

Abstract
We have investigated the structure and unfolding kinetics of the human telomeric intramolecular G quadruplex by using single-molecule fluorescence resonance energy transfer. An exploration of conformational heterogeneity revealed two stable folded conformations, in both sodium- and potassium-containing buffers, with small differences between their enthalpies and entropies. Both folded conformations can be opened by the addition of a 21-base complementary DNA oligonucleotide. The unfolding of both substates occurs at the same rate, which showed dependence on the monovalent metal cation present. Temperature-dependence studies in 100 mM KCl gave an apparent activation enthalpy and entropy of 6.4 ± 0.4 kcal·mol–1 and –52.3 ± 1.4 cal·mol–1·K–1, respectively, indicating that the unfolding is entropically driven and can occur easily. In contrast, in 100 mM NaCl the respective values are 14.9 ± 0.2 kcal·mol–1 and –23.0 ± 0.8 cal·mol–1·K–1, suggesting a more significant enthalpic barrier. Molecular modeling suggests that the two species are likely to be the parallel and antiparallel quadruplex structures. The unfolding free energy barrier is estimated to be between 3 and 15 kBT based on Kramers' theory. We conclude that under near-physiological conditions these structures coexist and can interconvert on a minute time scale.
DNA sequences with stretches of multiple guanines can form four-stranded structures called quadruplexes (1). The evidence is growing that quadruplexes may be important for several biological mechanisms (1–8). Evidence supports the occurrence of quadruplexes in the cell nucleus (9), and, furthermore, a recent study suggests that particular quadruplexes may have the potential to control gene expression (10). The intramolecular DNA quadruplex based on the human telomeric repeat sequence, d(TTAGGG), has been the subject of extensive biophysical and biological studies. It has been demonstrated, in vitro, that formation of this structure impedes the extension mechanism of human telomerase (3). Furthermore, stabilization of this quadruplex may be a natural or unnatural mechanism to influence the regulation of teleomeres at the ends of chromosomes. Because telomerase function and telomere maintenance are critical for the division of cancer cells, the human telomeric intramolecular quadruplex (HTIQ) is being seriously considered as a potential molecular target for the development of novel anticancer agents (7, 8). Detailed structural studies have provided evidence for two distinct folds for the HTIQ in the presence of sodium and potassium ions (11, 12). An NMR spectroscopic study, where the dominant monovalent cation was sodium, showed that the HTIQ exists predominantly with an antiparallel arrangement of strands with a diagonal loop across a terminal tetrad and edgewise loops (11) (see Fig. 7A, which is published as supporting information on the PNAS web site). In contrast, a recent x-ray crystal structure of the same oligonucleotide, in the presence of potassium, shows all four strands to be parallel with all loops located down the side of the quadruplex, leaving both terminal tetrads exposed (12) (see Fig. 7B). This apparent contradiction may actually be a reflection of a conformational preference for the HTIQ in sodium vs. potassium, or a direct consequence of other experimental differences between the two studies. Chaires and coworkers (13) have provided evidence, based on detailed analysis of absorbance spectra, that for a DNA quadruplex multiple species coexist in sodium buffer. Studies that probe the folding and unfolding processes of the quadruplex will provide insight into such structural heterogeneity.
Fluorescence resonance energy transfer (FRET) is a powerful tool for the study of the structure and dynamics of nucleic acids (14–16). In FRET a donor fluorophore nonradiatively transfers its excitation energy to an acceptor fluorophore, with an efficiency that depends on their separation and relative orientations (17, 18). FRET can be used to determine molecular conformations (19–21) and has been applied at the ensemble level to study DNA quadruplex formation (22–26). By operating in the single-molecule regime, where FRET is recorded from molecules one at a time, it is possible to determine the number and distribution of molecular conformations and study how these vary with changing conditions. It is also possible to observe the relative populations of these conformations as a function of time to monitor the progress of a reaction, and hence elucidate the reaction rates and pathway. Here we describe work using single-molecule FRET (27–29) to study the HTIQ structure. These studies have revealed the coexistence of two folded conformations, and we have studied the unfolding of these structures over a range of temperatures. These experiments have provided information on the kinetics and energetics of quadruplex unfolding.
