

Abstract
Kinetic and structure analysis of inducible nitric oxide synthase (iNOS) revealed that, in addition to the increase of iNOS expression in inflamed areas, the major pathway causing overproduction of NO is destabilization of the iNOS–nitrosyl complex(es) that form during steady-state catalysis. Formation of such a complex allows iNOS to operate at only a fraction (20–30%) of its maximum activity. Thus, bioavailability of NO scavengers at sites of inflammation may play an essential role in up-regulation of the catalytic activity of iNOS, by preventing the catalytic activity inhibition that is attributed to nitrosyl complex formation. Myeloperoxidase (MPO), a major NO scavenger, is a pivotal enzyme involved in leukocyte-mediated host defenses. It is thought to play a pathogenic role under circumstances such as acute inflammatory tissue injury and chronic inflammatory conditions. However, a detailed understanding of the interrelationship between iNOS and MPO at sites of inflammation is lacking. We used direct spectroscopic, HPLC, and selective NO-electrode measurements to determine the interdependent relationship that exists between iNOS and MPO and the role of the MPO/H2O2 system in up-regulating the catalytic activity of iNOS that occurs at sites of inflammation. Scavenging free NO from the iNOS milieu by the MPO/H2O2 system subsequently restores the full capacity of iNOS to convert l-aginine to product (NO), as judged by the increase in the rates of citrulline and nitrite/nitrate production. Studies of iNOS catalytic mechanisms and function are essential to a more fundamental understanding of these factors, which govern iNOS-dependent processes in human health and disease.
Keywords: inflammation, peroxidase
Nitric oxide (NO) is a ubiquitous signaling molecule that plays essential bioregulatory roles in a wide range of processes, including vasodilation, cell proliferation, nerve transmission, tumor surveillance, antimicrobial defense, and regulation of inflammatory responses (1–4). NO is generated enzymatically by three distinct isoforms of NO synthase (NOS): neuronal, inducible (iNOS), and endothelial. All of these isoforms typically use l-arginine, O2, and NADPH to generate NO and citrulline (5). The biological effects of NO are governed in part, by its intrinsic instability, reactivity, lipophilicity, and affinity toward iron; these characteristics make it ideal for both signal transduction and defense (6). NO is freely diffusible; its effect in a given circumstance depends on its diffusion to reach the target cell, and the bioavailability of NO scavengers such as superoxide (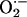 ) and oxyhemoglobin binding, which limit its ability to exert biologic effects (7). Although NO generated under normal conditions appear to serve a signaling function, under pathological conditions, such as during atherosclerosis, asthma, and other inflammatory processes, rates of NO production become excessive (8–15). Importantly, the cytokine-iNOS isoform that is present in many tissues, including lung, liver, kidney, heart, and smooth muscle, is implicated in most diseases involving overproduction of NO (14–16).
) and oxyhemoglobin binding, which limit its ability to exert biologic effects (7). Although NO generated under normal conditions appear to serve a signaling function, under pathological conditions, such as during atherosclerosis, asthma, and other inflammatory processes, rates of NO production become excessive (8–15). Importantly, the cytokine-iNOS isoform that is present in many tissues, including lung, liver, kidney, heart, and smooth muscle, is implicated in most diseases involving overproduction of NO (14–16).
The ability of NO to react with the NOS heme iron at nearly diffusion-controlled rates suggests that the enzyme is a potential target for NO feedback inhibition during steady-state catalysis (17–23). Indeed, our laboratory and others have demonstrated that NO down-regulates the catalytic activity of iNOS by competing with O2 at the catalytic site of the enzyme during catalysis, generating inactive NOS–nitrosyl complexes (18–22, 24). Formation of such a complex is associated with a significant instantaneous decrease in both the rate of citrulline accumulation and the rate of NO production. The inhibitory impact of the formation of the iNOS–nitrosyl complex on the enzyme catalytic activity can be partially or completely prevented depending on the bioavailability of NO scavengers, such as the superoxide-generating system or oxyhemoglobin (19, 20, 22).
Sites of chronic inflammation are also associated with significant elevation of expression of myeloperoxidase (MPO) and other members of the mammalian peroxidase superfamily [lactoperoxidase and eosinophil peroxidase (EPO)], as well as elevation of the level of their natural substrate, H2O2. The mammalian peroxidase superfamily typically uses H2O2, in combination with halides or pseudo halides, to generate the corresponding hypohalous acid (25–28). Recently, we have demonstrated that NO modulates the catalytic activity of mammalian peroxidases in two distinct ways depending on the experimental conditions, either by serving as a substrate or as a ligand (29–32). Detailed kinetic analysis of mammalian peroxidases has revealed that low NO concentration enhanced the catalytic activity of MPO by increasing the formation and decay of MPO, lactoperoxidase, and EPO compound II, the rate-limiting step in the peroxidase catalytic cycle (29–31). These reactions presumably generate NO cation (NO+), a labile nitrosating species that is rapidly hydrolyzed in aqueous solutions, forming nitrite (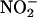 ) as an end product (33). NO+ is characterized by its capacity to undergo addition and substitution reactions with nucleophiles, such as electron-rich bases and aromatic compounds (33, 34). The propensity for thiol nitrosation under physiological conditions and in biological systems has been recognized (33–38). It has been suggested that NO is stored and carried as RS–NO adducts, and that S-nitrosoglutathione (GS-NO), among other RS-NO adducts, might serve as a reservoir for NO in cells and tissues (39, 40). In contrast, at higher levels NO reacts with the ferrous and ferric forms of MPO, lactoperoxidase, and EPO, thereby generating the corresponding low-spin six-coordinated complexes and limiting the catalytic activity of these enzymes by the release of NO from the complex (29–32). Similarly, the mammalian peroxidase superfamily can use H2O2 and nitrite (
) as an end product (33). NO+ is characterized by its capacity to undergo addition and substitution reactions with nucleophiles, such as electron-rich bases and aromatic compounds (33, 34). The propensity for thiol nitrosation under physiological conditions and in biological systems has been recognized (33–38). It has been suggested that NO is stored and carried as RS–NO adducts, and that S-nitrosoglutathione (GS-NO), among other RS-NO adducts, might serve as a reservoir for NO in cells and tissues (39, 40). In contrast, at higher levels NO reacts with the ferrous and ferric forms of MPO, lactoperoxidase, and EPO, thereby generating the corresponding low-spin six-coordinated complexes and limiting the catalytic activity of these enzymes by the release of NO from the complex (29–32). Similarly, the mammalian peroxidase superfamily can use H2O2 and nitrite (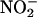 ), a major end product of NO metabolism, to promote the formation of a microbicidal oxidant (NO2) capable of nitrating phenolic compounds and proteins (41–45). For example, recent studies with EPO-deficient mice have demonstrated the crucial role of EPO in the formation of nitrotyrosine in a mouse model of asthma.§ MPO may also indirectly contribute to formation of NO-derived oxidants through secondary oxidation of
), a major end product of NO metabolism, to promote the formation of a microbicidal oxidant (NO2) capable of nitrating phenolic compounds and proteins (41–45). For example, recent studies with EPO-deficient mice have demonstrated the crucial role of EPO in the formation of nitrotyrosine in a mouse model of asthma.§ MPO may also indirectly contribute to formation of NO-derived oxidants through secondary oxidation of 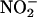 by HOCl (42, 46). Collectively, mammalian peroxidases can catalyze the oxidation of NO and/or its metabolite
by HOCl (42, 46). Collectively, mammalian peroxidases can catalyze the oxidation of NO and/or its metabolite 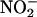 to more reactive-nitrogen species, and thereby contribute to thiol nitrosation and protein tyrosine nitration.
