

Abstract
Action potential generation is governed by the opening, inactivation, and recovery of voltage-gated sodium channels. A channel's voltage-sensing and pore-forming α subunit bears an intrinsic fast inactivation particle that mediates both onset of inactivation upon membrane depolarization and rapid recovery upon repolarization. We describe here a novel inactivation particle housed within an accessory channel subunit (A-type FHF protein) that mediates rapid-onset, long-term inactivation of several sodium channels. The channel-intrinsic and tethered FHF-derived particles, both situated at the cytoplasmic face of the plasma membrane, compete for induction of inactivation, causing channels to progressively accumulate into the long-term refractory state during multiple cycles of membrane depolarization. Intracellular injection of a short peptide corresponding to the FHF particle can reproduce channel long-term inactivation in a dose-dependent manner and can inhibit repetitive firing of cerebellar granule neurons. We discuss potential structural mechanisms of long-term inactivation and potential roles of A-type FHFs in the modulation of action potential generation and conduction.
Introduction
Voltage-gated sodium channels carry the inward flow of sodium ions driving generation of action potentials in excitable cells. Rising membrane potential leads to sodium channel opening and also induces fast inactivation, which facilitates membrane repolarization and fast channel recovery. The inactivation–recovery cycle of sodium channels permits repetitive firing in muscle and nerve cells. A membrane-embedded sodium channel α subunit harbours the channel's ion selectivity pore, the voltage-gated activation mechanism, and the fast inactivation gating particle residing in a short cytoplasmic loop (West et al. 1992; Eaholtz et al. 1994; Caterall, 2000). Other mechanisms of channel inactivation have been described. Upon membrane depolarization, a rapid open channel block can be conferred on neuronal sodium channel Nav1.6 by certain associated channel β subunits, and unblocking upon repolarization accounts for the transient resurgent current of this channel in certain cells (Raman & Bean, 1997, 2001; Grieco et al. 2005). A quite distinct mode of inactivation, so-called slow inactivation, requires a far longer period of membrane depolarization to induce, and recovery from slow inactivation may take on the order of seconds or minutes. Slow inactivation is thought to reflect conformational changes to the external portion of the alpha subunit pore (Ulbricht, 2005).
Sodium channel fast inactivation is modulated by alpha subunit interaction with a family of cytoplasmic proteins termed fibroblast growth factor homologous factors (FHFs) (Smallwood et al. 1996; Hartung et al. 1997; Wang et al. 2000; Goldfarb, 2005). Several FHFs have been shown to delay channel inactivation by raising the voltage at which inactivation occurs (Liu et al. 2003; Wittmack et al. 2004; Lou et al. 2005; Rush et al. 2006; Goldfarb et al. 2007). FHF modulation of sodium channel fast inactivation enhances the excitability of cerebellar granule and Purkinje neurons, lowering the voltage threshold for action potential onset and allowing for repetitive firing upon depolarizing current injection (Goldfarb et al. 2007; Shakkottai et al. 2009).
Two FHFs, FHF2A and FHF4A, have also been shown to induce a distinct mode of long-term inactivation of Nav1.6 (Rush et al. 2006; Laezza et al. 2009). In this paper, we report that all A-type FHFs exert rapid onset long-term inactivation on Nav1.6 and other sodium channels. A-type FHFs accomplish long-term inactivation by providing an independent, N-terminally situated cytoplasmic gating particle that competes with the channel's intrinsic inactivation particle for blockade of the channel upon membrane depolarization. We further show that injection of a synthetic peptide corresponding to the A-type FHF particle reproduces long-term sodium channel inactivation and concomitantly acts to oppose sustained firing of excitable cells.
