

Abstract
The mitogen-activated protein kinase phosphatase Dusp1 (also known as MKP-1) is essential for control of the inflammatory response to systemic challenge with the lipopolysaccharide of Gram-negative bacteria. Here, we have investigated the consequences of Dusp1-deficiency in colon ascendens stent peritonitis (CASP) and caecal ligation and puncture (CLP), two mouse models of septic peritonitis. Following CASP, Dusp1−/− mice had increased serum levels of CCL4, interleukin-10 (IL-10) and IL-6, with differences from wild-type mice being dependent on severity of sepsis. These cytokines, along with inducible nitric oxide synthase messenger RNA, were also expressed at higher levels in spleen and liver. Similar over-production of these cytokines was detected in the CLP model, with even larger differences from wild-type mice. Despite the increased inflammatory response, bacterial clearance was impaired in Dusp1−/− mice subjected to CASP and CLP. Dusp1−/− mice suffered increased lethality in both peritonitis models. Together our data indicate that exaggerated inflammatory responses to gut bacteria introduced into the peritoneum in the absence of Dusp1 do not help to control bacterial replication but are detrimental for the host.
Keywords: cytokines, sepsis, signalling
Introduction
The robust activation of innate immune cells following microbial challenge during infection requires efficient negative feedback control mechanisms. The action of these regulatory host factors is evident in sepsis patients suffering from a systemic inflammatory response, characterized by the initial production of chemokines, tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6) and IL-1β, or a compensatory anti-inflammatory response, that includes IL-10, transforming growth factor-β and IL-1 receptor a.1 Triggering of innate immune cells’ pattern recognition receptors, such as the Toll-like receptors (TLRs), strongly activates the p38, Jun N-terminal kinase and extracellular signal-regulated kinase 1/2 mitogen-activated protein kinase (MAPK) signalling module that contributes essentially to inflammatory gene expression through activation of transcription factors like activating protein-1 and cAMP response element binding protein.2 Negative feedback control at the level of MAPK signalling is exerted by the action of MAPK phosphatases of the dual specificity phosphatase family (Dusp), that bind to activated MAPK and dephosphorylate them at tyrosine and threonine residues.3 It is known that Dusp1 (also known as MKP-1 for MAPK phosphatase-1) is induced in lipopolysaccharide (LPS) -treated mice and limits p38 MAPK activation. Dusp1 is required in vivo to prevent overwhelming release of a subset of inflammatory cytokines that includes IL-6, TNF-α, CCL3 and CCL4.4–7 Although the immunoregulatory cytokine IL-10 is also strongly over-produced, Dusp1−/− mice die when given LPS doses that are sublethal in wild-type (WT) mice. These data suggested that MAPK phosphatases in general, and Dusp1 in particular, play a major role in balancing the immune response to microbial stimulation and during sepsis.
In contrast to the experimental LPS challenge model, clinical sepsis is not caused by a bolus injection of a purified TLR ligand, but develops from acute or persisting bacterial infection. The limitations of the LPS shock model for the study of sepsis are well recognized, and include the immediate induction of a hypodynamic cardiovascular state after LPS injection; the high levels of inflammatory mediators observed after LPS, which are usually not seen in septic patients where cytokine elevation are much lower but of longer duration. It is therefore important to extend the investigation of the regulatory role of Dusp1 on the host response to models of infection and sepsis. Recently, Liu and colleagues investigated the phenotype of Dusp1−/− mice in infection with Staphylococcus aureus8 and Escherichia coli,9 and found a similarly increased inflammatory response with increased lethality as seen after LPS challenge.
The gold standard model of sepsis research in rodents has been caecal ligation and puncture (CLP), which mimics ruptured appendicitis or perforated diverticulitis in humans.1 Survival in the CLP model requires TNF-dependent abscess formation that walls off and contains infection in the peritoneum.10–12 The colon ascendens stent peritonitis (CASP) model has been used increasingly; insertion of a stent leads to continued release of faecal content into the peritoneum, creating acute polymicrobial septic peritonitis.13 In contrast to CLP, the leakage created by the stent is not sealed off by formation of an abscess but creates on ongoing, diffuse polymicrobial peritonitis.14 Consistent with this difference in the course and pathology of the disease models, differences in the major host-protective immunological factors have been identified. In the CASP model TNF is not protective, instead a lack of interferon-γ (IFN-γ) and its inducer IL-12 are detrimental.13,15,16 On the other hand, the production of the anti-inflammatory IL-10 early after sepsis induction is required in both CLP and CASP to prevent excess death probably as the result of acute hyper-inflammation.17,18
Here, we have investigated the phenotype of Dusp1−/− mice in the CLP and CASP models of polymicrobial peritonitis. Similar to the results obtained in LPS shock, we found strongly increased levels of IL-6, IL-10, CCL3 and CCL4 in Dusp1−/− mice, which correlated with excess lethality. Our results therefore strengthen the concept that regulation of MAPK activity by Dusp1 is an important control element preventing adverse hyper-inflammation during systemic infection.
Materials and methods
Mice
Dusp1−/− mice have been described previously.19 Breeding pairs on a mixed C57/129 background and backcrossed for seven generations to C3H were kindly provided by Dr Andrew Cato (FZ Karlsruhe, Germany). For some experiments mice were used that have been backcrossed to C57BL/6 for at least six generations (starting from the mixed C57/129 mice). In experiments employing mice on the mixed background, WT and heterozygous littermate controls were used. When Dusp1−/− mice backcrossed onto C3H or C57BL/6 background were used, the respective control mice were purchased from a commercial breeder (Harlan-Winkelmann, Borchen, Germany), as indicated in the figure legends, and housed in the same environment as Dusp1−/− mice for at least 1 week before experimental challenge. Ethical approval for all mouse experiments was obtained from the appropriate governmental authorities.