to more reactive-nitrogen species, and thereby contribute to thiol nitrosation and protein tyrosine nitration.
Although evidence supporting increased NO, thiol nitrosation, and protein tyrosine nitration at sites of inflammation is substantial, the factors regulating NO and its role in inflammation are still unclear. Our current results suggest that the MPO/H2O2 system, like other NO scavengers, consumed NO released by iNOS during steady-state catalysis, thereby preventing the NO-induced inhibition attributed to the formation of the iNOS–nitrosyl complex. Thus, removal of NO from the iNOS milieu by the MPO/H2O2 system during steady-state catalysis causes a significant increase in iNOS catalytic activity, production of citrulline, and presumably production of NO.
Materials and Methods
Materials. NO gas was purchased from Matheson Gas Products (Montgomeryville, PA) and used without further purification. Hypoxanthine manganese superoxide dismutase and hemoglobin were obtained from Sigma. Xanthine oxide from cow milk was purchased from Roche Molecular Biochemicals. Other chemicals and reagents were of the highest purity grades available and obtained from either Sigma or Aldrich. Full-length mouse iNOS was expressed in Escherichia coli and purified to homogeneity in the presence of tetrahydrobiopterin (H4B) as described (17–21). MPO was initially purified from detergent extracts of human leukocytes by sequential lectin affinity and gel filtration chromatography as described (47). Trace levels of contaminating EPO were then removed by passing over a sulfopropyl Sephadex column (48). Purity of the isolated MPO was estimated by demonstrating a purity number of 0.9 (A430/A280). Absence of EPO activity contamination was confirmed by SDS/PAGE analysis with Coomassie blue staining and gel tetramethylbenzidine peroxidase staining. Enzyme concentration was determined spectrophotometrically by using an extinction coefficient of 112,000 M–1·cm–1 per heme of MPO (49).
Optical Spectroscopy. Optical spectra of iNOS during catalysis were recorded in a final volume of 1-ml cuvettes on a Cary 100 Bio UV-visible spectrophotometer at 15°C, by monitoring the change in absorbance at 440 over time. For a typical reaction, iNOS was diluted to 1 μM in air-saturated 40 mM Hepes buffer, pH 7.4, containing 4 μM H4B, 0.1 mM DTT, and 0.2 mM l-arginine; 20 μM NADPH was added later to initiate NO synthesis. iNOS concentration was determined spectrophotometrically based on an estimated extinction coefficient of 76 mM–1·cm–1 per heme.
Electrochemical Detection of NO. NO was measured by using an NO meter (ISO-NO Mark II 4000) connected with an ISO-NOP 200 or Apollo 4000 (World Precision Instruments, Sarasota, FL) sensor, as reported (18, 19, 30, 31). NO measurements were performed in a 5-ml cell at 15°C. iNOS was incubated for 10 min in 40 mM Hepes buffer, pH 7.4 containing 0.2 mM arginine, 0.1 mM DTT, and 4 μM H4B, and the total reaction volume was 2 ml. The electrode current was monitored on a chart recorder with continuous stirring of the reaction solution. Calibration of the electrode was performed by the stepwise addition of a NO solution (1 μM each) to 3 ml of argon-deoxygenated buffer (40 mM Hepes, pH 7.4, at 15°C) as reported. After initiating NO synthesis, 10-μl aliquots were removed from the reactions at timed intervals, and iNOS activity was immediately quenched by mixing with 1 μl of 5 M HCl. Citrulline in the samples was derivatized with o-phthalaldehyde and quantitated by using fluorometric HPLC methods with standard citrulline samples as a reference (18–20). Nitrite (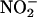 )/nitrate (
)/nitrate (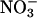 ) accumulation in the presence and absence of an MPO/H2O2 system was performed by using anion exchange HPLC methods (30).
) accumulation in the presence and absence of an MPO/H2O2 system was performed by using anion exchange HPLC methods (30).
Results
Absorbance spectroscopy was used to confirm the formation of an iNOS–nitrosyl complex during steady-state catalysis (19, 20, 22) and determine whether MPO and H2O2 could affect its formation and decay. Fig. 1 Left depicts an experiment in which a cycle of complex formation and decay was promoted by adding a limiting amount of NADPH (20 μM) to trigger NO synthesis in a sample of iNOS (1 μM). The percentage of complex formation (amplitude of the signal) and the time of the reaction (duration of the signal) were monitored at 440 nm. Fig. 1 Center and Right shows the effect of H2O2 and MPO on iNOS–ferric–nitrosyl complex formation during NO synthesis. In Fig. 1 Center, both H2O2 (20 μM) and NADPH (20 μM) were added to the reaction at time 0. In Fig. 1 Right, both MPO (40 nM) and NADPH (20 μM) were added to the reaction at time 0. The results show that the presence of H2O2 did not affect the amount of ferric–nitrosyl complex formed or the time required to complete the reaction. However, the presence of MPO did partially lower the amount of ferric–nitrosyl complex formation, as judged by the decrease in amplitude of iNOS–ferric–nitrosyl complex monitored at 440 nm.