Experimental Methods
Materials. The fluorophore-labeled DNA oligonucleotides I and II used in this study were purchased from Cruachem (Glasgow, U.K.). Unlabeled DNA oligonucleotides III, IV, and V were purchased from Oswel (Southampton, U.K.). The sequences of these oligonucleotides are as follows: I, 5′-Cy5-GGG TTA GGG TTA GGG TTA GGG AGA GGT AAA AGG ATA ATG GCC ACG GTG CGG ACG GC-3′; II, 5′-GCC GTC CGC ACC GTG GCC ATT ATC CTT *TTA CCT CT-3′ (*T represents the TAMRA-dT residue; see below); III, 5′-CCC TAA CCC TAA CCC TAA CCC-3′; IV, 5′-GGG TTA GGG TTA GGG TTA GGG AGA GGT AAA AGG ATA ATG GCC ACG GTG CGG ACG GC-3′; and V, 5′-AGA GGT AAA AGG ATA ATG GCC ACG GTG CGG ACG GC-3′.
I contains the human telomeric repeat motif d(GGG TTA GGG TTA GGG TTA GGG), which can form an intramolecular G quadruplex, with Cy5 coupled to the 5′ terminus by phosphoramidite methodology (Cy5 phosphoramidite, available from Glen Research, Sterling, VA). II is the complement of the 35-nucleotide overhang of I, with tetramethylrhodamine (TMR) coupled to a thymine (T28) by a six-carbon and acrylamide linkage (TAMRA-dT, available from Glen Research). III is the complement of the quadruplex-forming region of I. IV is an unlabeled analogue of I. V is the complement of II. DNA concentrations were determined by absorbance at 260 nm. The absorbance at 555 nm for TMR and 649 nm for Cy5 were used to check the purity of the samples.
Single-Molecule Measurements. A home-built dual-channel confocal fluorescence microscope was used to detect freely diffusing single molecules. For the details of this apparatus, refer to refs. 28 and 30. The donor, TMR, was excited by an argon ion laser (model 35LAP321–230, Melles Griot, Carlsbad, CA) with 150 μW at 514.5 nm. The confocal volume was measured to be 0.4 fL by fluorescence correlation spectroscopy. Donor and acceptor fluorescence were collected through an oil-immersion objective (Nikon Plan Fluor ×100, numerical aperture 1.4) and detected separately by two photon-counting modules (SPCMAQR14, Perkin–Elmer). The outputs of the two detectors were recorded by two computer-implemented multichannel scalar cards (MCS-plus, EG & G, ORTEC, Oak Ridge, TN). Sample solutions of 50 pM were used to achieve single-molecule detection. All the samples contained 200 μM sodium ascorbate and 0.01% Tween 20 to reduce photobleaching and adsorption of DNA molecules onto the glass surface, respectively. The temperature of the sample was controlled by a thermostage (PE60, Linkam Scientific Instruments, Surrey, U.K.). The temperature was monitored by using a thermocouple microprobe (0.025-inch diameter, model 1T-1E, World Precision Instruments, Sarasota, FL) placed close to the laser focus (≈1 mm), and regulated better than ± 0.2°C. This thermostage can control the sample temperature between 0 and 45°C. Hybridization kinetics were measured by mixing 50 pM I·II with different amounts of the complementary sequence III and monitoring the populations of both folded and unfolded quadruplex by sequential single-molecule FRET histograms. The concentration of added complementary DNA was high enough to ensure pseudo-first-order kinetics in the event of a second-order reaction.
A threshold of 35 counts per millisecond bin for the sum of the donor and acceptor fluorescence signals was used to differentiate single molecule bursts from the background. A background of between 2 and 3 counts per millisecond, obtained from independent measurements of buffer solutions without labeled samples, was subtracted from each burst. Apparent FRET efficiencies, Eapp, of each burst were calculated according to Eapp = nA/(nA + γnD), where nA and nD are the acceptor and donor counts, respectively. γ = (ϕAηA)/(ϕDηD) is a factor accounting for the difference in the quantum yields, ϕA and ϕD, and detection efficiencies, ηA and ηD, for the acceptor and donor channels, respectively. This factor has been previously measured to be close to 1 for our setup (28).
Molecular Modeling. Molecular modeling of I·II was carried out by using macromodel 5.5 (31). All modeling of DNA was performed with the amber* forcefield and GB/SA implicit solvation. Structures of the parallel and antiparallel intramolecular quadruplexes were obtained from the Protein DataBank, model 1KF1 and an arbitrarily chosen model from 143D, respectively. Further details are provided in Supporting Experimental Methods, which is published as supporting information on the PNAS web site.
