

Abstract
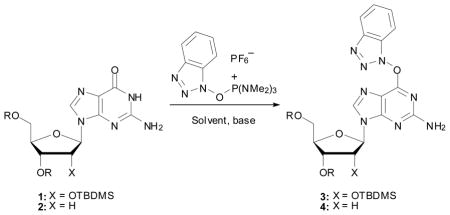
A facile method for the introduction of various substituents at the C-6 position of guanosine and 2′-deoxyguanosine is reported. In a simple, 1-step transformation, tert-butyldimethylsilyl protected guanosine and 2′-deoxyguanosine were converted to the O6-(benzotriazol-1-yl) derivatives via reaction with 1H-benzotriazol-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate (BOP) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU). The easily isolated, stable and storable, O6-(benzotriazol-1-yl) guanosine derivatives upon exposure to range of nucleophiles, under appropriate conditions, led to the C-6 modified 2-amino purine nucleoside analogues in good yields.
Introduction
The development of chemical methods to modify nucleosides continues to attract attention since unnatural and unusual nucleosides play a prominant role in biochemistry, biology and medicine.1 Typically, modification at the C-6 position of the guanine nucleus requires the introduction of a suitable leaving group at that position, followed by SNAr displacement with nucleophiles.
Although chlorination of guanosine can be readily accomplished from its triacetate derivative,2,3 chlorination of the 2′-deoxy analogue has been reported to be low yielding.4 Therefore, improved methods have been reported for C-6 chlorination of 2′-deoxyguanosine 3′,5′-diacetate with careful control of reaction conditions.5,6 In addition to C-6 chlorination of the guanine nucleosides, facile synthesis of the O6-arylsulfonates is also known.6–12 Thus, the 6-chloro and O6-arylsulfonyl derivatives have served as the principal electrophilic nucleoside derivatives used in displacement reactions leading to C-6 modified 2-amino purine nucleosides.
Recently, we have developed a new class of O6-(benzotriazol-1-yl)inosine analogues that are easily prepared via a reaction of either protected or unprotected inosine and 2′-deoxyinosine with 1H-benzotriazol-1-yloxy-tris(dimethylamino)phosphonium hexafluorophosphate (BOP).13 The basis for this chemistry resides in the postulated formation of C-6 phosphonium salts in the reaction of the hypoxanthine moiety with PPh3/I214 or BOP.15 We reasoned that in the absence of other nucleophiles, the hydroxybenzotriazole from BOP should react as a nucleophile on the nucleoside phosphonium salts. This was indeed the case and not only were we able to synthesize O6-(benzotriazol-1-yl)inosine analogues, but we have been able to utilize the underlying chemistry to load inosine and 2′-deoxyinosine onto a polymer linker.16,17 The intermediacy of nucleoside phosphonium salts has also proven important in our recently described N2-modification of 2′-deoxyguanosine,18 and in chemistry leading to N,N-modified adenosine analogues via reaction of protected inosine and 2′-deoxyinosine with hexaalkylphosphorus triamides produced in situ and I2.19 In this paper, we report our findings on the synthesis of O6-(benzotriazol-1-yl)guanosine and 2′-deoxyguanosine, and their applications leading to C-6 modified 2-amino purine nucleoside analogues. While this work was in progress, preparation of O6-(benzotriazol-1-yl)-3′,5′-di-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine and its use for the synthesis of C-8 acetylarylamine adducts was reported.20 However, no use of the O6-(benzotriazol-1-yl)guanine nucleosides for other types of functionalization is currently known.
Results and Discussion
In our initial work, we had shown that conversion of inosine and deoxyinosine to the O6-(benzotriazol-1-yl) derivatives could be accomplished via the use of BOP and i-Pr2NEt as base. Although the reactions of the hypoxanthine nucleosides proceeded to completion,13 similar reactions of the guanine nucleosides proved to be quite slow. In fact, the literature reports 72 h even in a solvent such as DMF.20 Therefore, we decided to reassess this reaction at the outset.
Table 1 summarizes the results from these inital experiments. In order to avoid formation of the N6,N6-dimethyl-2,6-diaminopurine derivate from decomposition of DMF, we chose to conduct our first experiment in DMSO (Table 1, entry 1). In this solvent, elevated temperature and a prolonged reaction time were necessary. Upon switching to THF as solvent (entry 2), practically no reaction was observed, but when DBU was used as base a significantly fast reaction was observed (entry 3) that reached completion within 4 h. Finally, replacing THF with CH3CN led to an improvement in reaction time and yield. In CH3CN as solvent and with DBU as base, the reaction was complete within 2 h with a 65% isolated yield of 3 (entry 4). Under these conditions the 2′-deoxy analogue 4 could be prepared in 85% yield (the previously reported yield for 4 was 67%20).
Table 1.
Initial studies at determining optimal conditions for synthesis of the O6-(benzotriazol-1-yl)guanosine analogues 3 and 4a
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | Solvent | Base | Temp | Time | Yieldb |
| 1 | 1 | DMSO | i-Pr2NEt | 55 °C | 24 h | 54% |
| 2 | 1 | THF | i-Pr2NEt | rt | 96 h | NRc |
| 3 | 1 | THF | DBU | rt | 4 h | 45% |
| 4 | 1 | CH3CN | DBU | rt | 2 h | 65% |
| 5 | 2 | CH3CN | DBU | rt | 1 h | 85% |
Reactions were performed using 2.0 molar equiv each of BOP and DBU at ~0.1 M nucleoside concentration.
Where reported, yield is of isolated and purified products.
No reaction was observed and only 1 was present.
Our observations on optimal solvent and base parallel those reported for the amination of 4-hydroxyquinazoline using BOP and n-BuNH2.21 Besides solubility considerations, one major contributor to the differences in the reactions of inosine and guanosine derivatives should be the pKa values of the acidic amide protons. Inosine is more acidic, with the amide pKa of ~8; in guanosine it is ~9.22,23 Nevertheless, both nucleosides can be conveniently converted to the O6-(benzotriazol-1-yl) derivatives under appropriate conditions.
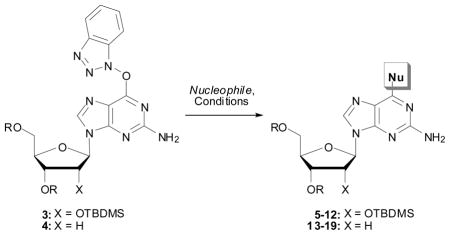
With the synthesis of the O6-(benzotriazol-1-yl)guanine derivatives 3 and 4 optimized, the next step was an evaluation of their use for modification at the C-6 position. For this amine, alcohol, phenol and thiol nucleophiles were selected. In addition, our prior work13 provided a basis for this analysis and the results are shown in Table 2.
Table 2.