Fig. 1.

Effect of catalytic amount of H2O2 and MPO on iNOS–ferric–nitrosyl complex formation and decay during steady-state catalysis. Generation of ferric–nitrosyl iNOS was monitored at 440 nm in the reaction run at 15°Cinthe absence of MPO and H2O2 (Left) or in the presence of either 40 nM MPO (Center)or20 μMH2O2 (Right). Both MPO and H2O2 were added to the iNOS solution mixture and to the blank at time 0. The experiment shown is representative of three.
We next used an NO-selective electrode to determine whether MPO/H2O2 system can use NO, generated by iNOS, as a general substrate and prevent iNOS feedback inhibition attributed to the formation of the iNOS–Fe(III)–NO complex during steady-state catalysis. After addition of NADPH (40 μM) to a continuously stirred reaction mixture of iNOS (0.5 μM) under air, the NO signal rose rapidly, achieved its maximum level after 30 s, and then gradually returned to baseline after all of the NADPH was consumed by the enzyme (Fig. 2). Addition of catalytic levels of H2O2 (20 μM) to the solution mixture had no significant effect on NO disappearance. Remarkably, subsequent addition of catalytic levels of MPO (40 nM) to the iNOS solution caused a rapid disappearance of free NO, indicating that NO is consumed as a substrate by MPO during steady-state catalysis (Fig. 2 Left). Reversal of the order of peroxidase and H2O2 addition demonstrated a modest brief decrease in NO concentration after addition of only MPO, and then a similar significant acceleration in NO consumption upon subsequent addition of H2O2 (Fig. 2 Right). Thus, removal of free NO from the iNOS milieu requires the presence of both H2O2 and MPO.
Fig. 2.

Consumption of NO generating by iNOS during steady-state catalysis by the MPO/H2O2 system. Solid lines, a typical recording by a NO-selective electrode, demonstrating the release of NO and its autoxidation from iNOS after addition of NADPH (40 μM) to stirred iNOS solution under air, at 15°C. (Left) Dashed line indicates addition of MPO followed by H2O2 results in a dramatic acceleration in the rate of NO consumption. (Right) Dashed line indicates addition of MPO results in slight acceleration in rate of NO consumption. Catalytic consumption of NO by MPO in the presence of H2O2 results in additive rate of NO removal.
To examine whether the removal of NO from the iNOS milieu is associated with an increase in the catalytic activity of iNOS, citrulline production was followed in the presence and absence of the MPO/H2O2 system by removing 10-μl aliquots from the reaction at identified time points and quenching with diluted HCl. Identical experiments were repeated in the presence of a superoxide-generating system. This system is known to scavenge NO and prevent feedback inhibition attributable to the formation of the iNOS–Fe(III)–NO complex. Fig. 3 shows the effect of both the MPO/H2O2 system and the xanthine oxide/hypoxanthine superoxide-generating system on citrulline accumulation during iNOS NO synthesis. Xanthine oxide was added to the reaction at time 0 to initiate concurrent NO synthesis by iNOS and 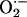 production by xanthine oxidase. The condition was such that the measured rate of
production by xanthine oxidase. The condition was such that the measured rate of 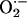 production by xanthine oxide in the reaction (25 μM/min) was approximately four times the rate of NO synthesis (8 μM/min) by iNOS.
production by xanthine oxide in the reaction (25 μM/min) was approximately four times the rate of NO synthesis (8 μM/min) by iNOS.
Fig. 3.

Effect of NO scavenger on the catalytic activity of iNOS. The amounts of citrulline formation in the absence (□) and the presence of the MPO/H2O2 system (*) or in the presence of superoxide-generating system (•) as determined by HPLC analysis on 10-μl aliquots removed and quenched with acid at the indicated times. The data shown are representative of three similar experiments.
We also measured the extent of 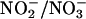 build-up in the reaction mixture in the presence and absence of the MPO/H2O2 system on the samples quenched at 1.6 min after initiating iNOS NO synthesis by using anion exchange HPLC. At this time point, we observed that the concentration of
build-up in the reaction mixture in the presence and absence of the MPO/H2O2 system on the samples quenched at 1.6 min after initiating iNOS NO synthesis by using anion exchange HPLC. At this time point, we observed that the concentration of 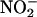 plus
plus 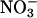 accumulated in the presence of the MPO/H2O2 system is ≈4-fold higher compared with its absence over the time course of use (data not shown).
accumulated in the presence of the MPO/H2O2 system is ≈4-fold higher compared with its absence over the time course of use (data not shown).
In control experiments, we used exogenous NO and a NO-selective electrode to determine whether the MPO/NADPH system is capable of removing NO from buffer solution and contributing partially or completely to the up-regulation of iNOS catalytic activity. After addition of an aliquot of NO-saturated buffer to the continuously stirred reaction, the NO signal rose rapidly, achieved a maximum after ≈30 s, before gradually returning to the origin as NO was depleted by autoxidation. Addition of NADPH to the reaction mixture had little or no effect on the rate of NO removal from the solution. Subsequent addition of MPO caused rapid removal of free NO from the solution mixture, indicating that NO is consumed nonenzymatically by MPO. NO removal by MPO/NADPH occurs in a concentration-dependent fashion. For example, when 40 μM NADPH was added to a continuously stirred 1 μM NO solution, only 10–15% of the amount of free NO was removed as occurs in the presence of a catalytic amount of MPO. In contrast, all of the added NO was instantly consumed when 150–200 μM NADPH was added to a buffer solution containing a catalytic amount of MPO (data not shown).
Discussion
Previously, we had shown that iNOS catalytic activity is down-regulated by NO feedback inhibition by a mechanism that constrains the enzyme to operate at only a fraction of its maximum activity (18–20). iNOS and MPO are both colocalized and secreted from the primary granules of activated leukocytes (50), and their overexpression is commonly associated with inflammatory diseases and disorders (51–53). However, specific interactions between iNOS and MPO at sites of inflammation have been infrequently explored. Understanding the mechanism of the mutual influences of individual enzyme activity should be useful for elucidation of disease mechanisms and development of clinically useful therapy. Our main aim in these studies is to understand the interplay of iNOS, NO, and mammalian peroxidases in the process of inflammation. In this article, the cross talks between iNOS and MPO during iNOS NO synthesis has been examined. Our central finding is that the MPO/H2O2 system scavenges iNOS-generated NO and prevents the feedback inhibition process attributed to the formation of the iNOS–Fe–NO complex, thereby allowing the enzyme to operate at its maximum catalytic activity. In this study, this hypothesis was directly demonstrated by monitoring citrulline accumulation as a function of time and by the buildup of free NO and its oxidative end products (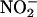 and
and 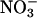 ) generated by iNOS during steady-state catalysis in the absence and presence of the MPO/H2O2 system.