Results
The dual-labeled quadruplex system I·II was designed as shown in Fig. 1. The quadruplex is based on the HTIQ and is connected to a 35-bp duplex. The design places the two dyes about 4.7 nm apart in the folded state. This value is close to the estimated Förster distance of 5.3 nm (32), which enhances the sensitivity of FRET changes caused by alterations in structure. Furthermore, positioning of the TMR dye in the duplex minimizes short-range quenching interactions with Cy5 or guanines in the quadruplex (23, 33). Unfolding the quadruplex of I·II in the presence of a complementary strand leads to a large decrease in FRET efficiency (from ≈70% to ≈5%) because of the increase in distance between the fluorophores.
Fig. 1.

Schematic representation of the unfolding of the quadruplex system I·II in the presence of III. This system was designed to show high FRET in its folded quadruplex form and low FRET when hybridized to III.
UV Melting and CD Spectroscopy. UV-melting analysis supports the proposed structure of I·II. Two distinct melting transitions can be observed for I·II (see Table 2 and Fig. 8, which are published as supporting information on the PNAS web site). One transition, seen at both 260 and 295 nm, shows hyperchromism on melting and has the same Tm (70 ± 2°C) in either 100 mM NaCl or KCl, consistent with duplex melting. The other transition, observed only at 295 nm, shows hypochromism on melting and has a higher Tm in KCl (67 ± 3°C) than in NaCl (50 ± 1°C), consistent with quadruplex melting (34). In 100 mM NaCl the melting temperatures of I (50 ± 1°C) and IV (52 ± 1°C) are similar, indicating that Cy5 does not alter the stability of the system. In the presence of 100 mM KCl, the melting temperature of I (67 ± 3°C) is higher than that of IV (59 ± 3°C), suggesting that Cy5 stabilizes the quadruplex. Because the TMR was positioned 8 bp along the duplex, its presence was assumed to have little effect on the stability of the quadruplex.
CD spectra of V·II are almost identical in NaCl and KCl, as expected for a DNA duplex. Spectra of I·II in NaCl and KCl show some differences, but clearly have contributions from the duplex and quadruplex. Subtraction of the V·II spectra from the I·II spectra gave traces with characteristics similar to those seen previously of antiparallel quadruplexes, i.e., peaks at 295 nm and troughs at 265 nm (35) (Fig. 9, which is published as supporting information on the PNAS web site).
Single-Molecule Conformational Analysis. Initial studies were performed at low salt concentration (10 mM). Fig. 2A shows the FRET histograms obtained at different temperatures in the presence of NaCl. Two subpopulations are clearly observed. The low-FRET species (X), centered at 0.35, is significantly broader than the high-FRET species (Y) centered at 0.85. The observed widths for X and Y are significantly higher than their calculated shot-noise-induced widths (0.08 and 0.06, respectively), suggesting that contributions are from conformational heterogeneity. Addition of 10 mM KCl to the sample at 43°C significantly increased the population of the high-FRET conformation.
Fig. 2.

Single-molecule histograms of apparent FRET efficiencies for DNA quadruplex I·II. (A) From top to bottom, all in 10 mM NaCl and 10 mM Tris·HCl (pH 7.4), at 7°C, 20°C, 43°C, and 43°C with additional 10 mM KCl. (B) From top to bottom, all in 10 mM Tris·HCl (pH 7.4), at 20°C, 100 mM NaCl; at 37°C, 100 mM NaCl; at 20°C, 100 mM KCl; and at 37°C, 100 mM KCl. Solid curves are the best fit to the Gaussian functions. The “zero” peaks, largely due to the inactive Cy5, have been subtracted for clarity (see Supporting Experimental Methods).