CASP prodedure
The CASP procedure used for induction of septic peritonitis was described in detail previously.13 Briefly, the colon ascendens was exteriorized under complete anaesthesia and a 7/0 ethilon thread was stitched through the anti-mesenteric portion of the colon ascendens approximately 10 mm distal of the ileocaecal valve. An 18- or 20-gauge (G) venous catheter was inserted by puncture anti-mesenterically through the colonic wall into the intestinal lumen, directly proximal of the pre-tied knot, and fixed. To ensure proper intra-luminal position of the stent, stool was milked from the caecum into the colon ascendens until a small drop appeared. Fluid resuscitation of the animals was performed by flushing 0·5 ml sterile saline into the peritoneal cavity before closure of the abdominal wall. For investigation of the inflammatory response, mice were killed after 12 hr under anaesthesia. Blood was collected from the retro-orbital plexus, the peritoneal cavity was lavaged with phosphate-buffered saline (PBS), and separate samples of spleen and liver were removed for analysis of bacterial counts and for preparation of tissue RNA. Control animals were housed under identical conditions but were not laparotomized.
CLP procedure
The mice were anaesthetized by intraperitoneal injection of 75 mg/kg Ketanest® (Parke, Davis & Company, München, Germany) and 16 mg/kg Rompun® (Bayer AG, Leverkusen, Germany). The caecum was exteriorized and the distal end (20–30% of total length) was ligated and punctured once with a needle (27G, 0·4-mm diameter opening) to achieve a sublethal CLP as described elsewhere.20 For investigation of the inflammatory response, mice were killed after 12–16 hr. Blood was collected from the retro-orbital plexus, the peritoneal cavity was lavaged with thioglycollate, and separate samples of spleen and liver were removed for analysis of bacterial counts and for preparation of tissue RNA. Control animals were housed under identical conditions but were not laparotomized.
To test the role of IL-6 in the regulation of the inflammatory response to sepsis, mice were injected intraperitoneally directly after the CLP surgery with 250 μg anti-IL-6 antibody (clone MP5-20F3) or 250 μg sgp130-Fc (both in 100 μl). Mice treated with CLP and injected with 100 μl PBS served as controls.
Cytokine enzyme-linked immunosorbent assay determination
Serum concentrations of IL-6, IL-10, CCL3, CCL4, IL-1β and TNF-α were investigated in samples from untreated and CASP- or CLP-treated mice by enzyme-linked immunosorbent assay (ELISA) using antibody pairs (Duoset; R&D, Wiesbaden, Germany) following the manufacturer's instructions.
Quantitative real-time polymerase chain reaction measurements
Total RNA was prepared from spleen and liver samples using Trifast (Peqlab, Erlangen, Germany) or Qiagen RNAeasy columns (Qiagen, Hilden, Germany). After reverse transcription, the expression of cytokines, chemokines and inflammatory mediators was determined by quantitative real-time polymerase chain reaction (qPCR) on an ABI 7900 SDS system using the Roche Universal Probe library. Sequences for primers and probes are available on request. Threshold cycle values were normalized to Hprt, and fold changes were calculated by the ΔΔCT method using untreated WT or heterozygous control mice as calibrators.
Determination of bacterial loads in the peritoneum and spleen or liver
Peritoneal lavage fluid was serially diluted and plated on blood agar plates. Organs were homogenized in 5 ml PBS (CASP) or thioglycollate (CLP) using an Ultra-Turrax device, serially diluted and plated on blood agar plates. Colony-forming units (CFU) were counted after 24 hr of incubation at 37° and calculated per 1 ml peritoneal lavage or organ, respectively.
Uptake and killing of E. coli by macrophages in vitro
Bone-marrow-derived macrophages were generated by differentiation for 7 days in macrophage colony-stimulating factor containing medium as described previously.21 The capacity of Dusp1−/− bone-marrow-derived macrophages to ingest and kill the Gram-negative bacterium E. coli (strain HB101) was determined essentially as described by Wiese et al.22 In brief, bacteria were added to the cell culture plates at the indicated multiplicity of infection (MOI) and centrifuged for 5 min. After 1 hr of infection, cells were washed twice and extracellular bacteria were killed by addition of gentamicin (100 μg/ml for 1 hr, followed by 25 μg/ml for the rest of the experiment). Macrophages were lysed with 0·1% Triton X-100 in PBS and serial dilutions were plated on agar plates for counting of intracellular CFU. Relative survival after 24 hr is given as percentage compared to the bacterial numbers after 2 hr (the 1-hr infection period plus 1 hr of high-dose gentamicin).
Statistical analysis
The ELISA and qPCR data were analysed with two-sided, unpaired Student's t-test for significant differences (P < 0·05). Statistical analysis of survival data was done with the log rank test (http://bioinf.wehi.edu.au/cgi-bin/russell/logrank/logrank.pl).
Results
Increased expression of cytokines and chemokines in Dusp1−/− mice after CASP
Dusp1−/− mice respond with over-shooting production of IL-6 and IL-10 to TLR stimulation in vivo. As the inflammatory response in the CASP model of septic peritonitis depends on TLR signalling through MyD88,23 we asked whether cytokine production in Dusp1−/− mice is dysregulated after induction of polymicrobial peritonitis. Systemic levels of cytokines were determined in the serum 12 hr after the CASP procedure, a time when the inflammatory response shows a maximum level for most cytokines.13,24 The levels of IL-6 and IL-10 induced by insertion of an 18G stent were significantly higher in Dusp1−/− mice (Fig. 1a). Use of the smaller 20G stent for the CASP procedure led to lower concentrations of IL-6 and IL-10 in WT mice; again, Dusp1−/− mice showed a significant increase with a larger difference from WT than in the 18G procedure (Fig. 1b). In addition, serum levels of CCL4 and TNF-α were also significantly higher in Dusp1−/− mice (Fig. 1b). Next, we analysed changes in gene expression in spleen and liver, two anatomical sites responding to the polymicrobial challenge in CASP with a differential requirement for MyD88 signalling.25 The expression of Dusp1 was increased in spleen and liver following the CASP procedure (Fig. 1c). Consistent with the serum cytokine levels, the induced expression of IL-6 and IL-10 was significantly higher in the absence of Dusp1 in both spleen and liver (Fig. 1d,e). Expression of IL-1β was induced in the spleens of Dusp1−/− but not WT mice (Fig. 1d); IL-1β protein was also detectable in the serum of some Dusp1−/− mice but not in WT mice following 20G CASP (not shown). The expression of genes relevant for anti-microbial defence was also investigated in spleen and liver RNA. CASP caused no induction of IFN-γ expression after 12 hr; the expression of the IFN-γ-induced GTPase Lrg47 was weakly induced in the spleen of WT but not Dusp1−/− mice (Fig. 1d). In contrast, expression of Nos2, encoding the NO-producing enzyme inducible nitric oxide synthase (iNOS), was up-regulated in the liver with a significantly stronger expression in Dusp1−/− mice (Fig. 1e). Together, Dusp1−/− mice responded to the polymicrobial peritonitis induction by CASP with uncontrolled release of a subset of inflammatory mediators, reminiscent of the phenotype observed previously in the LPS shock model. In addition, we determined the expression of the neutrophil marker gene myeloperoxidase after CASP (Fig. 1d,e). While splenic expression did not change much, in the livers of CASP-treated WT mice increased myeloperoxidase messenger RNA (mRNA) levels were found; again, Dusp1−/− mice responded much more strongly than WT mice, indicating increased granulocyte infiltration in the liver.