) generated by iNOS during steady-state catalysis in the absence and presence of the MPO/H2O2 system.
Our laboratory and others have carried out comprehensive studies to investigate the susceptibility of each NOS isoform to NO autoinhibition, the mechanism by which this inhibition occurs, and the implications of this inhibition on the biological functions of each isoform (18–22, 24). Further work in our laboratory indicates that neuronal NOS generates a Fe(II)–NO complex, iNOS generates both Fe(II)- and Fe(III)–NO complexes during catalysis, and endothelial NOS may not generate any detectable Fe–NO complex from l-arginine during catalysis (18–21). However, endothelial NOS generated the Fe–NO complex by replacement of l-arginine with the intermediate Nω-hydroxy-l-arginine (18). For each of the three NOS isoforms, the build-up of the Fe–nitrosyl complexes is associated with a significant decrease in the rate of citrulline production and NADPH consumption (18–22). Stopped-flow, resonance Raman spectroscopy, and UV-visible studies showed significant differences in the nature and properties of the iNOS NO complex, as compared with the neuronal NOS isoform (18–22, 24). For example, formation of the iNOS NO complex is inhibited by added NO scavengers, indicating its formation depends on external NO (Fig. 3) (19, 22). Accordingly, the degree to which NO complex formation affected the O2 response curve of iNOS also depends on the level of external NO, creating a more complicated biological setting (19). Studies with neuronal NOS indicated that neither enzyme dilution nor NO scavengers diminished formation of the Fe(II)–NO complex or prevented the catalytic inhibition attributed to its formation (20, 21). Thus, NO binding to the NOS heme is a fundamental feature of catalysis and functions to down-regulate NO synthesis by the enzyme (20, 21).
Understanding how iNOS functions at sites of inflammation and the factors that modulate its productivity will help develop plans for pharmacological modulation. Based on our prior extensive studies characterizing NOS, the current results, and previous published studies, we have generated a comprehensive kinetic model describing unique features of the iNOS reactions that accommodate the NO feedback inhibition process (5, 17–24) (Fig. 4). In this model, iNOS catalyzes a NADPH- and O2-dependent oxidation of l-arginine to generate NO and l-citrulline. NOS flavins accept electrons from NADPH and transfer them immediately to the heme iron. Heme iron reduction is thought to enable the iron to bind and activate molecular oxygen and catalyze stepwise NO synthesis from l-arginine. Importantly, self-produced NO first equilibrates in the solution and then binds to the heme iron, which acts as a negative feedback modulator by: (i) competing with O2 for binding to the heme catalytic site, and (ii) forming an inactive ferric and/or ferrous–nitrosyl complex that can only rejoin the productive cycle through a breakdown reaction that is itself O2 dependent. Thus, the catalytic activity of the enzyme will be limited by the relative proportions of iNOS–Fe(II) intermediate available during steady-state catalysis. In the absence of substrate, reduction of the iNOS heme iron simply leads to 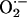 generation (Fig. 4).
generation (Fig. 4).
Fig. 4.

Simplified kinetic model of iNOS that describes the formation of the iNOS–nitrosyl complex during steady-state catalysis.
Subsequently, circumstances that influence the rate of NO elimination after its synthesis by iNOS and other NOS isoforms are of marked significant interest. One common and biologically significant pathway for NO removal in vivo is through interaction with oxyhemoglobin, yielding methemoglobin and 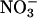 (54). In addition, NO reacts with
(54). In addition, NO reacts with 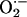 or molecular oxygen to form oxidation products, such as peroxynitrite (ONOO–),
or molecular oxygen to form oxidation products, such as peroxynitrite (ONOO–), 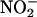 ,
, 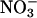 , and other reactive nitrogen species as primary end products, which leads to nitration of tyrosine residues in proteins (55–60). NO may also be consumed through oxidative pathways in airways during asthmatic episodes or by radical-coupling reactions with lipid alkoxyl and peroxyl radicals, species formed during lipid peroxidation (61–63). In addition, O'Donnell and Freeman (64) have also shown that NO can be consumed by lipoxygenases through interaction of NO with an intermediate radical species generated during lipoxygenase catalysis.
, and other reactive nitrogen species as primary end products, which leads to nitration of tyrosine residues in proteins (55–60). NO may also be consumed through oxidative pathways in airways during asthmatic episodes or by radical-coupling reactions with lipid alkoxyl and peroxyl radicals, species formed during lipid peroxidation (61–63). In addition, O'Donnell and Freeman (64) have also shown that NO can be consumed by lipoxygenases through interaction of NO with an intermediate radical species generated during lipoxygenase catalysis.
Further studies in our laboratory have indicated that mammalian peroxidases (MPO, EPO, and lactoperoxidase) may operate as alternative pathways for catalytic removal of NO at sites of inflammation and cardiovascular diseases (29–31). Mammalian peroxidases catalyze the oxidation of H2O2, generating a ferryl π cation radical [Fe(IV) = O•+π] intermediate named compound I, which is capable of oxidizing halides and pseudo halides through a 2 e– transition generating ground state and the corresponding hypohalous acid (25–28). Similarly, compound I can oxidize various organic and inorganic compounds by two consecutive 1 e– transitions, yielding radical species and the peroxidase intermediates compound II [Fe(IV) = O] and ground state, respectively (25–28). Enhancement in peroxidase catalysis caused by reduction of compound II has been noted with a series of physiological reductants like 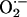 , NO, and ascorbic acid (27–32). Superoxide and NO also can serve as a ligand for Fe(III) to generate an inactive ferrous–dioxy complex and ferric–nitrosyl complex, respectively (27–32).
, NO, and ascorbic acid (27–32). Superoxide and NO also can serve as a ligand for Fe(III) to generate an inactive ferrous–dioxy complex and ferric–nitrosyl complex, respectively (27–32).