Reactions of 3 and 4 with various nucleophilesa
 | ||||
|---|---|---|---|---|
| Entry | Substrate | Nucleophile | Conditions | Yieldb |
| 1 | 3 | 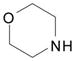 |
DME, 4 molar equiv morpholine, rt, 2 h | 5: 87% |
| 2 | 3 | 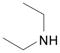 |
DME, 4 molar equiv Et2NH, rt, 19 h | 6: 69% |
| 3 | 3 | 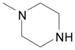 |
DME, 4 molar equiv 1-methylpiperazine, 55 °C, 3.5 h | 7: 82% |
| 4 | 3 | 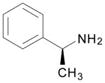 |
DME, 4 molar equiv S-(−)-α-methylbenzylamine, rt, 20.5 h | 8: 92% |
| 5 | 3 | Methanol | CH3OH solvent, 2 molar equiv Cs2CO3, 55 °C, 2 h | 9: 95% |
| 6 | 3 | Ethanol | CH3CH2OH solvent, 2 molar equiv Cs2CO3, 55 °C, 2.5 h | 10: 71% |
| 7 | 3 | Allyl alcohol | CH2=CHCH2OH solvent, 2 molar equiv Cs2CO3, rt, 20 h | 11: 88% |
| 8 | 3 | DME, 2 molar equiv benzyl mercaptan, 2 molar equiv Cs2CO3, rt, 24 h | 12: 89% | |
| 9 | 4 | 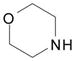 |
1-pot reaction: CH3CN, 2 molar equiv BOP, 2 molar equiv DBU, 4 molar equiv morpholine, rt, 3 h | 13: 91% (94%)d |
| 10 | 4 | 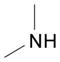 |
DME, 4 molar equiv Me2NH,c rt, 22 h | 14: 83% (86%)d |
| 11 | 4 | 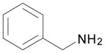 |
DME, 4 molar equiv benzyl amine, rt, 4 h | 15: 81% (80%)d |
| 12 | 4 | CH3OH | CH3OH solvent, 2 molar equiv Cs2CO3, rt, 4 h | 16: 79% (80%)d |
| 13 | 4 | Allyl alcohol | CH2=CHCH2OH solvent, 2 molar equiv Cs2CO3, rt, 7 h | 17: 82% (71%)d |
| 14 | 4 | 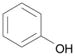 |
DME, 2 molar equiv phenol, 2 molar equiv, Cs2CO3, 60 °C, 2.5 h | 18: 89% (86%)d |
| 15 | 4 | 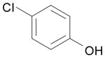 |
DME, 2 molar equiv 4-chlorophenol, 2 molar equiv, Cs2CO3, Cs2CO3 60 °C, 2.5 h | 19: 88% (90%)d |
Reaction times have not been optimized for each entry.
Yields of isolated and purified products.
A 40 wt% solution of Me2NH in water was used.
Yields in parentheses are those obtained from 3′,5′-di-O-(tert-butyldimethylsilyl)-O6-[(2,4,6-trimethylphenyl)sulfonyl]-2′-deoxyguanosine (reported in reference 24).
Our first examples on the SNAr displacement involved reactions of the ribose derivative 3 and secondary amines (morpholine, diethylamine and 1-methylpiperazine) in 1,2-dimethoxyethane (DME) as solvent, without additional base. These reactions were complete at room temperature or at 55 °C, with good to high product yields (Table 2, entries 1–3). Reacton with S-(−)-α-methylbenzyl amine also proceeded smoothly (entry 4), indicating that steric bulk proximal to the nitrogen atom does not interfere with the reaction. Displacements with alcohols (CH3OH, CH3CH2OH and CH2=CHCH2OH) were conducted using the alcohol as solvent with Cs2CO3 as base, and each proceeded smoothly (entries 5–7). Reaction with benzyl mercaptan was conducted in DME with Cs2CO3 as base (entry 8). These results clearly demonstrated the utility of the O6-(benzotriazol-1-yl)guanosine derivative 3 for the synthesis of C-6 modified 2-amino purine nucleosides.
With the disilyl 2′-deoxyguanosine 2, we initially explored the possibility of a 2-step, 1-pot transformation where the O6-(benzotriazol-1-yl) derivative was generated in situ. Thus, 2 was exposed to BOP and DBU in CH3CN. After disapparance of 2 (1 h), 4 molar equiv of morpholine was added. This reaction was complete within an additional 2 h and yielded the 6-morpholinyl product 13 in 91% yield. This is consistent with the reported 1-pot reaction of inosine with BOP and i-Pr2NEt, followed by displacement with morpholine.15 Similarly, we attempted a 2-step, 1-pot synthesis of the methyl ether 16 under identical conditions. However, this reaction only showed a trace of product after 52 h at room temperature. Thus it appears that the 1-pot protocol is currently only feasible with amines, although at this time we have not performed any extensive experiments on the etherification. In this context, reaction of 4 with MeNH2 was performed using a 40 wt% solution in water (entry 10). The good product recovery indicates that any competing reaction with water was minimal in the presence of the amine.
Reactions with phenols (entries 14 and 15) also proceed smoothly. On the basis of these results, it becomes clear that isolation of the nucleoside O6-(benzotriazol-1-yl) derivatives is important for fully exploiting their reactivity with a wide range of nucleophiles. The isolated compounds 3 and 4 possess excellent reactivity towards the range of nucleophiles as exemplified in Table 2 (entries 14 and 15).
We then became interested in evaluating the mechanistic pathway involved in the formation of 3 and 4 for comparison to the reactions of inosine and 2′-deoxyinosine.13 For this we utilized 31P{1H} NMR (Figure 1). The resonances for BOP appear at 45.1 ppm and −143.1 ppm (PF6−) in CD3CN (panel A in Figure 1). Upon addition of 1, no change was observed (panel B). However, two new signals at 35.7 ppm and 26.1 ppm were observed immediately upon addition of DBU (panel C). The resonance at 26.1 ppm corresponding to HMPA continued to grow (pure HMPA in CH3CN appears at 25.6 ppm) as the one at 35.7 ppm was depleted (panel D), indicating the likely intermediacy of a nucleoside phosphonium salt (I in Scheme 1). Finally, this room temperature reaction appears to be complete at 120 minutes (panel E). Therefore, the plausible mechanism is one which parallels that reported previously for the hypoxanthine nucleosides,13 involving formation of a nucleoside phosphonium salt.
Fig. 1.

Monitoring the course of the reaction between 1 and BOP at room temperature using 31P{1H} NMR: (A) BOP in 0.5 mL CD3CN; (B) after addition of 1 (0.1 M in CD3CN); (C) immediately after addition of DBU; (D) 60 minutes after addition of DBU; (E) 120 minutes after addition of DBU; (F) pure HMPA in CD3CN.
Scheme 1.