Methods
Plasmids
Murine Nav1.6 cDNA was amplified in segments by reverse transcription-PCR from mouse brain RNA and cloned into bicistronic vector pIRESneo3 (Clontech). The cDNA insert was sequenced in its entirety, and the plasmid-bearing E. coli was cultured at 25°C with 50 μg ml−1 ampicillin to avoid selection for deletions in the plasmid. Point mutations were introduced using complementary mutagenic primers and PfuTurbo DNA polymerase (Stratagene). TTX resistance was introduced by Y371S substitution, as shown previously (Rush et al. 2006). Further substitutions into Nav1.6TTXr included F1478Q in the DIII/DIV inactivation loop (West et al. 1992; Eaholtz et al. 1994; Caterall, 2000) or A1317Q in the loop's docking site (Smith & Goldin, 1997). All FHF cDNAs were cloned into bicistronic vector pIRES2-ZsGreen1 (Clontech) to enable expression of untagged FHFs along with fluorescent protein. FHF2A mutations I5A, LL/IA (L9A/I10A), 5Q (K13Q/R14Q/R17Q/R19Q/K21Q), and 7Q (K55Q/K56Q/R57Q/R58Q/R59Q/R60Q/R61Q) were generated using complementary mutagenic primers. The 8xMT octamutant version of FHF2A cDNA (Goetz et al. 2009) was shuttled into pIRES2-ZsGreen1. Human Nav1.5 cDNA in the pCDNA3.1 expression vector was the generous gift of R. Kass.
Cells
Neuro2A (N2A) cells and the sodium channels they express were described elsewhere (Lou et al. 2005). N2A cells were transfected with pNav1.6TTXr-IRESneo3 by the calcium phosphate method, selected with 1.4 mg ml−1 G418, and colonies maintained in 1 mg ml−1 G418 and screened electrophysiologically in the presence of TTX (see below) to obtain cells stably expressing Nav1.6TTXr. Cerebellar granule neurons were prepared from P9 mouse pups and cultured for 16 days, as previously described (Goldfarb et al. 2007), with the procedure modified to include 5 ng ml−1 brain-derived neurotrophic factor (BDNF) in the culture medium. As predicted from earlier studies (Courtney et al. 1997; Tucker & Fadool, 2002), BDNF enhanced viability and expression of potassium channels, allowing the neurons to generate action potentials with strong afterhyperpolarizations necessary for repetitive firing. Mice used for preparation of neuronal cultures were housed and treated in compliance with Hunter College's Institutional Animal Care and Use Committee (IACUC).
Peptides
Peptides were custom synthesized and purified commercially (Invitrogen Corp.). All peptides were N-terminally acetylated and corresponded to FHF2A residues 2–21, 2–12, and 11–21, purified by HPLC, and identity confirmed by mass spectroscopy.
Antibodies
An N-terminally acetylated synthetic peptide corresponding to FHF2A residues 2–21 with additional GlyGlyCys at C-terminus was directionally coupled via the sulfhydryl group to the carrier and used as immunogen in rabbits. Antisera were precleared of antibodies against the coupling moiety by taking flow-through from affinity column bearing an unrelated peptide, and specific antibodies were then purified on agarose bearing the coupled immunizing peptide. Rabbit antibodies against the C-termini of either FHF2 or FHF1 were described previously (Schoorlemmer & Goldfarb, 2001; Wittmack et al. 2004).
Transient DNA transfections
FHF bicistronic vectors were transiently expressed in N2A or N2A-Nav1.6TTXr cells. Semiconfluent cultures on 12-multiwell dishes were treated with a mixture of 2 μl Lipofectamine 2000 (Invitrogen) and 3 μg plasmid in 0.8 ml medium for 4 h, then refed with fresh medium. For immunoblotting experiments, cells were maintained another two days before lysate preparation. For electrophysiology, cells were trypsinized, plated at lower density onto polylysine-coated borosilicate coverslips, and maintained for 2–3 days, and green fluorescent cells were selected for recordings. For electrophysiological analyses of Nav1.5 and mutant forms of Nav1.6TTXr, N2A cells were cotransfected with 2.5 μg channel-expressing plasmid and 0.5 μg pIRES-ZsGreen plasmid with or without FHF cDNA inserts.
Immunoblotting
Immunoblotting was used to compare expression levels for different FHF vectors. Culture layers were directly lysed in cracking buffer (6 m urea, 2% SDS, 125 mm Tris pH 7, bromophenol blue) and equal portions of lysates from parallel transfected wells were electrophoresed through 12% polyacrylamide SDS gels. After transfer to PVDF membranes and blocking in 5% dehydrated milk, blots were incubated overnight with antibodies (0.5 μg ml−1) and subsequently with peroxidase-conjugated anti-rabbit IgG secondary antibodies, and detection performed by enhanced chemiluminescence. This analysis demonstrated that all FHF2 expression vectors gave comparable levels of expression (Fig. 3D), and that both FHF1A and FHF1B vectors gave comparable expression (Supplemental Fig. 1). Following immunoblot detection with antibodies against FHF2 C-terminus, antibodies were stripped using guanidine-containing solution at 50°C, and the filter was reblocked and probed with rabbit antibodies against the N-terminus of FHF2A.