Fig. 2B shows data acquired at high salt concentration (100 mM). In this case, the mean apparent FRET efficiency of X has increased from 0.35 to 0.52. The mean FRET efficiency of Y is unaffected by the change of salt concentration. This difference may be more obvious for X than Y because of the higher sensitivity of FRET to distance changes for FRET efficiencies near 50%. From the relative populations of conformations X and Y, it is possible to evaluate the temperature-dependent equilibrium constants by calculating the ratio of the number of events that contribute to X and Y, respectively (Table 1). Thermodynamic parameters for the transformation of X to Y were determined by using a van't Hoff analysis. In the presence of 100 mM NaCl, ΔH = –7.3 ± 0.7 kcal·mol–1 and ΔS = –25 ± 2 cal·mol–1·K–1. In the presence of 100 mM KCl, ΔH = 3.6 ± 0.9 kcal·mol–1 and ΔS = 10 ± 3 cal·mol–1·K–1.§ Both of these give relatively small values of ΔG (<1 kcal·mol–1) for the interconversion of X and Y in the temperature range 0–55°C.
Table 1. Equilibrium constants, K = [Y]/[X], for the interconversion of the two conformations of I·II derived from subpopulation analysis.
|
K
|
||||
|---|---|---|---|---|
| Monovalent cation | 12°C | 20°C | 28°C | 37°C |
| 100 mM NaCl | 0.63 ± 0.03 | 0.67 ± 0.03 | 0.51 ± 0.02 | 0.35 ± 0.02 |
| 100 mM KCl | 0.34 ± 0.02 | 0.36 ± 0.03 | 0.46 ± 0.03 | 0.50 ± 0.03 |
Hybridization Kinetics. Because we observed two conformations in sodium and potassium, we decided to investigate their kinetic stability by studying the rate of opening by hybridization to a complementary DNA (oligonucleotide III). The opening rate is relatively slow, allowing histograms to be measured at different times to follow the progress of the reaction. Fig. 3A shows representative histogram data for the hybridization of III to I·II in 10 mM Tris·HCl (pH 7.4)/100 mM NaCl. X and Y disappeared at the same rate within experimental error (796 ± 52 and 768 ± 35 s–1, respectively), as shown in Fig. 3B. Fig. 4 shows kinetic traces for the hybridization at 20 and 37°C. The rise of the zero FRET population (i.e., the open conformation) and decline of the sum of populations X and Y were both fitted with single exponentials and had the same time constant within experimental error (742 ± 29 and 791 ± 41 s–1, respectively). The rate of the reaction linearly depended on the concentration of III over two orders of magnitude (Fig. 10, which is published as supporting information on the PNAS web site).
Fig. 3.

(A) Changes in single-molecule FRET histograms during the opening of the DNA quadruplex by hybridization to 7 nM III. The “zero” peak at t = 0 has been subtracted from all histograms for clarity. The experiment was carried out at 20°C in 100 mM NaCl and 10 mM Tris·HCl (pH 7.4). (B) Kinetic traces for the low- and high-FRET subpopulations. Data were fit to a single exponential, y = A e–t/τ + C.
Fig. 4.

Single-molecule kinetics of unfolding by hybridization to 7 nM III in 100 mM NaCl and 10 mM Tris buffer. (A) Kinetic traces at 37°C. Time constants are 184 ± 7 s for the rise of the duplex population and 168 ± 5 s for the decay of the quadruplex population. (B) Kinetic traces at 20°C. Time constants are 742 ± 29 s for the rise of the duplex population and 791 ± 41 s for the decay of the quadruplex population.
Fig. 5 shows an Arrhenius plot of the rate constants in 10 mM Tris·HCl (pH 7.4) and either 100 mM NaCl or 100 mM KCl. The derived activation enthalpies and entropies are 14.9 ± 0.2 kcal·mol–1 and –23.0 ± 0.8 cal·mol–1·K–1, respectively, for the NaCl-containing buffer and 6.4 ± 0.4 kcal·mol–1 and –52.3 ± 1.4 cal·mol–1·K–1, respectively, for the KCl-containing buffer.
Fig. 5.

Arrhenius plot of the observed single-molecule kinetic rates in 100 mM NaCl (○) and in 100 mM KCl (▪). The derived activation energies and entropies are 14.9 ± 0.2 kcal·mol–1 and –23.0 ± 0.8 cal·mol–1 and 6.4 ± 0.4 kcal·mol–1·K–1 and –52.3 ± 1.4 cal·mol–1·K–1 for sodium- and potassium-containing buffer, respectively.