Plausible reaction pathway and chemical shifts from 31P{1H} NMR
It is also instructive to compare the reactions of 4 with those of 3′,5′-bis-O-(tert-butyldimethylsilyl)-O6-[(2,4,6-trimethylphenyl)sulfonyl]-2′-deoxyguanosine (yields shown in parentheses in Table 2).24 In this comparison 4 performs just as well as the nucleoside O6-arylsulfonate, although synthesis and utilization of the O6-(benzotriazol-1-yl) guanosine derivatives are generally operationally simpler.
Conclusions
In this paper we have demonstrated that silyl-protected guanosine (1) and 2′-deoxyguanosine (2) can be conveniently converted to the O6-(benzotriazol-1-yl) derivatives 3 and 4. Use of DBU as base in CH3CN as solvent leads to fast conversion in good to high yield. These O6-(benzotriazol-1-yl) derivatives are excellent reagents for SNAr displacement and a variety of C-6 modified 2-amino purine nucleosides can be efficiently synthesized. It appears that amination reactions can be conducted as a 2-step, 1-pot procedure but a similar reaction with an alcohol was not successful. Extensive experimentation has not presently been conducted on a 1-pot synthesis of C-6 ethers via amide group activation by BOP, and work is in progress on this aspect. However, the readily isolated and stable O6-(benzotriazol-1-yl) guanosine derivatives undergo efficient conversion to the wide variety of products using relatively simple methods.
Experimental
General experimental considerations
All reactions were carried out in oven-dried glassware. Reactions were monitored on glass-backed TLC plates coated with silica gel (250 μm), containing a fluorescent indicator. Column chromatographic purifications were performed on 200–300 mesh silica gel. CH3CN and i-Pr2NEt were distilled over CaH2, CH2Cl2 was distilled over CaCl2, THF was distilled over LiAlH4 and then over Na. Commercially available anhydrous DMSO and DME were used without further purification. All other reagents were obtained from commercial sources and used without further purification. 1H NMR spectra were recorded at 500 MHz and are referenced to residual protonated solvent. Chemical shifts are reported in parts per million (δ) and coupling constants (J) are in hertz. 13C NMR spectra were obtained at 125 MHz and are referenced to the solvent resonance. The imidazolyl proton in the nucleoside is labeled H–8 and the sugar protons are numbered 1′-5′ beginning at the anomeric carbon atom and proceeding to the primary carbinol carbon atom.
General procedure for the synthesis of O6-(benzotriazol-1-yl)-2′,3′,5′-tri-O-(tert-butyldimethylsilyl)guanosine (3) and O6-(benzotriazol-1-yl)-3′,5′-di-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (4)
In a clean, dry round-bottomed flask equipped with a stirring bar were placed the silylated nucleoside (1 or 2), BOP (2.0 molar equiv), and DBU (2.0 molar equiv) in dry CH3CN (8.5 mL/mmol). The reaction mixture was flushed with nitrogen gas, stoppered and stirred at room temperature for 1 to 2 h. The reaction progress was monitored by TLC. Upon completion, the mixture was diluted with EtOAc and washed with deionized water containing a small amount of NaCl. The aqueous layer was back extracted (2x) with EtOAc. The combined organic layer was dried over Na2SO4, filtered and evaporated to leave a yellowish oil that was dried under oil pump vacuum. Chromatographic purification then provided products 3 and 4.
O6-(Benzotriazol-1-yl)-2′,3′,5′-tri-O-(tert-butyldimethylsilyl)guanosine (3)
Chromatography on a silica gel column using 20% EtOAc in hexanes gave a white solid (65% yield). Rf (silica/20% EtOAc in hexanes) = 0.46; δH (500 MHz; CDCl3) 8.23 (1H, s, H–8), 8.11 (1H, d, J 8.3, Ar–H), 7.54-7.48 (2H, m, Ar–H), 7.44 (1H, dt, J 1.5, 8.3, Ar–H), 5.95 (1H, d, J 4.4, H–1′), 4.75 (2H, s, NH2), 4.47 (1H, t, J 4.2, H–2′), 4.30 (t, 1H, J 4.2, H–3′), 4.12 (1H, q, J 3.9, H–4′), 4.00 (1H, dd, J 3.4, 11.3, H–5′), 3.80 (1H, dd, J 3.0, 11.3, H–5′), 0.97, 0.93 and 0.84 (27H, 3s, tert-Bu), 0.16, 0.15, 0.11, 0.09, 0.00 and −0.11 (18H, 6s, SiCH3);δC (125 MHz; CDCl3) 159.6, 159.0, 156.2, 143.7, 140.2, 129.1, 128.7, 124.9, 120.6, 113.3, 109.2, 87.8, 85.9, 77.1, 72.3, 62.9, 26.4, 26.0, 25.9, 18.8, 18.3, 18.2, −4.1, −4.6, −4.4, −4.8, −5.1; HRMS (ESI) calcd for C34H59N8O5Si3 [M + H]+: 743.3911, found: 743.3898.
O6-(Benzotriazol-1-yl)-3′,5′-di-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (4)20
Chromatography on a silica gel column using 30% EtOAc in hexanes gave a white solid (85% yield). Rf (silica, 30% EtOAc in hexanes) = 0.24; δH (500 MHz; CDCl3): 8.12 (1H, s, H–8), 8.11 (1H, d overlapping with the singlet at 8.12, J 7.3, Ar–H), 7.54-7.48 (2H, m, Ar–H), 7.44 (1H, t, J 6.8, Ar–H), 6.35 (1H, t, J 6.6, H–1′), 4.76 (2H, s, NH2), 4.60 (1H, m, H–3′), 4.00 (1H, app q, J 3.4, H–4′), 3.84 (1H, dd, J 3.9, 11.2, H–5′), 3.77 (1H, dd, J 2.9, 11.2, H–5′), 2.57 (1H, app quint, J 6.5, H–2′), 2.40 (1H, ddd, J 3.4, 6.3, 13.7, H–2′), 0.924 and 0.916 (18H, 2s, tert-Bu), 0.108, 0.101, 0.098 (12H, 3s, SiCH3); δC (125 MHz; CDCl3) 159.7, 158.8, 156.0, 143.6, 140.4, 129.1, 128.8, 124.9, 120.6, 113.7, 109.1, 88.1, 84.3, 72.0, 63.0, 41.4, 26.2, 26.0, 18.7, 18.3, −4.4, −4.5, −5.1, −5.2.
General procedure for the reaction of 3 with amines
In a clean, dry reaction vial equipped with a stirring bar were placed 3 (50.0 mg, 67.3 μmol), amine (4.0 molar equiv) and anhydrous DME (0.5 mL). The reaction mixture was flushed with nitrogen gas, sealed with a Teflon-lined cap and stirred either at room temperature or at 55 °C for 2–20.5 h. Progress of the reaction was monitored by TLC. Upon completion, the mixture was diluted with EtOAc and washed with deionized water containing a small amount of NaCl. The aqueous layer was back extracted (2x) with EtOAc. The combined organic layer was dried over Na2SO4, filtered and evaporated. Purification on a silica gel column using 20% EtOAc in hexanes then provided the C-6 amino derivatives 5–8. Additional details and any deviations from this general procedure are noted under the individual compound headings.