Although NO biosynthesis is regulated at multiple levels in cells including NOS gene transcription, mRNA processing, protein expression and dimerization, enzyme modification, and enzyme reaction kinetics, the concentration of NO in any biologic system at specific time point is a sum of its rate of enzymatic formation and exhaustion by other biological molecules (65). In the context of increased scavenging of NO by the MPO/H2O2 system, increased enzymatic synthesis is a likely mechanism that governed the increased NO levels at sites of inflammation. Fig. 5 summarizes the tridirectional relationship that exists between MPO, iNOS, and NO. Using a combination of biochemical, physiological, and kinetic approaches, we have shown that local accumulation of NO and its overproduction can alter the bioavailability of the reduced form of iNOS by forming the inhibitory complex iNOS–Fe–nitrosyl, leaving the enzyme able to operate at only fraction of its maximum activity. If a sink for NO were nearby (e.g., MPO/H2O2 system), especially in situations of inflammation where iNOS and MPO may be elevated, this is akin to NO scavenging. NO scavenging could potentially increase the iNOS activity by decreasing the iNOS–nitrosyl complex, thereby demonstrating that NO itself regulates NOS activity. The MPO/H2O2 system, like the superoxide-generating system and oxyhemoglobin (18–22), serves as a major catalytic sink for NO, limiting its bioavailability and function. This process allows iNOS to operate at its maximum catalytic activity, as judged by the significant increase in the rates of citrulline and 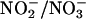 production during steady-state catalysis. Indeed, we observed that the concentration of citrulline and
production during steady-state catalysis. Indeed, we observed that the concentration of citrulline and 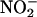 plus
plus 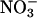 accumulated in the presence of the MPO/H2O2 system is ≈4-fold higher compared with its absence over the time course of use. Our current and recently published results strongly suggest that NO interacts directly with MPO compounds I and II and leads to reduction of the respective heme groups (29–31). In addition, we looked for evidence of Nω-hydroxy-l-arginine accumulation during iNOS steady-state catalysis in the presence and absence of the MPO/H2O2 system. None was seen; rather, only citrulline was formed. Our data may suggest that the Nω-hydroxy-l-arginine decomposition to NO and citrulline may be much faster than its apparent rate of formation in our system. This observation is also consistent with NADPH and O2 being provided in excess relative to l-arginine and with iNOS undergoing multiple rounds of heme reduction and catalysis. To determine whether NO removal by the MPO/H2O2 system alters the Km O2 of iNOS, we measured NADPH oxidation versus O2 concentration in the presence of the MPO/H2O2 system to scavenge NO (H.M.A.-S., unpublished results). This process gave an apparent Km O2 value of 50 μM, which is ≈3-fold lower than the value observed in the absence of NO scavenger, but is similar to that obtained in the presence of oxyhemoglobin (19). This finding is consistent with our previous work with iNOS and other NOS isoforms that suggests NO complex formation can have a major impact on NOS O2 tension (18–21). Removal of NO by the MPO/H2O2 system is mediated through direct interaction of NO as a peroxidase substrate. NO could serve to modulate peroxidase catalytic activity by acting as both a ligand and a substrate (29–32). As a substrate, NO accelerates the rate-limiting step in the peroxidase cycle and the reduction of compound II and enhances the overall rates of catalysis (29–31). Finally, NO can serve as a ligand for both MPO and iNOS, rendering them catalytically inactive (17–24, 29–32).
accumulated in the presence of the MPO/H2O2 system is ≈4-fold higher compared with its absence over the time course of use. Our current and recently published results strongly suggest that NO interacts directly with MPO compounds I and II and leads to reduction of the respective heme groups (29–31). In addition, we looked for evidence of Nω-hydroxy-l-arginine accumulation during iNOS steady-state catalysis in the presence and absence of the MPO/H2O2 system. None was seen; rather, only citrulline was formed. Our data may suggest that the Nω-hydroxy-l-arginine decomposition to NO and citrulline may be much faster than its apparent rate of formation in our system. This observation is also consistent with NADPH and O2 being provided in excess relative to l-arginine and with iNOS undergoing multiple rounds of heme reduction and catalysis. To determine whether NO removal by the MPO/H2O2 system alters the Km O2 of iNOS, we measured NADPH oxidation versus O2 concentration in the presence of the MPO/H2O2 system to scavenge NO (H.M.A.-S., unpublished results). This process gave an apparent Km O2 value of 50 μM, which is ≈3-fold lower than the value observed in the absence of NO scavenger, but is similar to that obtained in the presence of oxyhemoglobin (19). This finding is consistent with our previous work with iNOS and other NOS isoforms that suggests NO complex formation can have a major impact on NOS O2 tension (18–21). Removal of NO by the MPO/H2O2 system is mediated through direct interaction of NO as a peroxidase substrate. NO could serve to modulate peroxidase catalytic activity by acting as both a ligand and a substrate (29–32). As a substrate, NO accelerates the rate-limiting step in the peroxidase cycle and the reduction of compound II and enhances the overall rates of catalysis (29–31). Finally, NO can serve as a ligand for both MPO and iNOS, rendering them catalytically inactive (17–24, 29–32).
Fig. 5.

Model showing the influence of the MPO/H2O2 system on the formation of the iNOS–nitrosyl complex during steady-state catalysis. Formation of the iNOS–nitrosyl complex is associated with the release of NO from iNOS unless the MPO/H2O2 system is present.
NO synthesis depends on posttranslational modifications to generate active NOS. Specifically, NOSs are synthesized as monomers and must dimerize to generate NO. Scavenging NO has also a remarkable impact on iNOS dimer assembly, the only active form of the enzyme, which enhances the capacity of iNOS to generate NO (66). Previous studies by Albakri and Stuehr (66) have demonstrated that scavenging NO or inhibition of NO synthesis increases the dimer assembly rate. Other possible pathways, which allow iNOS to overproduce NO, are iNOS mutations that may cause destabilization of the iNOS–nitrosyl complex formation during steady-state catalysis, enhancement in the rate of heme reduction, flavin reduction, and/or buildup of iNOS–dioxy complex (67). However, implication of NOS's mutation in inflammatory or other diseases has not yet been reported.