Modeling. We hypothesized that species X and Y may be the parallel and antiparallel folded HTIQ that have been described (11, 12). To test this hypothesis would necessitate that both structures be differentiated by FRET in the context of our experimental system. We constructed molecular models of I·II, with the quadruplex-forming region folded as either a parallel or antiparallel quadruplex. The quadruplex regions were obtained by modifying structures from the Protein Data Bank, and the duplex regions were constructed within the modeling program. The quadruplex and duplex were connected by a phosphodiester linkage, and the resulting structures were energy-minimized. A conformational search around three bonds of the linker with a maximum resolution of 30° was carried out, and the lowest energy conformation found for the parallel and antiparallel quadruplex-containing structures was subjected to energy minimization. The results of this modeling are shown in Fig. 6. Distances were measured between the attachment points of the TMR (C7 of T28 on II) and Cy5 (C5′ of G1 on I), giving 3.0 and 4.8 nm for the parallel and antiparallel quadruplex-containing models, respectively.
Fig. 6.

Energy-minimized models of I·II in the parallel (A) and antiparallel (B) quadruplex conformations. The DNA bases are represented as sticks (A, blue; C, green; G, red; and T, yellow) and the fluorophores are shown in space-filling models (TMR, orange and Cy5, magenta). The fluorophores are shown for illustrative purposes, and the conformations of their linkers are arbitrary. The distances between the fluorophore attachment sites as measured on the models are shown by double-headed arrows.
Discussion
Thermodynamics. Single-molecule FRET measurements showed the presence of two clearly resolvable species. The free energy difference between these structures was small (<1 kcal·mol–1) over the temperature range investigated. Clear differences also exist in quadruplex stability, depending on the identity of the monovalent metal ions present. We considered the possibility that structures X and Y are due to the presence of fully folded and (partially) unfolded quadruplex. If this were so, the sign of ΔS would be the same in both Na+ and K+, which is not the case. This result suggests that species X and Y are more likely to be folded quadruplex structures. We can put lower and upper limits on the rate of interconversion between X and Y; it is slower than 1 ms, because we can resolve the two species during their transit across the confocal volume, but it occurs within a few minutes, because the changes have taken place between runs when we raise the temperature. NMR spectroscopic hydrogen/deuterium exchange studies on the HTIQ in the presence of sodium (11) showed exchange of all imino protons with solvent within minutes, suggesting that this may be the time scale for a rearrangement of this structure. This finding is entirely consistent with our data.
Kinetics. We have observed that both conformations X and Y apparently unfold at the same rate in a second-order hybridization reaction. In the presence of potassium, the activation free energy of unfolding (ΔG = 22.6 kcal·mol–1 at 37°C) is largely entropic, suggesting that the transition state is preorganized in the correct conformation for hybridization to occur. However, in the presence of sodium ions, although the free energy of activation (ΔG = 22.3 kcal·mol–1 at 37°C) is similar, a relatively larger enthalpic contribution occurs. It therefore seems likely that different transition-state structures exist in the presence of sodium or potassium ions.
Conformations. We have observed the same two structural subpopulations, X and Y, based on FRET, in both sodium- and potassium-containing buffers. Therefore, either the same two structures are formed in the presence of sodium and potassium ions, or we cannot distinguish between the two pairs of structures by FRET. We hypothesize that structures X and Y are in fact parallel and antiparallel folds of the quadruplex.¶ Because detailed structures for parallel and antiparallel conformations of the HTIQ have been determined (11, 12), we used molecular modeling to predict possible distances between the fluorophores, and hence FRET efficiencies, for each case. Assuming a Förster distance, R0, of 7.1 nm for the TMR/Cy5 pair,∥ we can estimate the contribution of the linkers to the interfluorophore distance according to
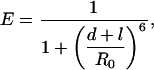 |
[1] |
where d is the distance between two labeling sites, as measured from the modeling, and l is the distance contribution from the linkers. Both sets of linker-length contributions were evaluated. If X is antiparallel, l was found to be 2.3 and 2.2 nm for X and Y, respectively. This outcome gives a difference, Δl, of 0.1 nm. On the other hand, if Y is antiparallel, then l would be 4.0 and 0.5 nm for X and Y, respectively, giving a Δl of 3.5 nm. The distance between the TMR, placed in an arbitrary location relative to its attachment point, and the Cy5 attachment point can be determined by using vector arithmetic (see Supporting Experimental Methods). Assuming that the TMR vector is the same with respect to the duplex, an upper limit for Δl of 2.6 nm was worked out. This determination means that, of the two possible assignments given above, only the former is compatible with this model.†† Therefore, we propose X corresponds to the antiparallel quadruplex, whereas Y corresponds to the parallel quadruplex.