2-Amino-9-[2,3,5-tri-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl]-6-(morpholin-4-yl)purine (5)
Light orange solid, prepared in 87% yield using 3 (70.0 mg, 94.2 μmol) and morpholine (33 μL, 0.38 mmol) in DME (0.7 mL), in a reaction time of 2 h at room temperature. Rf (silica, 5% MeOH in CH2Cl2) = 0.46; δH (500 MHz; CDCl3) 7.75 (1H, s, H–8), 5.88 (1H, d, J 5.4, H–1′), 4.61 (1H, t, J 4.7, H–2′), 4.54 (2H, s, NH2), 4.28 (1H, t, J 3.9, H–3′), 4.21 (4H, br s, morpholinyl–CH2), 4.08 (1H, app q, J 3.9, H–4′), 3.97 (1H, dd, J 4.4, 11.2, H–5′), 3.80 (4H, t, J 4.9, morpholinyl–CH2), 3.76 (1H, dd, J 3.0, 11.2, H–5′), 0.935, 0.925 and 0.830 (27H, 3s, tert-Bu), 0.11, 0.106, 0.10, 0.09, −0.03 and −0.13 (18H, 6s, SiCH3); δC (125 MHz; CDCl3) 159.3, 154.5, 153.3, 135.3, 115.6, 88.0, 85.1, 75.6, 72.2, 67.3, 62.9, 45.7 (br), 26.4, 26.1, 26.0, 18.8, 18.4, 18.2, −4.1, −4.4, −4.5, −4.6, −5.0, −5.1; HRMS (ESI) calcd for C32H63N6O5Si3 [M + H]+: 695.4163, found: 695.4173.
9-[2,3,5-Tri-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl]-N6,N6-diethyl-2,6-diaminopurine (6)
Yellow solid, prepared in 69% yield using Et2NH (28 μL, 0.27 mmol) in a reaction time of 19 h at room temperature. Rf (silica, 20% EtOAc in hexanes) = 0.47; δH (500 MHz; CDCl3) 7.68 (1H, s, H–8), 5.85 (1H, d, J 5.4, H–1′), 4.68 (1H, t, J 4.7, H–2′), 4.48 (2H, br s, NH2), 4.29 (1H, t, J 4.2, H–3′), 4.07 (1H, app q, J 3.9, H–4′), 3.96 (1H, dd, J 4.4, 11.0, H–5′), 3.90 (4H, br, NCH2), 3.75 (1H, dd, J 3.4, 11.0, H–5′), 1.23 (6H, t, J 7.0, CH3) 0.93, 0.92 and 0.82 (27H, 3s, tert-Bu), 0.103, 0.097, 0.090, −0.03 and −0.15 (18H, 5s, SiCH3); δC (125 MHz; CDCl3) 159.4, 154.4, 152.8, 135.2, 115.5, 88.1, 85.0, 75.0, 72.2, 62.9, 42.8 (br), 26.3, 26.1, 26.0, 18.8, 18.4, 18.2, 13.8, −4.1, −4.4, −4.5, −4.8, −5.0, −5.1; HRMS (ESI) calcd for C32H65N6O4Si3 [M + H]+: 681.4370, found: 681.4380.
2-Amino-9-[2,3,5-tri-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl]-6-(4-methylpiperazin-1-yl)purine (7)
White solid, prepared in 82% yield using 1-methylpiperazine (30 μL, 0.27 mmol) in a reaction time of 3.5 h at 55 °C. Rf (silica, 20% EtOAc in hexanes) = 0.05; δH (500 MHz; CDCl3) 7.74 (1H, s, H–8), 5.86 (1H, d, J 5.4, H–1′), 4.61 (1H, t, J 4.7, H–2′), 4.51 (2H, s, NH2), 4.29 (1H, t, J 4.2, H–3′), 4.24 (4H, br s, piperazinyl–CH2), 4.08 (1H, app q, J 3.9, H–4′), 3.99 (1H, dd, J 4.4, 11.2, H–5′), 3.78 (1H, dd, J 3.0, 11.2, H–5′), 2.51 (4H, br s, piperazinyl–CH2), 2.34 (3H, s, CH3), 0.93, 0.925 and 0.830 (27H, 3s, tert-Bu), 0.103, 0.096, 0.087, −0.03 and −0.13 (18H, 5s, SiCH3); δC (125 MHz; CDCl3) 159.3, 154.5, 153.3, 135.2, 115.7, 88.1, 85.1, 75.4, 72.2, 62.9, 55.4, 46.5, 45.0 (br), 26.4, 26.1, 26.0, 18.8, 18.4, 18.2, −4.1, −4.4, −4.5, −4.6, −5.0, −5.1; HRMS (ESI) calcd for C33H66N7O4Si3 [M + H]+: 708.4479, found: 708.4486.
9-[2,3,5-tri-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl]-N6-[(1S)-phenylethyl]-2,6-diaminopurine (8)
Light yellow solid, prepared in 92% yield using (S)-(−)-α-methylbenzylamine (35 μL, 0.27 mmol) in a reaction time of 20.5 h at room temperature. Rf (silica, 20% EtOAc in hexanes) = 0.13; δH (500 MHz; CDCl3) 7.85 (1H, s, H–8), 7.42 (2H, d, J 7.3, Ar–H), 7.31 (2H, t, J 7.3, Ar–H), 7.22 (1H, t, J 7.3, Ar–H), 6.53 (1H, br s, NH), 5.84 (1H, d, J 4.4, H–1′), 5.50 (1H, br s, NCH), 4.71 (2H, br s, NH2), 4.53 (1H, t, J 4.4, H–2′), 4.28 (1H, t, J 3.9, H–3′), 4.07 (1H, app q, J 3.9, H–4′), 3.99 (1H, dd, J 4.4, 11.5, H–5′), 3.78 (1H, dd, J 3.0, 11.5, H–5′), 1.60 (3H, d, J 6.8, CH3), 0.94, 0.92 and 0.83 (27H, 3s, tert-Bu), 0.119, 0.114, 0.090, 0.00 and −0.11 (18H, 5s, SiCH3); proton assignments were made by 1H-1H COSY of this compound at 60 °C, where the NH sharpened to a br d (5.97 ppm, J 7.8), the NCH became a sharper br m (5.56 ppm) and the CH3 d sharpened (1.61 ppm, J 7.3); δC (125 MHz; CDCl3) 160.0, 154.5,144.5, 136.4, 128.7, 127.2, 126.5, 115.0, 88.4, 84.9, 75.7, 71.8, 62.6, 26.4, 26.1, 26.0, 22.9, 18.8, 18.4, 18.2, −4.1, −4.4, −4.5, −4.6, −5.1; HRMS (ESI) calcd for C36H65N6O4Si3 [M + H]+: 729.4370, found: 729.4374.