Figure 3. Nav1.6 long-term inactivation requires channel-binding and cytoplasmic effector domains of FHF2A.
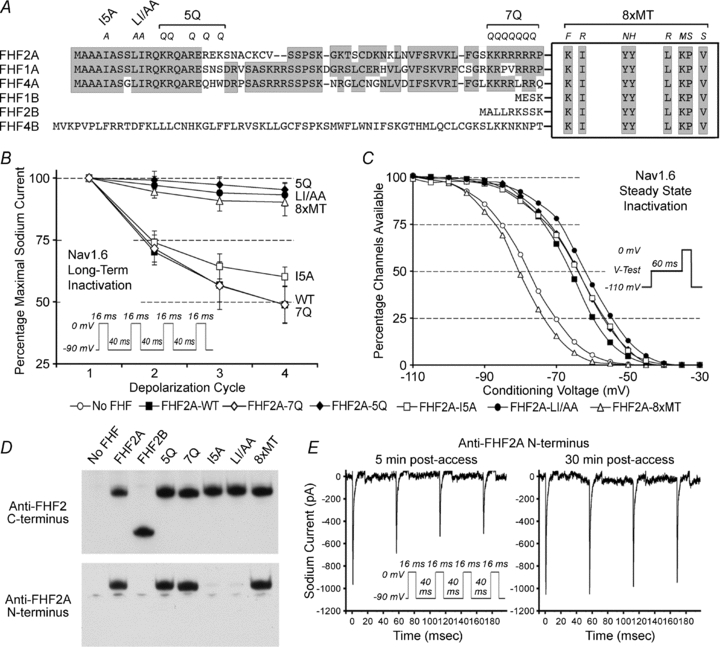
A, FHF sequence alignment and engineered FHF2A mutations. N-terminal sequences of FHF isoforms are shown attached to the core homology domain for all FHFs (box). Eight core residues required for FHF binding to channel tail (Goetz et al. 2009) are indicated. Sequence similarity is shaded. Individual and combined amino acid substitutions (italicized) and names of mutant FHF2A proteins are marked at the top. B, accumulating long-term inactivation of Nav1.6TTXr mediated by mutant FHF2A proteins. Cells were subjected to four 0 mV 16 ms depolarizations separated by −90 mV 40 ms recovery intervals. The 7Q mutant was as effective as wild-type FHF2A, I5A mutant had reduced potency, and mutants 8xMT, 5Q and LI/AA were incapable of inducing long-term inactivation. See Suppl. Table 3 for supporting statistical data. C, steady-state (fast) inactivation of Nav1.6TTXr mediated by mutant FHF2A proteins. Several mutants impaired for long-term inactivation (5Q, LI/AA, I5A) were still able to shift V1/2 steady-state inactivation. See Suppl. Table 3 for supporting statistical data. D, characterization of rabbit polyclonal antibodies reactive to N-terminal FHF2A epitope. Lysates from N2A cells transfected with FHF2 expression vectors were assayed by immunoblotting with antibodies reactive with C-terminus of FHF2 (top). All FHF2 isoforms and mutants were detectable. The blot was then stripped and reprobed with antibodies against the N-terminus of FHF2A (bottom). The epitope for antibodies to the FHF2A N-terminus includes Ile-5, Leu-9 and Ile-10. E, antibodies to the FHF2A N-terminus block FHF2A-induced Nav1.6 long-term inactivation. Sodium current traces from an N2A-Nav1.6TTXr cell expressing FHF2A were recorded 5 and 30 min after break-in, using patch pipette solution containing rabbit anti-FHF2A (200 μg ml−1). Nav1.6 accumulating long-term channel inactivation is initially detected, but is inhibited following 30 min diffusion of antibodies into the cell. See supporting data in Suppl. Table 3.