The observation that the parallel and antiparallel structures are in equilibrium is consistent with the results of other workers. A switching between parallel and antiparallel quadruplexes has been observed by Raman spectroscopy and depends on the concentrations of Na+ and K+ (36). More recently, multiple species for a human telomeric sequence have been discovered by UV spectroscopy (13). The antiparallel structure for HTIQ was determined by NMR spectroscopy at high (≈millimolar) concentration, so it is possible that intermolecular interactions may help stabilize one particular structure. Our experiments, carried out at 50 pM, indicate that the antiparallel conformation is not the sole structure in solution but the dominant conformation in the presence of either Na+ or K+. One advantage of working at the single-molecule level is that the likelihood of intermolecular interactions is low. It is possible that the conjugated 35-bp duplex affects the relative populations of the parallel and antiparallel conformations. A very recent platinum cross-linking study of the human telomeric sequence AG3(T2AG3)3 and (T2AG3)4 also suggested that the antiparallel structure exists in the presence of both sodium and potassium ions (37). However, this study cannot rule out the presence of the parallel quadruplex, which may not have the same potential to form cross-links.
Energy Landscape. Kramers' theory can be used to obtain limits on the free energy barrier between the folded and unfolded states (38). Recent studies suggest that the essential features of nucleic acid-folding dynamics, by analogy with protein folding, can be described as a diffusive process on a low-dimensional free energy surface (30, 39). With use of Kramers' equation (40), the folding time τf is given by
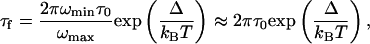 |
[2] |
where ωmin and ωmax are the frequencies that characterize the curvature of the 1D free energy surface in the harmonic well of the unfolded state and at the barrier top, respectively, Δ is the height of the folding free energy barrier, kB is the Boltzmann constant, T is the absolute temperature, τ0 = kBT/(mωmin2D) is the reconfiguration time in the unfolded well, and D is the diffusion constant for motion along the coordinate. In the simplest case, in which ωmin and ωmax are equal, we can estimate Δ from τ0 and τf (41, 42). Because the loop conformations of the antiparallel and parallel structures are very different, we propose that interconversion between them would go through an unfolded or partially folded intermediate. The fact that our single-molecule FRET efficiency histogram can resolve subpopulations at 1-ms time resolution gives the quadruplex DNA unfolding time a lower limit of 1 ms. On the other hand, second-order hybridization kinetics restrain the unimolecular unfolding time constant to an upper limit of 200 s. Experimental observations of the minimal chain-diffusion times for single-stranded DNA are of the order of 10–100 μs, based on fluorescence correlation and temperature-jump measurements (30, 39, 43). This finding gives upper and lower limits for the free energy barrier of unfolding as 15 and 3 kBT, respectively.
In addition, the relative free energies for both quadruplex conformations and the unfolded quadruplex can be derived from equilibrium constants obtained from data from single-molecule measurements and bulk thermal melting. The antiparallel structure has the lowest free energy in the presence of either 100 mM Na+ or 100 mM K+. The free energy difference between the antiparallel and parallel forms in 100 mM K+ at 37°C is ≈0.7 kBT, whereas that between the antiparallel and unfolded forms is ≈3.7 kBT. The hybridization reaction of the quadruplex to a complementary C-rich strand is second-order, and similar reaction rates were observed for the two conformations. This outcome strongly suggests that, although the reaction occurs by means of an open intermediate, the rate-determining step is the hybridization rather than the unfolding of the quadruplex. The result for this system differs from our kinetics investigation of another quadruplex system using peptide nucleic acid as a trapping reagent, in which first-order hybridization kinetic values were observed (26). This apparent discrepancy is simply due to a change in the rate-determining step from the previous system, where quadruplex unfolding was much slower than the hybridization.
Biological Implication. The HTIQ structure appears to coexist in two major forms that interconvert on a time scale faster than minutes. This interconversion time may well turn out to be a general feature of other intramolecular quadruplex structures. The recent discovery of other candidate quadruplexes in the human genome sequence with structural polymorphism (10) suggests that stabilization of particular quadruplex structures may have functional consequences. In this case, the ability for quadruplexes to interconvert between structures on a reasonably fast time scale may be essential.