General procedure for the reaction of 3 with alcohols
In a clean, dry reaction vial equipped with a stirring bar were placed 3 and Cs2CO3 (2.0 molar equiv) in the appropriate alcohol as solvent. The reaction mixture was flushed with nitrogen gas, sealed with a Teflon-lined cap and stirred at 55 °C for 2–20 h. Progress of the reaction was monitored by TLC. Upon completion, the mixture was diluted with EtOAc and washed with deionized water containing a small amount of NaCl. The aqueous layer was back extracted (2x) with EtOAc. The combined organic layer was dried over Na2SO4, filtered and evaporated. Purification on a silica gel column using 20% EtOAc in hexanes then provided the guanosine O6-alkyl ethers 9–11. Additional details are listed under the specific compound headings.
2′,3′,5′-Tri-O-(tert-butyldimethylsilyl)-O6-methylguanosine (9)
Light orange solid, prepared in 95% yield using 3 (45.3 mg, 61.0 μmol) and Cs2CO3 (42.9 mg, 0.131 mmol) in MeOH (0.45 mL), in a reaction time of 2 h at 55 °C. Rf (silica, 20% EtOAc in hexanes) = 0.11; δH (500 MHz; CDCl3) 7.98 (1H, s, H–8), 5.90 (1H, d, J 4.9, H–1′), 4.79 (2H, s, NH2), 4.50 (1H, t, J 4.4, H–2′), 4.28 (1H, t, J 4.2, H–3′), 4.08 (1H, app q, J 3.0, H–4′), 4.05 (3H, s, OCH3), 3.97 (1H, dd, J 3.9, 11.2, H–5′), 3.77 (1H, dd, J 2.4, 11.2, H–5′), 0.94, 0.92 and 0.82 (27H, 3s, tert-Bu), 0.12, 0.11, 0.09, 0.08, −0.04 and −0.16 (18H, 6s, SiCH3); δC (125 MHz; CDCl3) 161.7, 159.5, 153.8, 138.2, 116.2, 88.0, 85.4, 76.4, 72.1, 62.8, 54.0, 26.4, 26.1, 26.0, 18.8, 18.4, 18.2, 14.8, −4.1, −4.4, −4.5, −4.7, −5.1; HRMS (ESI) calcd for C29H58N5O5Si3 [M + H]+: 640.3741, found: 640.3745.
2′,3′,5′-Tri-O-(tert-butyldimethylsilyl)-O6-ethylguanosine (10)
White solid, prepared in 71% yield using 3 (50.0 mg, 67.3 μmol) and Cs2CO3 (43.8 mg, 0.134 mmol) in EtOH (0.45 mL), in a reaction time of 2.5 h at 55 °C. Rf (silica, 20% EtOAc in hexanes) = 0.20; δH (500 MHz; CDCl3) 7.98 (1H, s, H–8), 5.91 (1H, d, J 4.9, H–1′), 4.76 (2H, br s, NH2), 4.55 (2H, q, J 7.0, OCH2), 4.52 (1H, t, J 4.4, H–2′) 4.29 (1H, t, J 4.3, H–3′), 4.09 (1H, app q, J 2.9, H–4′), 3.99 (1H, dd, J 3.4, 11.5, H–5′), 3.76 (1H, dd, J 3.0, 11.5, H–5′), 1.45 (3H, t, J 7.0, CH3), 0.95, 0.93 and 0.83 (27H, 3s, tert-Bu), 0.13, 0.12, 0.095, 0.09, −0.03 and −0.15 (18H, 6s, SiCH3); δC (125 MHz; CDCl3) 161.5, 159.5, 153.9, 138.0, 116.1, 88.0, 85.4, 76.3, 72.2, 62.8, 26.4, 26.1, 26.0, 18.8, 18.4, 18.2, 14.8, −4.1, −4.4, −4.5, −4.7, −5.1; HRMS (ESI) calcd for C30H60N5O5Si3 [M + H]+: 654.3897, found: 654.3890.
O6-Allyl-2′,3′,5′-tri-O-(tert-butyldimethylsilyl)guanosine (11)
Orange solid, prepared in 88% yield using 3 (70.0 mg, 94.2 μmol) and Cs2CO3 (61.4 mg, 0.188 mmol) in allyl alcohol (0.7 mL), in a reaction time of 20 h at room temperature. Rf (silica, 20% EtOAc in hexanes) = 0.31; δH (500 MHz; CDCl3) 8.00 (1H, s, H–8), 6.15 (1H, m, =CH), 5.93 (1H, d, J 4.6, H–1′), 5.45 (1H, dd, J 1.5, 18.6, =CHtrans), 5.29 (1H, dd, J 1.5, 11.7, =CHcis), 5.02 (2H, d, J 5.9, OCH2), 4.79 (2H, s, NH2), 4.54 (1H, t, J 4.7, H–2′), 4.31 (1H, t, J 3.4, H–3′), 4.11 (1H, app q, J 3.9, H–4′), 4.01 (1H, dd, J 3.9, 11.2, H–5′), 3.80 (1H, dd, J 2.4, 11.2, H–5′), 0.97, 0.95 and 0.85 (27H, 3s, tert-Bu), 0.15, 0.14, 0.12, 0.11, 0.00 and −0.13 (18H, 6s, SiCH3); δC (125 MHz; CDCl3) 161.0, 159.4, 154.1, 138.1, 133.1, 118.4, 116.0, 87.9, 85.4, 76.4, 72.2, 67.4, 62.8, 26.4, 26.1, 26.0, 18.8, 18.4, 18.2, −4.1, −4.4, −4.5, −4.7, −5.11, −5.14; HRMS (ESI) calcd for C31H60N5O5Si3 [M + H]+: 666.3897, found: 666.3912.