Another fascinating pathway that emerges for catalytic consumption of NO by MPO is through a nonenzymatic pathway that required the combined presence of MPO and NADPH (H.M.A.-S., unpublished work). In this pathway, NADPH may interact with MPO, facilitate NO consumption through a mechanism similar to that previously reported for cytochrome P450NOR and fungal nitric oxide reductase (NOR), and has been extensively studied with regard to its catalytic and structural properties (68, 69). Cytochrome P450NOR and fungal NOR have been known to participate in fungal denitrification by converting NO to N2O in a multiple-step sequential mechanism (68, 69). Under our current experimental condition, this nonenzymatic pathway has little or no impact on iNOS catalytic activity and function because NO consumption by this system requires the presence of higher concentrations of NADPH.
In biological systems, enhancement of NO production by iNOS may display beneficiary or harmful roles, depending on the timing, rate of NO diffusion, circumstances that stimulate its overproduction, and local chemistry of NO degradation. Therefore, understanding the circumstances that lead to iNOS up-regulation is an approach that provides important information and a general framework for establishing the effect of related diseases. One of the most common and biologically significant functions of NO is its ability to bind reversibly to many hemoproteins and nonhemoproteins and to act as an activator or inhibitor (70). For example, the activation of soluble guanylate cyclase takes place through the interaction with NO in vascular smooth muscle, initiating a series of actions that lead to vasodilation (71, 72). Inactivation of hemoproteins and nonhemoproteins can occur through the ligation of heme iron or through the direct reaction with thiol- and iron-sulfer-containing enzymes, respectively. NO and its oxidative end products, such as 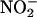 and
and 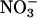 , may lead to protein inactivation through nitration of tyrosine residues in proteins or nitrosation of biologic constituents to form S-nitrosothiols. An increase in iNOS and peroxidase mRNA expressions and NO level is associated with many inflammatory diseases, and the creation of increased metabolic end products has been used as a marker for chronic diseases such as astherosclerosis and asthma (8–16, 51–53). The present studies confirm another pathway for enhancement of both iNOS and MPO catalytic activity that involves a cross linkage between the two enzymes. Further evidence for the involvement of catalytic levels of the peroxidases–H2O2 system in scavenging NO comes from our previous findings, which demonstrated multiple NO–peroxidase interactions (29–31). In these studies, we have shown that catalytic amounts of peroxidase were capable of scavenging NO and subsequently preventing NO-mediated smooth muscle relaxation of the preconstricted organ chamber of vascular and tracheal rings dilation (31).¶ These earlier findings were recently confirmed by Eiserich et al. (73).
, may lead to protein inactivation through nitration of tyrosine residues in proteins or nitrosation of biologic constituents to form S-nitrosothiols. An increase in iNOS and peroxidase mRNA expressions and NO level is associated with many inflammatory diseases, and the creation of increased metabolic end products has been used as a marker for chronic diseases such as astherosclerosis and asthma (8–16, 51–53). The present studies confirm another pathway for enhancement of both iNOS and MPO catalytic activity that involves a cross linkage between the two enzymes. Further evidence for the involvement of catalytic levels of the peroxidases–H2O2 system in scavenging NO comes from our previous findings, which demonstrated multiple NO–peroxidase interactions (29–31). In these studies, we have shown that catalytic amounts of peroxidase were capable of scavenging NO and subsequently preventing NO-mediated smooth muscle relaxation of the preconstricted organ chamber of vascular and tracheal rings dilation (31).¶ These earlier findings were recently confirmed by Eiserich et al. (73).
In summary, iNOS binding self-generated NO significantly constrains its rate and increases its apparent Km O2 value (19, 22). Thus, disturbance in cellular levels of NO caused by exposure to NO scavengers such as peroxidases, 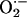 , and oxyhemoglobin, locally where iNOS is expressed or by the neighboring cells, would subsequently promote potential change in iNOS activity and O2 response via an effect on heme–NO complex formation. This is an important task in biologic setting, because the unbalanced ratio between NO and NO scavengers might modulate iNOS catalytic activity and function. Thus, understanding how iNOS–NO binding regulates the catalytic activity of the enzyme may provide potential new strategies for the pharmacological modulation of NO synthesis.
, and oxyhemoglobin, locally where iNOS is expressed or by the neighboring cells, would subsequently promote potential change in iNOS activity and O2 response via an effect on heme–NO complex formation. This is an important task in biologic setting, because the unbalanced ratio between NO and NO scavengers might modulate iNOS catalytic activity and function. Thus, understanding how iNOS–NO binding regulates the catalytic activity of the enzyme may provide potential new strategies for the pharmacological modulation of NO synthesis.
Acknowledgments
This work was supported by National Institutes of Health Grant 5 RO1 HL066367-04 (to H.M.A.-S.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: NOS, NO synthase; iNOS, inducible NOS; MPO, myeloperoxidase; EPO, eosinophil peroxidase.
Footnotes
Duguet, A., Iijima, H. & Eidelman, D. H. (2000) Am. J. Respir. Crit. Care Med. 161, A920 (abstr).
Abu-Soud, H. M. & Hazen, S. L. (2000) Nitric Oxide 4, 179 (abstr.).