Electrophysiological analysis of sodium channels in Neuro2A cells
General set-up and methods
All recordings were performed on cells grown on coverslips. For analysis of sodium currents in transiently transfected N2A and derivative cell lines, the recording chamber was filled with carbogen-bubbled extracellular solution: 120 mm NaCl, 26 mm NaHCO3, 3 mm KCl, 10 mm glucose, 4 mm MgCl2, 2 mm CaCl2, 0.2 mm CdCl2, 3 mmmyo-inositol, 2 mm sodium pyruvate (pH 7.2). For analyses on Nav1.5 and Nav1.6TTXr, 1 mm TTX was added to fully block currents from endogenous sodium channels (Suppl. Fig. 2). The intracellular pipette solution for recording sodium currents contained 104 mm CsF, 50 mm tetraethylamine chloride, 10 mm Hepes pH 7.2, 5 mm glucose, 2 mm MgCl2, 10 mm EGTA, 2 mm ATP, 0.2 mm GTP, with pipettes pulled to give electrical resistance of 1–2 MΩ. In some experiments, pipette solution was supplemented with peptides or antibodies. Voltage and current commands and recordings utilized an Axopatch 200B amplifier, Digidata 1322 digital/analog interface, and pCLAMP9 software (Molecular Devices, Sunnyvale, CA, USA). After obtaining a pipette/cell seal of >5 GΩ on green fluorescent cells, whole cell access was obtained by brief gentle suction. Recordings were initiated at least 5 min after break-in to allow cellular dialysis and dissipation of junctional potential between pipette and cytoplasm. In N2A cells, recordings with peptides in pipettes were initiated at least 10 min after break-in, while recordings with antibodies in pipettes were generally initiated at least 30 min after break-in to allow for macromolecule diffusion into cells. Signals were filtered at 5 kHz and digitized at 10 or 20 kHz. Passive properties of recorded cells (membrane capacitance, leak resistance, and series/access resistance) were calculated from current responses to a voltage-clamped step depolarization from −90 mV to −80 mV (see Suppl. Table 6). For measurements of evoked sodium currents, electrotonic capacitative and leak currents in voltage clamp pCLAMP protocols were subtracted during data acquisition using the pre-sweep hyperpolarizing P/N method in the software; series resistance (3.5 ± 0.9 MΩ) was not compensated. For each accessed cell, a full set of command/record protocols (see below) were attempted, although loss of access sometimes occurred prior to set completion.
Two-pulse protocol for sodium channel recovery
For Nav1.6TTXr, mutant derivatives, and N2A endogenous channels, the 40-sweep protocol used −90 mV holding command preceding two 16 ms 0 mV pulses separated by a variable length −90 mV interpulse recovery phase (0.5–20 ms). For Nav1.5, the interpulse and holding command voltages used was −130 mV and depolarization pulses were to −30 mV. Fraction channels recovered for each sweep equalled INa-peak(2)/INa-peak(1).
Four-pulse protocol for accumulation of sodium channel long-term inactivation
For Nav1.6TTXr, mutant derivatives, and N2A endogenous channels, the 8-sweep protocol used −90 mV holding command, four 0 mV pulses of varying duration (2–16 ms), and 40 ms −90 mV interpulse recovery phases. For Nav1.5, the interpulse and holding command voltages used was −130 mV and depolarization pulses were to −30 mV. Fraction of channels recovered 40 ms after n depolarization cycle equalled INa-peak(n+ 1)/INa-peak(1).
Recovery from long-term inactivation
For Nav1.6TTXr and mutant derivatives, the 20-sweep protocol used −90 mV hold, three 10 ms 0 mV pulses with 20 ms interpulse recoveries, followed by a variable −90 mV recovery phase of 20 + 100(n− 1) ms and a final 0 mV pulse. In each of the n sweeps, the first three pulses allowed for accumulation into long-term inactivation, and channel recovery calculated as INa-peak(4)/INa-peak(1); sweep 1 recovery time was defined as t= 0, thereby filtering out most fast-recovering channels. Values for recovered channels in all sweeps were then fitted to the exponential recovery equation: 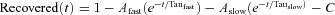 . At least 70% of total sodium conductance was associated with slow recovering channels.
. At least 70% of total sodium conductance was associated with slow recovering channels.
For Nav1.5, several two-pulse depolarization protocols were run with variable −130 mV recovery phases spanning from 1–400 ms. Percentage recovered channels for all time points were fitted to equation: 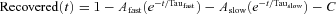 . In the presence of FHF2A or F2A(2–21) peptide, 15–20% of channels displayed slow recovery.