Figure 1.
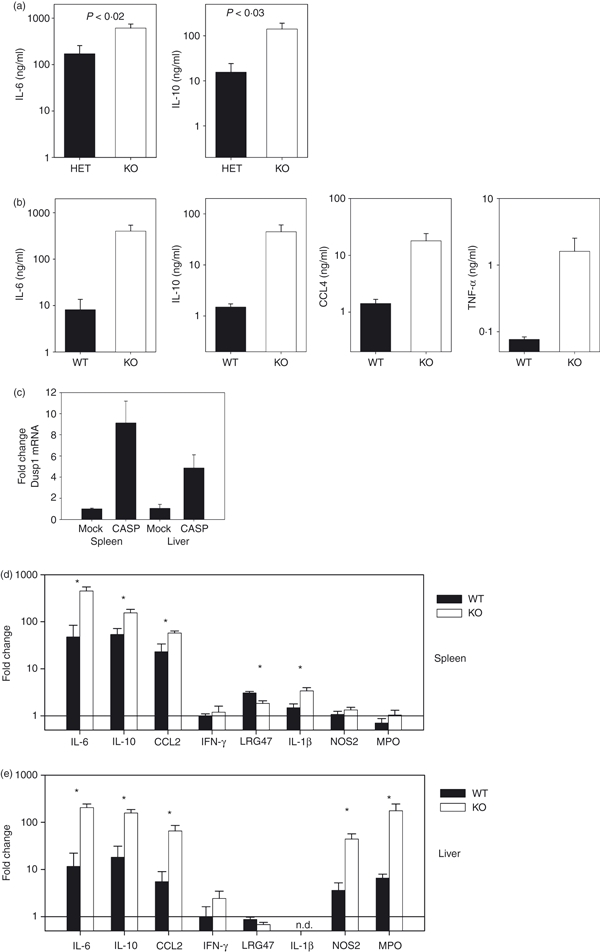
Increased expression of cytokines and chemokines in Dusp1−/− mice after colon ascendens stent peritonitis (CASP). Mice were subjected to the CASP procedure and killed 12 hr later. Serum levels of cytokines were determined by enzyme-linked immunosorbent assay. (a) Interleukin-6 (IL-6) and IL-10 levels after 18G CASP. Mice were on mixed 129 × C57BL/6 background, heterozygote littermate controls; data are pooled from two experiments, mean and SEM (n = 10). (b) IL-6 and IL-10 after 20G CASP. Mice backcrossed to C57BL/6, controls were from Harlan (n = 10) or wild-type (WT) littermates (n = 5). Data are pooled from three experiments. (c) Changes in expression of Dusp1 in spleen and liver 12 hr after 20G CASP. (d, e) Changes in gene expression in spleen (d) or liver (e) 12 hr after 20G CASP. (c–e) Data are pooled from two experiments, mean and SEM (n = 10 for each genotype for treated, n = 2 for untreated control mice). Asterisks indicate P < 0.05 in Student's t-test.
Increased cytokine and chemokine expression of Dusp1−/− mice in the CLP model
The classical CLP model of septic peritonitis is characterized by more transient bacterial dissemination in the peritoneum and peripheral organs than is found in the CASP model, and by the pivotal role that TNF-α plays in host survival.1,10 We next asked whether Dusp1−/− mice also over-produce cytokines and chemokines in the CLP model. Mice were subjected to a mild form of CLP that induces only up to 20% lethality, followed by analysis of systemic cytokine and chemokine protein levels as well as spleen and liver mRNA abundance at the 12-hr time-point, similar to the experiments in the CASP model. Consistent with the literature,1 the serum levels of IL-6, IL-10, CCL3 and CCL4 in WT mice were lower after CLP than in the CASP model (Fig. 2a, compare with Fig. 1a). However, comparable to the CASP model, Dusp1−/− mice responded more vigorously to CLP than their WT or heterozygous littermate controls. Protein levels of IL-6, IL-10, CCL3 and CCL4 were significantly higher in Dusp1−/− serum. In contrast to the CASP model, TNF-α was not detectable by ELISA in the serum of WT or Dusp1−/− mice (not shown). Consistent with the local containment of the polymicrobial infection in the CLP model, the expression of inflammatory mediators and the induction of Dusp1 mRNA was less pronounced than after CASP in the spleen (Fig. 2b,c) and was almost unaffected in the liver (Fig. 2b,d) of WT mice 12 hr after CLP. CCL4, IL-6 and IL-10 were induced more strongly in Dusp1−/− spleens and livers, whereas only minor differences were seen for CCL2, CCL3, Lrg47 and Nos2 at the level of gene expression (Fig. 2c,d).
Figure 2.