S6-Benzyl-2′,3′,5′-tri-O-(tert-butyldimethylsilyl)thioguanosine (12)
In a clean, dry reaction vial equipped with a stirring bar were placed 3 (50.0 mg, 67.0 μmol), Cs2CO3 (43.8 mg, 0.134 mmol) and dry DME (0.5 mL). Benzyl mercaptan (16.0 μL, 0.134 mmol) was added, the reaction mixture was flushed with nitrogen gas, sealed with a Teflon-lined cap and allowed to stir at room temperature for 24 hrs. The mixture was diluted with EtOAc and washed with deionized water containing a small amount of NaCl. The aqueous layer was back extracted (2x) with EtOAc. The combined organic layer was dried over Na2SO4, filtered and evaporated. Purification on a silica gel column using 20% EtOAc in hexanes then provided 12 as a light yellow solid (89% yield). Rf (silica, 10% EtOAc in hexanes) = 0.47; δH (500 MHz; CDCl3) 7.98 (1H, s, H–8), 7.43 (2H, d, J 7.3, Ar–H), 7.31-7.21 (3H, m, Ar–H), 5.90 (1H d, J 4.9, H–1′), 4.81 (s, 2H, NH2), 4.57 (2H, ABquart, J 13.7, SCH2), 4.54 (1H, t, J 4.4, H–2′), 4.29 (t, 1H, J 4.2, H–3′), 4.10 (1H, app q, J 2.9, H–4′), 3.97 (1H, dd, J 3.9, 11.2, H–5′), 3.77 (1H, dd, J 2.9, 11.2, H–5′), 0.93, 0.927 and 0.83 (27H, 3s, tert-Bu), 0.12, 0.11, 0.010, 0.09, −0.02, −0.16 (18H, 5s, SiCH3); δC (125 MHz; CDCl3) 161.1, 159.0, 150.8, 139.0, 138.1, 129.3, 128.7, 127.4, 126.2, 88.1, 85.3, 76.1, 72.1, 62.7, 32.9, 26.4, 26.1, 26.0, 18.8, 18.4, 18.2, −4.1, −4.4, −4.5, −4.7, −5.0, −5.1; HRMS (ESI) calcd for C35H62N5O4SSi3 [M + H]+: 732.3825, found: 732.3831.
9-[3,5-Di-O-(tert-butyldimethylsilyl)-2-deoxy-β-D-ribofuranosyl]-6-(morpholin-4-yl)purine (13)24
One-pot procedure
In a clean, dry, round-bottomed flask equipped with a stirring bar were placed 2 (70.0 mg, 0.141 mmol) and BOP (0.125 g, 0.283 mmol) in anhydrous CH3CN (1.20 mL). DBU (47.3 μL, 0.316 mmol) was added to the stirring mixture, the flask was flushed with nitrogen gas and stoppered. After being stirred at room temperature for 1 h TLC showed consumption of 2. At this time morpholine (49.7μl, 0.568 mmol) was added. The flask was again flushed with nitrogen gas, stoppered and the mixture was allowed to stir at room temperature for an additional 2 hours at which time the reaction was complete as assessed by TLC. The mixture was diluted with EtOAc and washed with deionized water containing a small amount of NaCl. The aqueous layer was back extracted (2x) with EtOAc. The combined organic layer was dried over Na2SO4, filtered and evaporated to leave a yellowish oil. Purification on a silica gel column using 30% EtOAc in hexanes then provided 13 as a yellow solid (91% yield). Rf (silica, 30% EtOAc in hexanes) = 0.11; δH (500 MHz; CDCl3) 7.72 (1H, s, H–8), 6.31 (1H, t, J 6.8, H–1′), 4.60 (2H, s, NH2), 4.57 (1H, m, H–3′), 4.21 (4H, br s, morpholinyl CH2), 3.96 (1H, app q, J 4.1, H–4′), 3.79 (4H, t, J 4.4, morpholinyl–CH2), 3.76 (1H, dd, J 4.6, 11.1, H–5′), 3.75 (1H, dd, J 3.6, 11.1, H–5′), 2.55 (1H, app quint, J 6.8, H–2′), 2.33 (1H, ddd, J 3.4, 6.0, 13.0, H–2′), 0.91 and 0.90 (18H, 2s, tert-Bu), 0.10, 0.07 and 0.06 (12H, 3s, SiCH3); δC (125 MHz; CDCl3) 159.4, 154.4, 153.1, 134.6, 115.4, 87.8, 83.6, 72.4, 67.3, 63.2, 45.7 (br), 40.9, 26.2, 26.0, 18.7, 18.3, −4.4, −4.5, −5.1, −5.2.
General procedure for the reaction of 4 with amines
In a clean, dry reaction vial equipped with a stirring bar were placed 4 (50.0 mg, 81.6 μmol), amine (4.0 molar equiv) and anhydrous DME (0.5 mL). The reaction mixture was flushed with nitrogen gas, sealed with a Teflon-lined cap and stirred at room temperature for 4–22 h (see details under specific compound headings). Progress of the reaction was monitored by TLC. Upon completion, the mixture was diluted with EtOAc and washed with deionized water containing a small amount of NaCl. The aqueous layer was back extracted (2x) with EtOAc. The combined organic layer was dried over Na2SO4, filtered and evaporated. Purification on a silica gel column using 30% EtOAc in hexanes then provided the C-6 amino derivatives 14 and 15. Additional details and any deviations from this general procedure are noted under the individual compound headings.
9-[3,5-Di-O-(tert-butyldimethylsilyl)-2-deoxy-β-D-ribofuranosyl]-N6,N6-dimethyl-2,6-diaminopurine (14)24
Light yellow solid, prepared in 83% yield using a 40 wt% solution of Me2NH in water (37.3 μL, 0.331 mmol) in a reaction time of 2 h. Rf (silica, 5% MeOH in CH2Cl2) = 0.43;δH (500 MHz; CDCl3) 7.69 (1H, s, H–8), 6.30 (1H, t, J 6.1, H–1′), 4.67 (2H, s, NH2), 4.56 (1H, m, H–3′), 3.95 (1H, app q, J 4.0, H–4′), 3.78-3.71 (2H, m, H–5′), 3.43 (6H, br s, NCH3), 2.54 (1H, app quint, J 6.6, H–2′), 2.32 (1H, ddd, J 3.4, 6.0, 13.1, H–2′), 0.90 and 0.896 (18H, 2s, tert-Bu), 0.09, 0.06 and 0.05 (12H, 3s, SiCH3); δC (125 MHz; CDCl3) 159.4, 155.5, 152.7, 134.2, 115.7, 87.8, 83.5, 72.4, 63.2, 40.8, 38.5 (br), 26.2, 26.0, 18.7, 18.3, −4.4, −4.5, −5.1, −5.2.
N6-Benzyl-9-[3,5-di-O-(tert-butyldimethylsilyl)-2-deoxy-β-D-ribofuranosyl]purine (15)24
Light yellow solid, prepared in 81% yield using benzylamine (35.6 μL, 0.33 mmol) in a reaction time of 4 h. Rf (silica, 30% EtOAc in hexanes) = 0.08; δH (500 MHz; CDCl3) 7.76 (1H, s, H–8), 7.37-7.27 (5H, m, Ar–H), 6.30 (1H, t, J 6.8, H–1′), 5.89 (1H, br s, NH), 4.79 (2H, s, NH2), 4.71 (2H, br s, NCH2), 4.58 (1H, m, H–3′), 3.97 (1H, app q, J 3.4, H–4′), 3.81 (1H, dd, J 4.4, 11.0, H–5′), 3.75 (1H, dd, J 2.9, 11.0, H–5′), 2.59 (1H, app quint, J 6.8, H–2′), 2.34 (1H, ddd, J 3.7, 6.0, 13.0, H–2′), 0.90 and 0.896 (18H, 2s, tert-Bu), 0.10, 0.08 and 0.07 (12H, 3s, SiCH3); δC (125 MHz; CDCl3) 160.0, 155.18, 139.0, 136.0, 128.8, 128.0, 127.6, 115.1, 87.9, 83.8, 72.3, 63.1, 44.7 (br), 41.0, 26.2, 26.0, 18.7, 18.3, −4.4, −4.5, −5.10, −5.2.
