:645–646. [Google Scholar]
- 7.Trost BM, Dake GR. Nitrogen pronucleophiles in the phosphine-catalyzed γ-addition reaction. J Org Chem. 1997;62:5670–5671. [Google Scholar]
- 8.Cowen BJ, Miller SJ. Enantioselective catalysis and complexity generation from allenoates. Chem Soc Rev. 2009;38:3102–3116. doi: 10.1039/b816700c. [DOI] [PubMed] [Google Scholar]
- 9.Trost BM, Dake GR. Nucleophilic α-addition to alkynoates. A synthesis of dehydroamino acids. J Am Chem Soc. 1997;119:7595–7596. [Google Scholar]
- 10.Liu B, Davis R, Joshi B, Reynolds DW. Phosphine-catalyzed annulation of thioamides and 2-alkynoates: A new synthesis of thiazolines. J Org Chem. 2002;67:4595–4598. doi: 10.1021/jo016154u. [DOI] [PubMed] [Google Scholar]
- 11.Virieux D, Guillouzic A-F, Cristau H-J. Phosphines catalyzed nucleophilic addition of azoles to allenes: Synthesis of allylazoles and indolizines. Tetrahedron. 2006;62:3710–3720. [Google Scholar]
- 12.Trost BM, Kazmaier U. Internal redox catalyzed by triphenylphosphine. J Am Chem Soc. 1992;114:7933–7935. [Google Scholar]
- 13.Kwong CKW, Fu MY, Lam CSL, Toy PH. The phosphine-catalyzed alkyne to 1,3-diene isomerization reaction. Synthesis. 2008:2307–2317. [Google Scholar]
- 14.Smith SW, Fu GC. Asymmetric carbon–carbon bond formation γ to a carbonyl group: Phosphine-catalyzed addition of nitromethane to allenes. J Am Chem Soc. 2009;131:14231–14233. doi: 10.1021/ja9061823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun J, Fu GC. Phosphine-catalyzed formation of carbon–sulfur bonds: Catalytic asymmetric synthesis of γ-thioesters. J Am Chem Soc. 2010;132:4568–4569. doi: 10.1021/ja101251d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Agostinho M, Kobayashi S. Strontium-catalyzed highly enantioselective Michael additions of malonates to enones. J Am Chem Soc. 2008;130:2430–2431. doi: 10.1021/ja710332h. [DOI] [PubMed] [Google Scholar]
- 17.Chen Z, et al. Asymmetric formation of quaternary carbon centers catalyzed by novel chiral 2,5-dialkyl-7-phenyl-7-phosphabicyclo[2.2.1]heptanes. J Org Chem. 1998;63:5631–5635. [Google Scholar]
- 18.Gladiali S, Dore A, Fabbri D, De Lucchi O, Manassero M. Novel atropisomeric phosphorus ligands: 4,5-Dihydro-3H-dinaphtho[2,1-c;1′,2′-e]phosphepine derivatives. Tetrahedron Asymmetr. 1994;5:511–514. [Google Scholar]
- 19.Wurz RP, Fu GC. Catalytic asymmetric synthesis of piperidine derivatives through the [4 + 2] annulation of imines with allenes. J Am Chem Soc. 2005;127:12234–12235. doi: 10.1021/ja053277d. [DOI] [PubMed] [Google Scholar]
- 20.Chung YK, Fu GC. Phosphine-catalyzed enantioselective synthesis of oxygen heterocycles. Angew Chem Int Edit. 2009;48:2225–2227. doi: 10.1002/anie.200805377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marinetti A, Voituriez A. Enantioselective phosphine organocatalysis. Synlett. 2010:174–194. [Google Scholar]
- 22.Balasubramaniam S, Aidhen IS. The growing synthetic utility of the Weinreb amide. Synthesis. 2008:3707–3738. [Google Scholar]
- 23.Fürstner A, Kattnig E, Lepage O. Total syntheses of amphidinolide X and Y. J Am Chem Soc. 2006;128:9194–9204. doi: 10.1021/ja061918e. [DOI] [PubMed] [Google Scholar]
- 24.Ugurchieva TM, Veselovsky VV. Advances in the synthesis of natural butano- and butenolides. Russ Chem Rev. 2009;78:337–373. [Google Scholar]
- 25.Seitz M, Reiser O. Synthetic approaches towards structurally diverse γ-butyrolactone natural-product-like compounds. Curr Opin Chem Biol. 2005;9:285–292. doi: 10.1016/j.cbpa.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Carballeira JD, et al. Directed evolution and axial chirality: Optimization of the enantioselectivity of Pseudomonas aeruginosa lipase towards the kinetic resolution of a racemic allene. Chem Commun. 2007:1913–1915. doi: 10.1039/b700849j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.