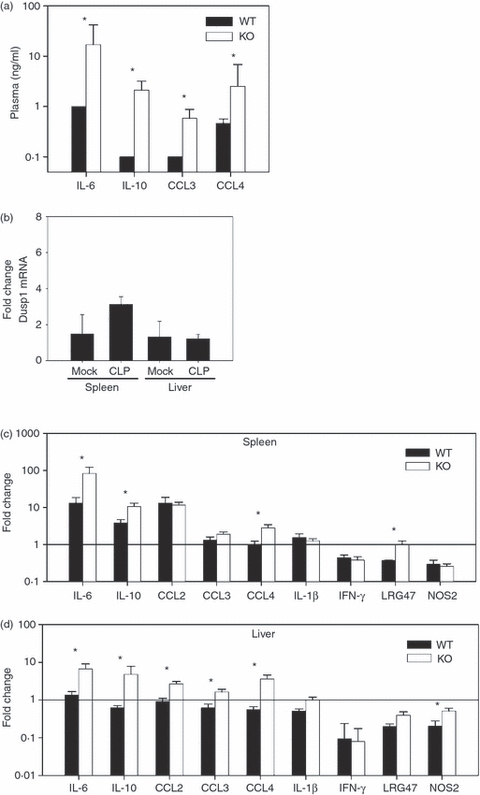
Increased expression of cytokines and chemokines in Dusp1−/− mice after caecal ligation and puncture (CLP). Mice were killed 12 hr after CLP. (a) Serum levels of interleukin-6 (IL-6), IL-10, CCL3 and CCL4. Mice were backcrossed to C57BL/6. Mean and SEM (n = 5) are shown. (b) Dusp1 expression in spleen and liver after CLP. (c, d) Induction of inflammatory gene expression 12 hr after CLP in the spleens (c) and livers (d) of wild-type (WT) and Dusp1−/− mice (n = 5). Mean and SEM from one experiment out of two with similar results are depicted. Asterisks indicate P < 0.05 in Student's t-test.
Blockade of IL-6 does not reduce dysregulated cytokine production in Dusp1−/− mice
Interleukin-6 is required for survival in experimental sepsis,26,27 but may also contribute to exaggerated inflammation and pathology.28 We therefore explored the possible contribution of elevated IL-6 levels to the increase in inflammatory responses in Dusp1−/− mice in peritonitis. In addition to direct effects on cells expressing the IL-6Ra (macrophages, lymphocytes and hepatocytes), IL-6 can also affect other cell types through so-called trans-signalling mediated through binding of IL-6/sIL-6Ra complexes to the ubiquitously expressed gp130 signalling chain. Directly after CLP surgery, Dusp1−/− mice were injected with a neutralizing anti-IL-6 antibody28 or with the recombinant sgp130-Fc fusion protein that abrogates IL-6 trans-signalling.29 Investigation of serum cytokine levels and gene expression in spleen and liver after 16 hr revealed no significant effect of blockade of IL-6 by antibodies or sgp130-Fc on IL-10, CCL3, CCL4 or IL-6 itself (data not shown).
Effects of Dusp1-deficiency on control of bacterial growth in peritonitis models and in vitro
We next asked whether the unrestrained production of cytokines and chemokines after CASP or CLP in the absence of Dusp1 benefits the host through more efficient and faster elimination of the polymicrobial infection. First, the bacterial load was determined in the peritoneum and liver 12 hr after CASP induction. Although no significant difference was found following the 18G CASP procedure (not shown), Dusp1−/− mice had elevated numbers of bacteria in the peritoneal lavage and in the liver when the smaller 20G stent was implanted (Fig. 3a). In the CLP model, the CFU were moderately higher in the peritoneal lavage fluid of Dusp1−/− mice compared with WT (Fig. 3b). To determine whether Dusp1−/− macrophages have a cell-autonomous defect in the control of bacteria, we performed in vitro infections of macrophages with E. coli and analysed the killing of this prototypic Gram-negative enteric bacterium over time. The rate of uptake and killing of E. coli was similar between Dusp1−/− and control macrophages (Fig. 3c), indicating that the higher bacterial load in vivo was not the result of a lack of the macrophages’ capacity to phagocytose and digest bacteria.
Figure 3.
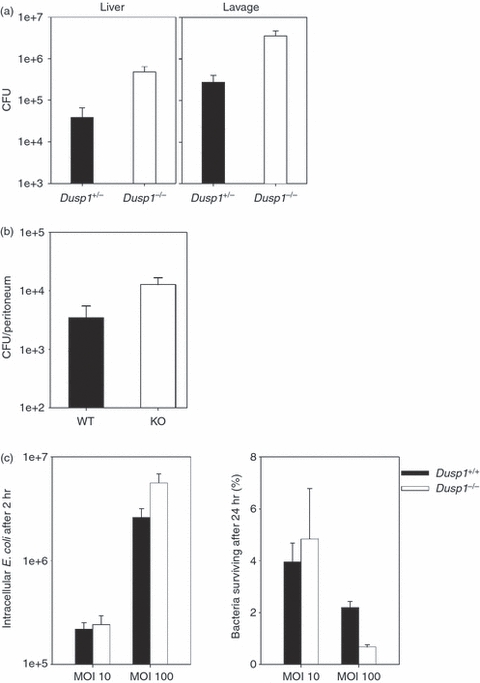
Increased bacterial load in colon ascendens stent peritonitis (CASP) and caecal ligation and puncture (CLP) models in vivo but intact bacterial killing by Dusp1−/− macrophages in vitro. (a) Bacterial load in liver and peritoneal lavage 12 hr after induction of 20G CASP. Data are pooled from two independent experiments (n = 10). Mean and SEM, P = 0.014 (liver) and 0.012 (lavage). (b) Bacterial load in the peritoneum of mice 12–16 hr after CLP. Results from three independent experiments were pooled (n = 15). Mean and SEM, P = 0.045. (c) Bone-marrow-derived macrophages were infected with Escherichia coli at the indicated multiplicity of infection for 1 hr. Number of E. coli recovered from the cells after extensive washing directly after infection and the percentage of bacteria recovered from the macrophages after 24 hr are shown. Mean and SD from duplicate measurements; representative of two experiments performed.