Supplementary Material
Acknowledgments
We thank Stephen Neidle for helpful discussions. This work was supported by the Biotechnology and Biological Sciences Research Council.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: HTIQ, human telomeric intramolecular quadruplex; FRET, fluorescence resonance energy transfer; TMR, tetramethylrhodamine.
Footnotes
The difference between the melting temperatures of I and IV means that we cannot rule out the possibility that the presence of Cy5 may influence the relative populations of the two conformations.
Another possibility considered for the observed populations was a “hinging motion” of the quadruplex/duplex linker, which is a stable “stacked” structure resulting from π interaction between the terminal base pair of the duplex and a terminal G tetrad. Molecular modeling predicted that the energy of such a stacked conformer was ≈10 kcal·mol–1 higher than that of the extended structure presented in Fig. 6A. We expect that a change from a stacked to an extended conformation would be accompanied by an increase in entropy due to the greater freedom of the linker in this conformation. However, our results show that the entropy difference between the species changes sign in different buffer conditions, which is inconsistent with this model.
The measured quantum yield of TMR in II·IV is 0.81, leading to a calculated Förster radius, R0, of 7.1 nm, assuming an orientational factor, κ2, of 2/3. This value is higher than commonly measured for the TMR/Cy5 pair, largely because of the high quantum yield of TMR in this system.
Our assignment of the two conformations is insensitive to R0 for values >4 nm.
References
- 1.Simonsson, T. (2001) Biol. Chem. 382, 621–628. [DOI] [PubMed] [Google Scholar]
- 2.Sen, D. & Gilbert, W. (1988) Nature 334, 364–366. [DOI] [PubMed] [Google Scholar]
- 3.Zahler, A. M., Williamson, J. R., Cech, T. R. & Prescott, D. M. (1991) Nature 350, 718–720. [DOI] [PubMed] [Google Scholar]
- 4.Fletcher, T. M., Sun, D., Salazar, M. & Hurley, L. H. (1998) Biochemistry 37, 5536–5541. [DOI] [PubMed] [Google Scholar]
- 5.Kerwin, S. M. (2000) Curr. Pharm. Des. 6, 441–471. [DOI] [PubMed] [Google Scholar]
- 6.Arthanari, H. & Bolton, P. H. (2001) Chem. Biol. 8, 221–230. [DOI] [PubMed] [Google Scholar]
- 7.Neidle, S. & Parkinson, G. (2002) Nat. Rev. Drug Discov. 1, 383–393. [DOI] [PubMed] [Google Scholar]
- 8.Hurley, L. H. (2002) Nat. Rev. Cancer 2, 188–200. [DOI] [PubMed] [Google Scholar]
- 9.Schaffitzel, C., Berger, I., Postberg, J., Hanes, J., Lipps, H. J. & Plückthun, A. (2001) Proc. Natl. Acad. Sci. USA 98, 8572–8577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siddiqui-Jain, A., Grand, C. L., Bearss, D. J. & Hurley, L. H. (2002) Proc. Natl. Acad. Sci. USA 99, 11593–11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang, Y. & Patel, D. J. (1993) Structure (London) 1, 263–282. [DOI] [PubMed] [Google Scholar]
- 12.Parkinson, G. N., Lee, M. P. H. & Neidle, S. (2002) Nature 417, 876–880. [DOI] [PubMed] [Google Scholar]
- 13.Ren, J. S., Qu, X. G., Trent, J. O. & Chaires, J. B. (2002) Nucleic Acids Res. 30, 2307–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lilley, D. M. J. & Wilson, T. J. (2000) Curr. Opin. Chem. Biol. 4, 507–517. [DOI] [PubMed] [Google Scholar]
- 15.Klostermeier, D. & Millar, D. P. (2001) Methods 23, 240–254. [DOI] [PubMed] [Google Scholar]
- 16.Ha, T. (2001) Curr. Opin. Struct. Biol. 11, 287–292. [DOI] [PubMed] [Google Scholar]