General procedure for the reaction of 4 with alcohols
In a clean, dry reaction vial equipped with a stirring bar were placed 4 (50.0 mg, 81.6 μmol) and Cs2CO3 (53.1 mg, 0.163 mmol) in the appropriate alcohol as solvent (0.5 mL). The reaction mixture was flushed with nitrogen gas, sealed with a Teflon-lined cap and stirred for 4–7 hours at room temperature. Progress of the reaction was monitored by TLC. Upon completion, the mixture was diluted with EtOAc and washed with deionized water containing a small amount of NaCl. The aqueous layer was back extracted (2x) with EtOAc. The combined organic layer was dried over Na2SO4, filtered and evaporated. Purification on a silica gel column using 30% EtOAc in hexanes then provided the 2′-deoxyguanosine O6-alkyl ethers 16 and 17. Additional details are listed under the specific compound headings.
3′,5′-Di-O-(tert-butyldimethylsilyl)-O6-methyl-2′-deoxyguanosine (16)24
Light yellow solid, prepared in 79% yield using MeOH, in a reaction time of 4 h. Rf (silica, 5% MeOH in CH2Cl2) = 0.41; δH (500 MHz; CDCl3) 7.91 (1H, s, H–8), 6.32 (1H, t, J 6.4, H–1′), 4.83 (2H, s, NH2), 4.59 (1H, m, H–3′), 4.08 (3H, s, OCH3), 3.97 (1H, app q, J 3.9, H–4′), 3.81 (1H, dd, J 4.4, 11.2, H–5′), 3.75 (1H, dd, J 3.4, 11.2, H–5′), 2.57 (1H, app quint, J 6.3, H–2′), 2.34 (1H, ddd, J 3.8, 6.1, 13.0, H–2′), 0.912 and 0.910 (18H, 2s, tert-Bu), 0.10, 0.08 and 0.07 (12H, 3s, SiCH3); δC (125 MHz; CDCl3) 161.8, 159.5, 153.7, 137.8, 116.2, 87.9, 83.9, 72.1, 63.1, 54.1, 41.2, 26.2, 26.0, 18.7, 18.3, −4.4, −4.5, −5.1, −5.2.
O6-Allyl-3′,5′-di-O-(tert-butyldimethylsilyl)-2′-deoxyguanosine (17)24
Light yellow solid, prepared in 82% yield using allyl alcohol, in a reaction time of 7 h. Rf (silica, 5% MeOH in CH2Cl2) = 0.59; δH (500 MHz; CDCl3) 7.90 (1H, s, H–8), 6.32 (1H, t, J 6.8, H–1′), 6.12 (1H, m, =CH), 5.42 (1H, dd, J 1.5, 18.5, =CHtrans), 5.26 (1H, dd, J 1.5, 11.7, =CHcis), 5.01 (2H, d, J 5.4, OCH2), 4.81 (2H, s, NH2), 4.59 (1H, m, H–3′), 3.97 (1H, app q, J 3.3, H–4′), 3.81 (1H, dd, J 4.4, 11.1, H–5′), 3.75 (1H, dd, J 2.9, 11.1, H–5′), 2.57 (1H, app quint, J 6.0, H–2′), 2.35 (1H, ddd, J 3.7, 6.1, 13.1, H–2′), 0.913 and 0.910 (18H, 2s, tert-Bu), 0.10, 0.08 and 0.07 (12H, 3s, SiCH3); δC (125 MHz; CDCl3) 161.1, 159.4, 154.0, 137.8, 133.0, 118.4, 116.2, 87.9, 83.9, 72.2, 67.5, 63.1, 41.2, 26.2, 26.0, 18.7, 18.3, −4.4, −4.5, −5.1, −5.2.
General procedure for the reaction of 4 with phenols
In a clean, dry reaction vial equipped with a stirring bar were placed 4 (50.0 mg, 81.6 μmol), the appropriate phenol (2 molar equiv) and Cs2CO3 (53.1 mg, 0.163 mmol) in anhydrous DME (0.5 mL). The reaction mixture was flushed with nitrogen gas, sealed with a Teflon-lined cap and stirred at 60 °C for 2–2.5 hours. Progress of the reaction was monitored by TLC. Upon completion, the mixture was diluted with EtOAc and washed with deionized water containing a small amount of NaCl. The aqueous layer was back extracted (2x) with EtOAc. The combined organic layer was dried over Na2SO4, filtered and evaporated. Purification on a silica gel column using 30% EtOAc in hexanes then provided the 2′-deoxyguanosine O6-aryl ethers 18 and 19.
3′,5′-Di-O-(tert-butyldimethylsilyl)-O6-phenyl-2′-deoxyguanosine (18)24
Yellowish solid, prepared in 89% yield using phenol (15.3 mg, 0.163 mmol), in a reaction time of 2.5 h. Rf (silica, 5% MeOH in CH2Cl2) = 0.26; δH (500 MHz; CDCl3) 8.01 (1H, s, H–8), 7.40 (2H, t, J 7.8, Ar–H), 7.25-7.23 (3H, m, Ar–H), 6.34 (1H, t, J 6.4, H–1′), 4.82 (2H, s, NH2), 4.60 (1H, m, H–3′), 3.99 (1H, app q, J 3.4, H–4′), 3.83 (1H, dd, J 3.9, 11.2, H–5′), 3.77 (1H, dd, J 2.9, 11.2, H–5′), 2.58 (1H, app quint, J 6.8, H–2′), 2.37 (1H, ddd, J 3.9, 6.4, 13.1, H–2′), 0.92 and 0.91 (18H, 2s, tert-Bu), 0.10 and 0.09 (12H, 2s, SiCH3); δC (125 MHz; CDCl3) 160.7, 159.4, 154.7, 152.8, 138.7, 129.5, 125.5, 122.2, 116.2, 88.0, 84.0, 72.1, 63.1, 41.3, 26.3, 26.0, 18.7, 18.3, −4.4, −4.5, −5.1, −5.2.