Impact of Dusp1-deficiency on survival in polymicrobial peritonitis models
Finally, the outcome of peritonitis in Dusp1−/− mice in terms of survival was investigated. In the CASP model, the use of the larger 18G stent caused the death of 80% of littermate control mice within 3 days, whereas all Dusp1−/− mice died (Fig. 4a). The use of the smaller 20G stent for CASP caused a milder polymicrobial peritonitis that was survived by 60% of Dusp1+/− mice, but only 20% of all Dusp1−/− mice (P = 0·0341) (Fig. 4b). To test whether Dusp1−/− mice develop a higher lethality also after CLP, cohorts of Dusp1−/− mice together with heterozygous littermate controls (Fig. 5a) or WT controls from a commercial breeder (Fig. 5b) were subjected to CLP in two independent sets of experiments and their survival was recorded. Control mice showed 0–15% lethality over a period of 2 weeks under the mild CLP conditions used here. In contrast, 80–95% of all Dusp1−/− mice died within 1 week, resulting in a highly significant increase in the susceptibility of Dusp1−/− mice, independent of the genetic background of the mice used. Together, the data from the CASP and the CLP models showed that the increased inflammatory response in Dusp1−/− mice correlates with strongly increased lethality.
Figure 4.
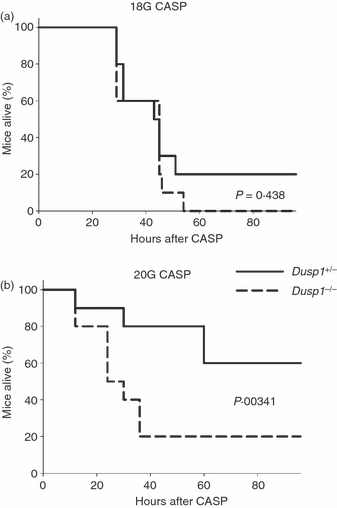
Survival of Dusp1−/− mice after colon ascendens stent peritonitis (CASP). Kaplan–Meier curves of mice after 18G (a) and 20G (b) CASP. Mice were on a mixed 129 × C57BL/6 background, heterozygote littermate controls were used. Data are pooled from two experiments (n = 10 for each condition). Significance was calculated using the log rank test.
Figure 5.
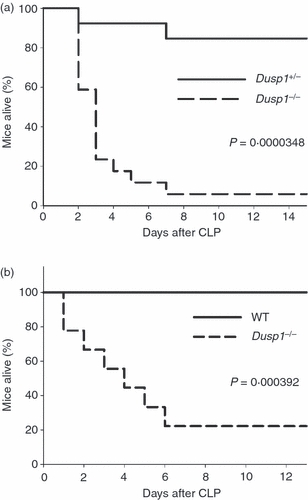
Increased lethality of Dusp1−/− mice after caecal ligation and puncture (CLP). Kaplan–Meier survival curves of mice after CLP. (a) Mixed 129 × C57BL/6 background, heterozygous littermate controls; data are pooled from two experiments (n = 17 for Dusp1−/−, n = 13 for Dusp1−/−). (b) C57BL/6 background, wild-type (WT) controls from Harlan. Pooled from two experiments (n = 10 for both genotypes. Statistical testing was done using the log rank test.
Discussion
This investigation of the phenotype of Dusp1−/− mice in the CASP and CLP models demonstrates that this MAPK phosphatase is essential for a balanced inflammatory response to the massive and acute polymicrobial challenge in abdominal sepsis. We observed a comparably exaggerated production of several cytokines and chemokines in Dusp1−/− mice after CASP or CLP, which was associated with a strong increase in lethality. Inflammatory gene expression was more pronounced after CASP than after CLP, which is consistent with the literature;1 however, the effect of the Dusp1 deficiency was comparable in both models. This phenotype was not unexpected given the high susceptibility of Dusp1−/− in the LPS shock model, where a similar set of dysregulated cytokines and chemokines had been previously observed.5 In addition, while this manuscript was under preparation, Liu's group reported that Dusp1−/− knockout mice are more susceptible to experimental infection with a high number of E. coli and over-produce TNF-α, IL-6 and IL-10.9 Hence, Dusp1 is essentially required to prevent over-shooting cytokine and chemokine production in response to the bolus release of a large quantity of bacterial pathogen-associated molecular patterns from the gut in CASP and CLP.
It is interesting to compare the phenotype in Dusp1−/− mice that fail to down-regulate MAPK activation with results obtained in polymicrobial peritonitis models using MyD88-deficient mice that lack a major pathway activated by bacterial TLR ligands. A mirror phenotype is observed in MyD88−/− mice after CASP induction; the attenuated signalling via TLRs causes reduced inflammatory responses and heightened resistance.23 In contrast, after CLP a split phenotype is observed in MyD88−/− mice that lack inflammatory cytokine production but show increased mortality.30 Therefore, although, depending on the model, the lack of full activation of innate responses through MyD88 is beneficial or detrimental, an unchecked MAPK activation in Dusp1−/− mice is deleterious for the host in both peritonitis models.