References
- 1.Ignarro, L. J. (1990) Annu. Rev. Pharmacol. Toxicol. 30, 535–560. [DOI] [PubMed] [Google Scholar]
- 2.Moncada, S., Palmer, R. M. J. & Higgs, E. A. (1991) Pharmacol. Rev. 43, 109–142. [PubMed] [Google Scholar]
- 3.Schmidt, H. H. H. W. & Walter, U. (1994) Cell 79, 919–925. [DOI] [PubMed] [Google Scholar]
- 4.Vincent, S. R. (1994) Prog. Neurobiol. 42, 129–160. [DOI] [PubMed] [Google Scholar]
- 5.Griffith, O. W. & Stuehr, D. J. (1995) Annu. Rev. Physiol. 57, 707–736. [DOI] [PubMed] [Google Scholar]
- 6.Lancaster, J. R., Jr. (1992) Am. Sci. 80, 248–259. [Google Scholar]
- 7.Lancaster, J. R., Jr. (1997) Nitric Oxide 1, 18–30. [DOI] [PubMed] [Google Scholar]
- 8.Barnes, P. J., Chung, K. F. & Page, C. P. (1998) Pharmacol. Rev. 50, 515–596. [PubMed] [Google Scholar]
- 9.Persson, M. G., Zetterstom, O., Agrenius, V, Ihre, E. & Gustafsson, L. E. (1994) Lancet 343, 146–147. [DOI] [PubMed] [Google Scholar]
- 10.Kharitonov, S. A., Yates, D., Robbins, R. A., Logan-Sinclair, R., Shinebourne, E. A. & Barnes, P. J. (1994) Lancet 343, 133–135. [DOI] [PubMed] [Google Scholar]
- 11.ten Hacken, N. H., van der Vaart, H., van der Mark, T. W., Koeter, G. H. & Postma, D. S. (1998) Am. J. Respir. Crit. Care Med. 158, 902–907. [DOI] [PubMed] [Google Scholar]
- 12.Alving, K., Weitzberg, E. & Lundberg, J. M. (1993) Eur. Respir. J. 6, 1368–1370. [PubMed] [Google Scholar]
- 13.Dweik, R. A., Comhair, S. A. A., Gaston, B., Thunnissen, F. B. J., Farver, C., Thomassen, M. J., Kavuru, M., Hammel, J., Abu-Soud, H. M. & Erzurum, S. C. (2001) Proc. Natl. Acad. Sci. USA 98, 2622–2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kroncke, K. D., Fehsel, K. & Kolb-Bachofen, V. (1995) Biol. Chem. Hoppe-Seyler 376, 327–343. [DOI] [PubMed] [Google Scholar]
- 15.Nathan, C. (1997) J. Clin. Invest. 100, 2417–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.MacMicking, J., Xie, Q. W. & Nathan, C. (1997) Annu. Rev. Immunol. 15, 323–350. [DOI] [PubMed] [Google Scholar]
- 17.Abu-Soud, H. M., Wu, C., Ghosh, D. K. & Stuehr, D. J. (1998) Biochemistry 37, 3777–2786. [DOI] [PubMed] [Google Scholar]
- 18.Abu-Soud, H. M., Ichimori, K., Presta, A. & Stuehr, D. J. (2000) J. Biol. Chem. 275, 17349–17357. [DOI] [PubMed] [Google Scholar]
- 19.Abu-Soud, H. M., Ichimori, K., Nakazawa, H. & Stuehr, D. J. (2001) Biochemistry 40, 6876–6881. [DOI] [PubMed] [Google Scholar]
- 20.Abu-Soud, H. M., Wang, J., Rousseau, D. L., Fukuto, J. M., Ignarro, L. J. & Stuehr, D. J. (1995) J. Biol. Chem. 270, 22997–23006. [DOI] [PubMed] [Google Scholar]
- 21.Abu-Soud, H. M., Rousseau, D. L. & Stuehr, D. J. (1996) J. Biol. Chem. 271, 32515–32518. [DOI] [PubMed] [Google Scholar]
- 22.Hurshman, A. R. & Marletta, M. A. (1995) Biochemistry 34, 5627–5634. [DOI] [PubMed] [Google Scholar]
- 23.Scheele, J. S., Bruner, E., Kharitonov, V. G., Martasek, P., Roman, L. J., Masters, B. S., Sharma, V. S. & Magde, D. (1999) J. Biol. Chem. 274, 13105–13110. [DOI] [PubMed] [Google Scholar]
- 24.Wang, J., Rousseau, D. L., Abu-Soud, H. M. & Stuehr, D. J. (1994) Proc. Natl. Acad. Sci. USA 91, 10512–10516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weiss, S. J., Klein, R., Slivka, A. & Wei, M. (1982) J. Clin. Invest. 70, 598–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Harrison, J. E. & Schultz, J. (1976) J. Biol. Chem. 25, 1371–1374. [PubMed] [Google Scholar]
- 27.Kettle, A. J. & Winterbourn, C. C. (1997) Redox Rep. 3, 3–15. [DOI] [PubMed] [Google Scholar]
- 28.Ortiz de Montellano, P. R. (1992) Annu. Rev. Pharmacol. Toxicol. 32, 89–107. [DOI] [PubMed] [Google Scholar]
- 29.Abu-Soud, H. M. & Hazen, S. L. (2000) J. Biol. Chem. 275, 5425–5430. [DOI] [PubMed] [Google Scholar]
- 30.Abu-Soud, H. M. & Hazen, S. L. (2000) J. Biol. Chem. 275, 37524–37532. [DOI] [PubMed] [Google Scholar]
- 31.Abu-Soud, H. M., Khassawneh, M. Y., Sohn, J. T., Murray, P., Haxhiu, M. A. & Hazen, S. L. (2001) Biochemistry 40, 11866–11875. [DOI] [PubMed] [Google Scholar]
- 32.Abu-Soud, H. M. & Hazen, S. L. (2001) Biochemistry 40, 10747–10755. [DOI] [PubMed] [Google Scholar]
- 33.Stamler, J. S., Singel, D. J. & Loscalzo, J. (1992) Science 258, 1898–1902. [DOI] [PubMed] [Google Scholar]
- 34.Stamler, J. S., Simon, D. I., Jaraki, O., Osborne, J. A., Francis, S., Mullins, M., Singel, D. & Loscalzo, J. (1992) Proc. Natl. Acad. Sci. USA 89, 8087–8091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stamler, J. S. (1994) Cell 78, 931–936. [DOI] [PubMed] [Google Scholar]
- 36.Ridd, J. H. (1978) Adv. Phys. Org. Chem. 16, 1–49. [Google Scholar]
- 37.Williams, D. C., ed. (1988) Nitrosation (Cambridge Univ. Press, New York).