3′,5′-Di-O-(tert-butyldimethylsilyl)-O6-(4-chlorophenyl)-2′-deoxyguanosine (19)24
Yellow solid, prepared in 88% yield using 4-chlorophenol (21.0 mg, 0.163 mmol), in a reaction time of 2 h. Rf (silica, 5% MeOH in CH2Cl2) = 0.28; δH (500 MHz; CDCl3) 8.02 (1H, s, H–8), 7.36 (2H, J 8.8, Ar–H), 7.19 (2H, d, J = 8.8, Ar H), 6.34 (1H, t, J 6.4, H–1′), 4.79 (2H, s, NH2), 4.61 (1H, m, H–3′), 3.99 (1H, app q, J 3.9, H–4′), 3.84 (1H, dd, J 4.4, 11.2, H–5′), 3.77 (1H, dd, J 2.9, 11.2, H–5′), 2.58 (1H, app quint, J 6.8, H–2′), 2.38 (1H, ddd, J 3.9, 5.9, 13.2, H–2′), 0.923 and 0.920 (18H, 2s, tert-Bu), 0.11 and 0.10 (12H, 2s, SiCH3); δC (125 MHz; CDCl3) 160.3, 159.3, 154.9, 151.3, 138.9, 130.8, 129.6, 123.6, 116.1, 88.0, 84.0, 72.1, 63.0, 41.3, 26.2, 26.0, 18.7, 18.3, −4.4, −4.5, −5.1, −5.2.
Supplementary Material
Acknowledgments
This work was partially supported by NSF grant CHE-0640417, NIGMS SCORE grant S06 GM008168-29 and a PSC CUNY-39 award. Infrastructural support at CCNY was provided by NIH/NCRR/RCMI grant G12 RR03060. We thank Dr. Cliff Soll (Hunter College) for high-resolution mass spectral analysis.
Footnotes
Electronic Supplementary Information (ESI) available: [copies of proton NMR spectra of compounds 3–19 and 1H-1H COSY spectrum of 3]. See DOI: 10.1039/b000000x/
Notes and references
- 1.Herdewijn P, editor. Modified Nucleosides in Biochemistry, Biotechnology and Medicine. Wiley-VCH; Weinheim: 2008. [Google Scholar]; Blackburn GM, Gait MJ, Loakes D, Williams DM, editors. Nucleic Acids in Chemistry and Biology. 3. RSC Publishing; Cambridge, UK: 2006. [Google Scholar]; Simons C. Nucleoside Mimetics: Their Chemistry and Biological Properties. Gordon and Breach; Amsterdam: 2001. [Google Scholar]; Kisakürek MV, Rosemeyer H, editors. Perspectives in Nucleoside and Nucleic Acid Chemistry. Verlag Helvetica Chimica Acta; Zurich: Wiley-VCH; Weinheim: 2000. [Google Scholar]; Suhadolnik RJ. Nucleosides and Nucleic Acids as Biological Probes. Wiley; New York: 1979. [Google Scholar]
- 2.Robins MJ, Uznanski B. Can J Chem. 1981;59:2601–2607. [Google Scholar]; Gerster JF, Jones JW, Robins RK. J Org Chem. 1963;28:945–948. [Google Scholar]
- 3.Nair V, Young DA. J Org Chem. 1984;49:4340–4344. [Google Scholar]
- 4.Mehta JR, Ludlum DB. Biochim Biophys Acta. 1978;521:770–778. doi: 10.1016/0005-2787(78)90316-7. [DOI] [PubMed] [Google Scholar]
- 5.Kamike K, Kinoshita K, Niwa K, Hirose K, Suzuki K, Ishido Y. Nucleosides Nucleotides Nucleic Acids. 2001;20:59–75. doi: 10.1081/NCN-100001437. [DOI] [PubMed] [Google Scholar]
- 6.Janeba Z, Francom P, Robins MJ. J Org Chem. 2003;68:989–992. doi: 10.1021/jo020644k. [DOI] [PubMed] [Google Scholar]
- 7.Bridson PK, Markiewicz WT, Reese CB. J Chem Soc, Chem Commun. 1977:791–792. [Google Scholar]
- 8.Nagatsugi F, Uemura K, Nakashima S, Maeda M, Sasaki S. Tetrahedron. 1997;53:3035–3044. [Google Scholar]
- 9.Daskalov HP, Sekine M, Hata T. Tetrahedron Lett. 1980;21:3899–3902. [Google Scholar]
- 10.Gaffney BL, Jones RA. Tetrahedron Lett. 1982;23:2257–2260. [Google Scholar]
- 11.Tanimura H, Sekine M, Hata T. Tetrahedron Lett. 1986;27:4047–4050. [Google Scholar]
- 12.Lakshman MK, Gunda P, Pradhan P. J Org Chem. 2005;70:10329–10335. doi: 10.1021/jo0513764. [DOI] [PubMed] [Google Scholar]
- 13.Bae S, Lakshman MK. J Am Chem Soc. 2007;129:782–789. doi: 10.1021/ja064682n. [DOI] [PubMed] [Google Scholar]
- 14.Zlatko J, Lin X, Robins MJ. Nucleosides, Nucleotides Nucleic Acids. 2004;23:137–147. doi: 10.1081/ncn-120027823. [DOI] [PubMed] [Google Scholar]; Lin X, Robins MJ. Org Lett. 2000;2:3497–3499. doi: 10.1021/ol000255h. [DOI] [PubMed] [Google Scholar]
- 15.Wan Z-K, Binnun E, Wilson DP, Lee J. Org Lett. 2005;7:5877–5880. doi: 10.1021/ol052424+. [DOI] [PubMed] [Google Scholar]
- 16.Bae S, Lakshman MK. J Org Chem. 2008;73:1311–1319. doi: 10.1021/jo7021795. [DOI] [PubMed] [Google Scholar]
- 17.Bae S, Lakshman MK. J Org Chem. 2008;73:3707–3713. doi: 10.1021/jo702558n. [DOI] [PubMed] [Google Scholar]
- 18.Bae S, Lakshman MK. Org Lett. 2008;10:2203–2206. doi: 10.1021/ol8006106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lakshman MK, Choudhury A, Bae S, Rochttis E, Pradhan P, Kumar A. Eur J Org Chem. 2009:152–159. doi: 10.1002/ejoc.200800752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Böge N, Krüger S, Schröder M, Meier C. Synthesis. 2007:3907–3914. [Google Scholar]
- 21.Wan Z-K, Wacharasindhu S, Levins CG, Lin M, Tabei K, Mansour TS. J Org Chem. 2007;72:10194–10210. doi: 10.1021/jo7020373. [DOI] [PubMed] [Google Scholar]
- 22.Saurina J, Hernández-Cassou S, Tauler R, Izquierdo-Ridorsa A. Anal Chim Acta. 2000;408:135–143. [Google Scholar]
- 23.Saenger W. Principles of Nucleic Acid Structure. Springer-Verlag; New York: 1984. [Google Scholar]
- 24.Lakshman MK, Ngassa FN, Keeler JC, Dinh YQV, Hilmer JH, Russon LM. Org Lett. 2000;2:927–930. doi: 10.1021/ol005564m. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.