An important question raised by our results is which of the over-produced inflammatory mediators are responsible for the higher lethality of Dusp1−/− mice. The increased expression of chemokines in spleen and liver following both CASP and CLP, and their high levels in the blood, probably cause enhanced infiltration by leucocytes and more severe organ damage than in other mice, similar to the findings after LPS challenge.7 Indeed, we observed a strong increase in the levels of hepatic myeloperoxidase gene expression in Dusp1−/− mice after CASP (Fig. 1e). In contrast, during systemic infection with E. coli the degree of neutrophilic infiltration was comparable between WT and Dusp1−/− mice.9 Another possibility is that the high systemic levels of chemokines may distract immune cells from the site of infection where they would be needed for control of bacteria in the peritoneal cavity. Such a mechanism has been shown elegantly in mice expressing TLR4 only on endothelial cells.30 In these animals, neutrophils were not trapped in the lungs, but the cells were recruited more efficiently to sites of infection and the animals were protected from an otherwise lethal infection with E. coli.31
Production of IL-6 is over 20-fold higher in Dusp1−/− mice with peritonitis than in WT mice. Interleukin-6 has long been used as a marker of sepsis severity in animal models32,33 and in human sepsis.34 High levels of IL-6 in the serum correlate with severity and predict a lethal outcome in sepsis. However, it is controversial whether IL-6 plays a beneficial or detrimental functional role during sepsis. Although IL-6 is required for optimal host defence against infection with Listeria monocytogenes,26Streptococcus pneumoniae,35E. coli36 and Klebsiella pneumoniae,37 the antibody-mediated blockade of IL-6 in CLP in mice improved survival.28 The pleiotropic effects of IL-6 are generated through signalling by way of the classical IL-6 receptor on leucocytes and hepatocytes; in addition, so-called IL-6 trans-signalling by binding of IL-6–sIL-6Ra complexes to ubiquitously expressed gp130 appears to play an important role in some inflammatory responses.38,39 The lack of an effect of blockade of trans-signalling with sgp130-Fc or of complete neutralization of IL-6 using a monoclonal antibody in Dusp1−/− on the elevated cytokine and chemokine levels after CLP suggests that IL-6 may also not be causally involved in the higher lethality. Consistent with our results, in a recent study of haemorrhagic shock and sepsis treatment of mice with sgp130-Fc had no significant effect on survival.40 In addition, early antibiotic treatment but not neutralization of IL-6 with antibodies protected mice from death in the CLP model.41 Therefore, despite robust expression of IL-6 in WT mice and excessive levels in Dusp1−/− mice, this cytokine does not appear to be the major determinant in the inflammatory response to polymicrobial peritonitis.
Over-production of IL-10 in Dusp1−/− mice was observed in both peritonitis models, similar to the findings after LPS challenge, and infection with S. aureus or E. coli.8,9 As IL-10 is an anti-inflammatory regulatory cytokine, the correlation of high levels of IL-10 production with increased lethality in endotoxic shock and septic peritonitis seems counter-intuitive. However, IL-10 and Dusp1 are connected through mutual control of expression,5,21 and IL-10 may not be fully active in the absence of Dusp1. Interleukin-10 strongly increases the TLR-induced expression of Dusp1 in macrophages and thereby leads to quicker down-regulation of p38 MAPK activity, which in turn contributes to the inhibitory effects of IL-10 on cytokine expression through effects on transcription as well as mRNA stability.21,42 However, even if IL-10 has lost some of its anti-inflammatory potential in Dusp1−/− mice, it is unlikely that it has pro-inflammatory effects and amplifies cytokine and chemokine production. Instead, uncontrolled IL-10 production in the absence of Dusp1 most likely affects the course of CASP and CLP by effects on bacterial load, as it impairs the containment of bacteria in mice infected with E. coli.9
The increased bacterial burden in Dusp1−/− mice following CLP and the CASP procedure with the smaller 20G stent is unlikely to be caused by a cell-autonomous defect of Dusp1−/− macrophages in the killing of gut bacteria, as suggested by our in vitro experiments using bone-marrow-derived macrophages. Instead, we consider it more likely that the high levels of IL-10 produced in Dusp1−/− mice in vivo during peritonitis impair the killing of bacteria in a similar way to their effects in E. coli infection.9 It is also important to note that the increase in bacterial load in Dusp1−/− mice showed some variability between experiments and was less pronounced than the exaggerated inflammatory cytokine and chemokine responses.
Finally, the deleterious effect of the Dusp1 knockout on the course of polymicrobial peritonitis in the mouse asks for a comparison with treatment strategies for human sepsis that target the MAPK signalling module. Pharmacological inhibition of p38 MAPK activity can decrease inflammatory cytokine and chemokine production43,44 as well as liver damage,43 in response to LPS or CLP, which is consistent with the results obtained here in Dusp1−/− mice. However, in mouse infection models with Mycobacterium tuberculosis or Streptococcus pneumoniae p38 MAPK inhibition led to higher levels of TNF-α and increased bacterial growth in the lungs.45 These differences demonstrate that the MAPK module and the DUSPs regulating it play a complex role in orchestrating the inflammatory and anti-microbial response in vivo. While in general the DUSPs can be considered attractive targets for potential pharmacological intervention,46 in the case of microbial peritonitis the results reported here suggest that Dusp1 is an essential endogenous attenuator of the inflammatory response whose absence has detrimental consequences for the host.
Acknowledgments
We thank Bristol Myers Squibb for permission to use Dusp1−/− mice and Dr Andrew Cato (Karlsruhe, Germany) who provided breeding pairs. We are grateful to Simone Kaiser-Moore and Gabriela Jusek for assistance with the CASP experiments. We thank Jonathan Jantsch and Melanie Wiese for help with killing assays, and Angela Servatius and Barbara Bodendorfer for managing the Dusp1−/− mouse colonies. This project has been funded by a grant from the Deutsche Forschungsgemeinschaft (SFB756 A11, and SFB 643 A11, RL).
Disclosures
The authors state no conflict of interest.