- 17.Stryer, L. & Haugland, R. P. (1967) Proc. Natl. Acad. Sci. USA 58, 719–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Selvin, P. R. (2000) Nat. Struct. Biol. 7, 730–734. [DOI] [PubMed] [Google Scholar]
- 19.Clegg, R. M., Murchie, A. I. H., Zechel, A. & Lilley, D. M. J. (1993) Proc. Natl. Acad. Sci. USA 90, 2994–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bassi, G. S., Murchie, A. I. H., Walter, F., Clegg, R. M. & Lilley, D. M. J. (1997) EMBO J. 16, 7481–7489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Furey, W. S., Joyce, C. M., Osborne, M. A., Klenerman, D., Peliska, J. A. & Balasubramanian, S. (1998) Biochemistry 37, 2979–2990. [DOI] [PubMed] [Google Scholar]
- 22.Simonsson, T. & Sjöback, R. (1999) J. Biol. Chem. 274, 17379–17383. [DOI] [PubMed] [Google Scholar]
- 23.Mergny, J.-L. & Maurizot, J.-C. (2001) ChemBioChem 2, 124–132. [DOI] [PubMed] [Google Scholar]
- 24.Ueyama, H., Takagi, M. & Takenaka, S. (2002) J. Am. Chem. Soc. 124, 14286–14287. [DOI] [PubMed] [Google Scholar]
- 25.Alberti, P. & Mergny J.-L. (2003) Proc. Natl. Acad. Sci. USA 100, 1569–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green, J. J., Ying, L. M., Klenerman, D. & Balasubramanian, S. (2003) J. Am. Chem. Soc. 125, 3763–3767. [DOI] [PubMed] [Google Scholar]
- 27.Deniz, A. A., Dahan, M., Grunwell, J. R., Ha, T., Faulhaber, A. E., Chemla, D. S., Weiss, S. & Schultz, P. G. (1999) Proc. Natl. Acad. Sci. USA 96, 3670–3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ying, L. M., Wallace, M. I., Balasubramanian, S. & Klenerman, D. (2000) J. Phys. Chem. B 104, 5171–5178. [Google Scholar]
- 29.Ha, T. (2001) Methods 25, 78–86. [DOI] [PubMed] [Google Scholar]
- 30.Wallace, M. I., Ying, L. M., Balasubramanian, S. & Klenerman, D. (2001) Proc. Natl. Acad. Sci. USA 98, 5584–5589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohamadi, F., Richards, N. G. J., Guida, W. C., Liskamp, R., Lipton, M., Caufield, C., Chang, G., Hendrickson, T. & Still, W. C. J. (1990) J. Comput. Chem. 11, 440–467. [Google Scholar]
- 32.Deniz, A. A., Laurence, T. A., Beligere, G. S., Dahan, M., Martin, A. B., Chemla, D. S., Dawson, P. E., Schultz, P. G. & Weiss, S. (2000) Proc. Natl. Acad. Sci. USA 97, 5179–5184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edman, L., Mets, U. & Rigler, R. (1996) Proc. Natl. Acad. Sci. USA 93, 6710–6715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mergny, J.-L., Phan, A. T. & Lacroix, L. (1998) FEBS Lett. 435, 74–78. [DOI] [PubMed] [Google Scholar]
- 35.Lu, M., Guo, Q. & Kallenbach, N. R. (1993) Biochemistry 32, 598–601. [DOI] [PubMed] [Google Scholar]
- 36.Miura, T., Benevides, J. M. & Thomas, G. J. (1995) J. Mol. Biol. 248, 233–238. [DOI] [PubMed] [Google Scholar]
- 37.Redon, S., Bombard, S., Elizondo-Riojas, M. A. & Chottard, J. C. (2003) Nucleic Acids Res. 31, 1605–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuler, B., Lipman, E. A. & Eaton, W. A. (2002) Nature 419, 743–747. [DOI] [PubMed] [Google Scholar]
- 39.Ansari, A, Kuznetsov, S. V. & Shen, Y. Q. (2001) Proc. Natl. Acad. Sci. USA 98, 7771–7776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kramers, H. A. (1940) Physica 7, 284–304. [Google Scholar]
- 41.Socci, N. D., Onuchic, J. N. & Wolynes, P. G. (1996) J. Chem. Phys. 104, 5860–5868. [Google Scholar]
- 42.Klimov, D. K. & Thirumalai, D. (1997) Phys. Rev. Lett. 79, 317–320. [Google Scholar]
- 43.Bonnet, G., Krichevsky, O. & Libchaber, A. (1998) Proc. Natl. Acad. Sci. USA 95, 8602–8606. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.