- 38.Wink, D. A., Kasprzak, K. S., Maragos, C. M., Elespuru, R. K., Misra, M., Dunams, T. M., Cebula, T. A., Koch, W. H., Andrews, A. W. & Allen, J. S. (1991) Science 25, 1001–1003. [DOI] [PubMed] [Google Scholar]
- 39.Mayers, P. R., Minor, R. L., Jr., Guerra, R., Jr., Bates, J. N. & Harrison, D. G. (1990) Nature 345, 161–163. [DOI] [PubMed] [Google Scholar]
- 40.Gaston, B., Reilly, J., Drazen, J. M., Fackler, J., Ramdev, P., Arnelle, D., Mullins, M. E., Sugarbaker, D. J., Chee, C., Singel, D. J., et al. (1993) Proc. Natl. Acad. Sci. USA. 90, 10957–10961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van der Vliet, A., Eiserich, J. P., Halliwell, B. & Cross, C. E. (1997) J. Biol. Chem. 272, 7617–7625. [DOI] [PubMed] [Google Scholar]
- 42.Eiserich, J. P., Hristova, M., Cross, C. E., Jones, A. D., Freeman, B. A., Halliwell, B. & van der Vliet, A. (1998) Nature 391, 393–397. [DOI] [PubMed] [Google Scholar]
- 43.Schmitt, D., Shen, Z., Zhang, R., Colles, S. M., Wu, W., Salomon, R. G., Chen, Y., Chisolm, G. M. & Hazen, S. L. (1999) Biochemistry 38, 16904–16915. [DOI] [PubMed] [Google Scholar]
- 44.Podrez, E. A., Schmitt, D., Hoff, H. F. & Hazen, S. L. (1999) J. Clin. Invest. 103, 1547–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sampson, J. B., Ye, Y., Rosen, H. & Beckman, J. S. (1998) Arch. Biochem. Biophys. 356, 207–213. [DOI] [PubMed] [Google Scholar]
- 46.Eiserich, J. P., Cross, C. E., Jones, A. D., Halliwell, B. & van der Vliet, A. (1996) J. Biol. Chem. 271, 19199–19208. [DOI] [PubMed] [Google Scholar]
- 47.Rakita, R. M., Michel, B. R. & Rosen, H. (1990) Biochemistry 29, 1075–1080. [DOI] [PubMed] [Google Scholar]
- 48.van Dalen, C. J., Whitehouse, M. W., Winterbourn, C. C. & Kettle, A. J. (1997) Biochem. J. 327, 487–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Agner, K. (1963) Acta Chem. Scand. 17, S332–S338. [Google Scholar]
- 50.Evans, T. J., Buttery, L. D., Carpenter, A., Springall, D. R., Polak, J. M. & Cohen, J. (1996) Proc. Natl. Acad. Sci. USA 93, 9553–9558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Van der Vliet, A., Eiserich, J. P. & Cross, C. E. (2000) Respir. Res. 1, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu, W., Chen, Y., d'Avignon, A. & Hazen, S. L. (1999) Biochemistry 38, 3538–3548. [DOI] [PubMed] [Google Scholar]
- 53.Ischiropoulos, H. (1988) Arch. Biochem. Biophys. 356, 1–11. [DOI] [PubMed] [Google Scholar]
- 54.Liu, X., Miller, M. J., Joshi, M. S., Sadowska-Krowicka, H., Clark, D. A. & Lancaster, J. R., Jr. (1998) J. Biol. Chem. 273, 18709–18713. [DOI] [PubMed] [Google Scholar]
- 55.Lancaster, J. R., Jr. (1994) Proc. Natl. Acad. Sci. USA 91, 8137–8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gryglewski, R. J., Palmer, R. M. & Moncada, S. (1986) Nature 320, 454–456. [DOI] [PubMed] [Google Scholar]
- 57.Hobbs, A. J. & Ignarro, L. J. (1996) Methods Enzymol. 269, 134–148. [DOI] [PubMed] [Google Scholar]
- 58.Moncada, S. (1997) Funct. Neurol. 12, 134–140. [PubMed] [Google Scholar]
- 59.Beckman, J. S., Chen, J., Ischiropoulos, H. & Crow, J. P. (1994) Methods Enzymol. 233, 229–240. [DOI] [PubMed] [Google Scholar]
- 60.Patel, R. P., McAndrew, J., Sellak, H., White, C. R., Jo, H., Freeman, B. A. & Darley-Usmar, V. M. (1999) Biochim. Biophys. Acta 1411, 385–400. [DOI] [PubMed] [Google Scholar]
- 61.O'Donnell, V. B., Chumley, P. H., Hogg, N., Bloodsworth, A., Darley-Usmar, V. M. & Freeman, B. A. (1997) Biochemistry 36, 15216–15223. [DOI] [PubMed] [Google Scholar]
- 62.Goss, S. P., Hogg, N. & Kalyanaraman, B. (1997) J. Biol. Chem. 272, 21647–21653. [DOI] [PubMed] [Google Scholar]
- 63.Jessup, W., Mohr, D., Gieseg, S. P., Dean, R. T. & Stocker, R. (1992) Biochim. Biophys. Acta 1180, 73–82. [DOI] [PubMed] [Google Scholar]
- 64.O'Donnell, V. B. & Freeman, B. A. (2001) Circ. Res. 88, 12–21. [DOI] [PubMed] [Google Scholar]
- 65.Stuehr, D. J. (1999) Biochim. Biophys. Acta 1411, 217–230. [DOI] [PubMed] [Google Scholar]
- 66.Albakri, Q. A. & Stuehr, D. J. (1996) J. Biol. Chem. 271, 5414–542178. [DOI] [PubMed] [Google Scholar]
- 67.Adak, S., Santolini, J., Tikunova, S., Wang, Q., Johnson, J. D. & Stuehr, D. J. (2001) J. Biol. Chem. 276, 1244–1252. [DOI] [PubMed] [Google Scholar]
- 68.Shimizu, H., Obayashi, E., Gomi, Y., Arakawa, H., Park, S.-Y., Nakamura, H., Adachi, S., Shoun, H. & Shir, Y. (2000) J. Biol. Chem., 275, 4816–4826. [DOI] [PubMed] [Google Scholar]
- 69.Takashi, K., Takaya, N., Park, S.-Y., Shiro, Y. & Shoun, H. (2001) J. Biol. Chem. 276, 5020–5026. [DOI] [PubMed] [Google Scholar]
- 70.Cooper, C. E. (1999) Biochim. Biophys. Acta 1441, 290–309. [DOI] [PubMed] [Google Scholar]
- 71.Waldman, S. A. & Murad, F. (1987) Pharmacol. Rev. 39, 163–196. [PubMed] [Google Scholar]
- 72.Furchgott, R. F., Zawadzki, J. V. & Cherry, P. D. (1981) in Vasodilation, eds. Vanhoutte, P. & Leusen, I. (Raven, New York), pp. 49–66.
- 73.Eiserich, J. P., Baldus, S., Brennan, M. L., Ma, W., Zhang, C., Tousson, A., Castro, L., Lusis, A. J., Nauseef, W. M., White, C. R. & Freeman, B. A. (2002) Science 296, 2391–2394. [DOI] [PubMed] [Google Scholar]