References
- 1.Buras JA, Holzmann B, Sitkovsky M. Animal models of sepsis: setting the stage. Nat Rev Drug Discov. 2005;4:854–65. doi: 10.1038/nrd1854. [DOI] [PubMed] [Google Scholar]
- 2.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 3.Lang R, Hammer M, Mages J. DUSP meet immunology: dual specificity MAPK phosphatases in control of the inflammatory response. J Immunol. 2006;177:7497–504. doi: 10.4049/jimmunol.177.11.7497. [DOI] [PubMed] [Google Scholar]
- 4.Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. Proc Natl Acad Sci USA. 2006;103:2274–9. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hammer M, Mages J, Dietrich H, Servatius A, Howells N, Cato AC, Lang R. Dual specificity phosphatase 1 (DUSP1) regulates a subset of LPS-induced genes and protects mice from lethal endotoxin shock. J Exp Med. 2006;203:15–20. doi: 10.1084/jem.20051753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. J Immunol. 2006;176:1899–907. doi: 10.4049/jimmunol.176.3.1899. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Q, Wang X, Nelin LD, et al. MAP kinase phosphatase 1 controls innate immune responses and suppresses endotoxic shock. J Exp Med. 2006;203:131–40. doi: 10.1084/jem.20051794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang X, Meng X, Kuhlman JR, Nelin LD, Nicol KK, English BK, Liu Y. Knockout of Mkp-1 enhances the host inflammatory responses to gram-positive bacteria. J Immunol. 2007;178:5312–20. doi: 10.4049/jimmunol.178.8.5312. [DOI] [PubMed] [Google Scholar]
- 9.Frazier WJ, Wang X, Wancket LM, Li XA, Meng X, Nelin LD, Cato AC, Liu Y. Increased inflammation, impaired bacterial clearance, and metabolic disruption after gram-negative sepsis in Mkp-1-deficient mice. J Immunol. 2009;183:7411–19. doi: 10.4049/jimmunol.0804343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Echtenacher B, Falk W, Mannel DN, Krammer PH. Requirement of endogenous tumor necrosis factor/cachectin for recovery from experimental peritonitis. J Immunol. 1990;145:3762–6. [PubMed] [Google Scholar]
- 11.Echtenacher B, Mannel DN, Hultner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. 1996;381:75–7. doi: 10.1038/381075a0. [DOI] [PubMed] [Google Scholar]
- 12.Echtenacher B, Weigl K, Lehn N, Mannel DN. Tumor necrosis factor-dependent adhesions as a major protective mechanism early in septic peritonitis in mice. Infect Immun. 2001;69:3550–5. doi: 10.1128/IAI.69.6.3550-3555.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zantl N, Uebe A, Neumann B, Wagner H, Siewert JR, Holzmann B, Heidecke CD, Pfeffer K. Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect Immun. 1998;66:2300–9. doi: 10.1128/iai.66.5.2300-2309.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maier S, Traeger T, Entleutner M, et al. Cecal ligation and puncture versus colon ascendens stent peritonitis: two distinct animal models for polymicrobial sepsis. Shock. 2004;21:505–11. doi: 10.1097/01.shk.0000126906.52367.dd. [DOI] [PubMed] [Google Scholar]
- 15.Echtenacher B, Freudenberg MA, Jack RS, Mannel DN. Differences in innate defense mechanisms in endotoxemia and polymicrobial septic peritonitis. Infect Immun. 2001;69:7271–6. doi: 10.1128/IAI.69.12.7271-7276.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Entleutner M, Traeger T, Westerholt A, Holzmann B, Stier A, Pfeffer K, Maier S, Heidecke CD. Impact of interleukin-12, oxidative burst, and iNOS on the survival of murine fecal peritonitis. Int J Colorectal Dis. 2006;21:64–70. doi: 10.1007/s00384-004-0707-0. [DOI] [PubMed] [Google Scholar]
- 17.Emmanuilidis K, Weighardt H, Maier S, et al. Critical role of Kupffer cell-derived IL-10 for host defense in septic peritonitis. J Immunol. 2001;167:3919–27. doi: 10.4049/jimmunol.167.7.3919. [DOI] [PubMed] [Google Scholar]
- 18.van der Poll T, Marchant A, Buurman WA, et al. Endogenous IL-10 protects mice from death during septic peritonitis. J Immunol. 1995;155:5397–401. [PubMed] [Google Scholar]
- 19.Dorfman K, Carrasco D, Gruda M, Ryan C, Lira SA, Bravo R. Disruption of the erp/mkp-1 gene does not affect mouse development: normal MAP kinase activity in ERP/MKP-1-deficient fibroblasts. Oncogene. 1996;13:925–31. [PubMed] [Google Scholar]
- 20.Echtenacher B, Hultner L, Mannel DN. Cellular and molecular mechanisms of TNF protection in septic peritonitis. J Inflamm. 1995;47:85–9. [PubMed] [Google Scholar]
- 21.Hammer M, Mages J, Dietrich H, Schmitz F, Striebel F, Murray PJ, Wagner H, Lang R. Control of dual-specificity phosphatase-1 expression in activated macrophages by IL-10. Eur J Immunol. 2005;35:2991–3001. doi: 10.1002/eji.200526192. [DOI] [PubMed] [Google Scholar]
- 22.Wiese M, Castiglione K, Hensel M, Schleicher U, Bogdan C, Jantsch J. Small interfering RNA (siRNA) delivery into murine bone marrow-derived macrophages by electroporation. J Immunol Methods. 2010;353:102–10. doi: 10.1016/j.jim.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 23.Weighardt H, Kaiser-Moore S, Vabulas RM, Kirschning CJ, Wagner H, Holzmann B. Cutting edge: myeloid differentiation factor 88 deficiency improves resistance against sepsis caused by polymicrobial infection. J Immunol. 2002;169:2823–7. doi: 10.4049/jimmunol.169.6.2823. [DOI] [PubMed] [Google Scholar]
- 24.Feterowski C, Novotny A, Kaiser-Moore S, Muhlradt PF, Rossmann-Bloeck T, Rump M, Holzmann B, Weighardt H. Attenuated pathogenesis of polymicrobial peritonitis in mice after TLR2 agonist pre-treatment involves ST2 up-regulation. Int Immunol. 2005;17:1035–46. doi: 10.1093/intimm/dxh282. [DOI] [PubMed] [Google Scholar]
- 25.Weighardt H, Mages J, Jusek G, Kaiser-Moore S, Lang R, Holzmann B. Organ-specific role of MyD88 for gene regulation during polymicrobial peritonitis. Infect Immun. 2006;74:3618–32. doi: 10.1128/IAI.01681-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dalrymple SA, Lucian LA, Slattery R, McNeil T, Aud DM, Fuchino S, Lee F, Murray R. Interleukin-6-deficient mice are highly susceptible to Listeria monocytogenes infection: correlation with inefficient neutrophilia. Infect Immun. 1995;63:2262–8. doi: 10.1128/iai.63.6.2262-2268.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sutherland RE, Olsen JS, McKinstry A, Villalta SA, Wolters PJ. Mast cell IL-6 improves survival from Klebsiella pneumonia and sepsis by enhancing neutrophil killing. J Immunol. 2008;181:5598–605. doi: 10.4049/jimmunol.181.8.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Riedemann NC, Neff TA, Guo RF, Bernacki KD, Laudes IJ, Sarma JV, Lambris JD, Ward PA. Protective effects of IL-6 blockade in sepsis are linked to reduced C5a receptor expression. J Immunol. 2003;170:503–7. doi: 10.4049/jimmunol.170.1.503. [DOI] [PubMed] [Google Scholar]
- 29.Rose-John S, Scheller J, Elson G, Jones SA. Interleukin-6 biology is coordinated by membrane-bound and soluble receptors: role in inflammation and cancer. J Leukoc Biol. 2006;80:227–36. doi: 10.1189/jlb.1105674. [DOI] [PubMed] [Google Scholar]
- 30.Peck-Palmer OM, Unsinger J, Chang KC, Davis CG, McDunn JE, Hotchkiss RS. Deletion of MyD88 markedly attenuates sepsis-induced T and B lymphocyte apoptosis but worsens survival. J Leukoc Biol. 2008;83:1009–18. doi: 10.1189/jlb.0807528. [DOI] [PubMed] [Google Scholar]
- 31.Andonegui G, Zhou H, Bullard D, et al. Mice that exclusively express TLR4 on endothelial cells can efficiently clear a lethal systemic Gram-negative bacterial infection. J Clin Invest. 2009;119:1921–30. doi: 10.1172/JCI36411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Remick DG, Bolgos G, Copeland S, Siddiqui J. Role of interleukin-6 in mortality from and physiologic response to sepsis. Infect Immun. 2005;73:2751–7. doi: 10.1128/IAI.73.5.2751-2757.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA. Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock. 2002;17:463–7. doi: 10.1097/00024382-200206000-00004. [DOI] [PubMed] [Google Scholar]
- 34.Frink M, van Griensven M, Kobbe P, Brin T, Zeckey C, Vaske B, Krettek C, Hildebrand F. IL-6 predicts organ dysfunction and mortality in patients with multiple injuries. Scand J Trauma Resusc Emerg Med. 2009;17:49. doi: 10.1186/1757-7241-17-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pajkrt D, Camoglio L, Tiel-van Buul MC, et al. Attenuation of proinflammatory response by recombinant human IL-10 in human endotoxemia: effect of timing of recombinant human IL-10 administration. J Immunol. 1997;158:3971–7. [PubMed] [Google Scholar]
- 36.Dalrymple SA, Slattery R, Aud DM, Krishna M, Lucian LA, Murray R. Interleukin-6 is required for a protective immune response to systemic Escherichia coli infection. Infect Immun. 1996;64:3231–5. doi: 10.1128/iai.64.8.3231-3235.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.van Enckevort FH, Sweep CG, Span PN, Netea MG, Hermus AR, Kullberg BJ. Reduced adrenal response and increased mortality after systemic Klebsiella pneumoniae infection in interleukin-6-deficient mice. Eur Cytokine Netw. 2001;12:581–6. [PubMed] [Google Scholar]
- 38.Mitsuyama K, Sata M, Rose-John S. Interleukin-6 trans-signaling in inflammatory bowel disease. Cytokine Growth Factor Rev. 2006;17:451–61. doi: 10.1016/j.cytogfr.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 39.Nowell MA, Richards PJ, Horiuchi S, Yamamoto N, Rose-John S, Topley N, Williams AS, Jones SA. Soluble IL-6 receptor governs IL-6 activity in experimental arthritis: blockade of arthritis severity by soluble glycoprotein 130. J Immunol. 2003;171:3202–9. doi: 10.4049/jimmunol.171.6.3202. [DOI] [PubMed] [Google Scholar]
- 40.Mees ST, Toellner S, Marx K, Faendrich F, Kallen KJ, Schroeder J, Haier J, Kahlke V. Inhibition of interleukin-6-transsignaling via gp130-Fc in hemorrhagic shock and sepsis. J Surg Res. 2009;157:235–42. doi: 10.1016/j.jss.2008.08.035. [DOI] [PubMed] [Google Scholar]
- 41.Vyas D, Javadi P, Dipasco PJ, Buchman TG, Hotchkiss RS, Coopersmith CM. Early antibiotic administration but not antibody therapy directed against IL-6 improves survival in septic mice predicted to die on basis of high IL-6 levels. Am J Physiol Regul Integr Comp Physiol. 2005;289:R1048–53. doi: 10.1152/ajpregu.00312.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schaljo B, Kratochvill F, Gratz N, et al. Tristetraprolin is required for full anti-inflammatory response of murine macrophages to IL-10. J Immunol. 2009;183:1197–206. doi: 10.4049/jimmunol.0803883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klintman D, Li X, Santen S, Schramm R, Jeppsson B, Thorlacius H. p38 mitogen-activated protein kinase-dependent chemokine production, leukocyte recruitment, and hepatocellular apoptosis in endotoxemic liver injury. Ann Surg. 2005;242:830–8. doi: 10.1097/01.sla.0000189132.86878.f7. discussion 838–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Asaduzzaman M, Wang Y, Thorlacius H. Critical role of p38 mitogen-activated protein kinase signaling in septic lung injury. Crit Care Med. 2008;36:482–8. doi: 10.1097/01.CCM.0B013E31816204FA. [DOI] [PubMed] [Google Scholar]
- 45.van den Blink B, Juffermans NP, ten Hove T, Schultz MJ, van Deventer SJ, van der Poll T, Peppelenbosch MP. p38 mitogen-activated protein kinase inhibition increases cytokine release by macrophages in vitro and during infection in vivo. J Immunol. 2001;166:582–7. doi: 10.4049/jimmunol.166.1.582. [DOI] [PubMed] [Google Scholar]
- 46.Jeffrey KL, Camps M, Rommel C, Mackay CR. Targeting dual-specificity phosphatases: manipulating MAP kinase signalling and immune responses. Nat Rev Drug Discov. 2007;6:391–403. doi: 10.1038/nrd2289. [DOI] [PubMed] [Google Scholar]