. In the presence of FHF2A or F2A(2–21) peptide, 15–20% of channels displayed slow recovery.
Voltage dependence of channel steady-state inactivation
The 21-sweep protocol used −110 mV holding command, a 60 ms variable test voltage step (−110 + 5(n− 1) mV), followed by a 0 mV pulse. For each of n sweeps, peak sodium current accompanying the 0 mV pulse was measured, and the fraction of channels available (not inactivated) at test voltage equalled INa-peak(Vtest)/INa-peak(−110 mV). To obtain V1/2 and k values for inactivation, data were fitted to a Boltzmann equation: 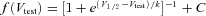 .
.
Voltage dependence of channel long-term inactivation
The 19-sweep protocol used −90 mV holding command, a 16 ms variable test voltage (−90 + 5(n− 1) mV) step, a 40 ms −90 mV recovery phase, followed by a 0 mV pulse. For each of n sweeps, peak sodium current accompanying the 0 mV pulse was measured, and the fraction of channels available (not long-term inactivated) at test voltage equalled INa-peak(Vtest)/INa-peak(−90 mV). Data were fitted to the Boltzmann equation shown above.
Voltage-dependent inhibition of channel long-term inactivation
The protocol assays for the ability of a variable test voltage to prevent subsequent 0 mV depolarization from inducing Nav1.6 long-term inactivation. The 19-sweep protocol used −90 mV holding command, a 60 ms variable test voltage (−90 + 5(n− 1) mV) step, a 16 ms 0 mV step, a 40 ms −90 mV recovery phase, followed by a second 0 mV pulse.
Voltage dependence of channel activation
The 17-sweep protocol used −90 mV holding command and a 30 ms variable test voltage (−60 + 5(n− 1) mV), and the peak sodium current induced by each test voltage measured. INa-peak was plotted against test voltage, and maximum sodium conductance computed from the ohmic linear portion of the plot (−5 to 20 mV). The fraction of activated channels at different test voltages was fitted to the Boltzmann equation shown above.
Electrophysiological analysis of cultured cerebellar granule neurons
For analysis of activated currents and excitability of cerebellar granule neurons, cells were placed in carbogen-bubbled physiological extracellular solution (120 mm NaCl, 26 mm NaHCO3, 3 mm KCl, 1.2 mm KH2PO4, 3 mm glucose, 3 mmmyo-inositol, 2 mm sodium pyruvate, 2 mm CaCl2, 1.2 mm MgSO4) and accessed with patch pipettes filled with 126 mm potassium gluconate, 4 mm NaCl, 5 mm Hepes, 15 mm glucose, 1 mm MgSO4, 3 mm ATP, 0.1 mm GTP, 0.15 mm BAPTA, 0.05 mm CaCl2 (buffered with KOH to pH 7.2) and pulled to have electrical resistance of 10–12 MΩ, as previously described (Goldfarb et al. 2007). Pipette solution was sometimes supplemented with F2A(2–21) peptide. Small diameter neurons that predominate in the cultures were whole cell patched, and their identity as mature granule cells confirmed by small membrane capacitance (3–6 pF), high input resistance (∼1 GΩ) and large inward (sodium) and outward (potassium) currents (>800 pA each). Passive properties of recorded cells (membrane capacitance, leak resistance, and series/access resistance) were calculated from current responses to a voltage-clamped step from −60 mV to −70 mV (see Suppl. Table 6). Each neuron was then voltage-clamped at −90 mV, and subjected to four 16 ms depolarizations at 0 mV separated by 40 ms recovery phases at −90 mV in order to record magnitude of induced currents and potential accumulation of long-term inactivation. The neuron was then switched to current clamp, sufficient current was manually injected to adjust membrane holding potential to −80 mV, and the cell subjected to a multisweep protocol with 800 ms current injections of variable magnitude. Injected current was varied among sweeps from below spiking threshold to above maximal spiking frequency in order to determine the maximum spike frequency of the neuron. Commanded and recorded voltages were adjusted by −10 mV to offset error caused by pipette/bath liquid junctional potential (Goldfarb et al. 2007).
Statistical analysis of electrophysiological data
Parameter values recorded from a cohort of comparable cells were expressed as means ± standard deviation. Significance of differences in parameter values between cohorts was tested by Student's two-tailed unpaired t test.
Computer modelling of sodium channel long-term inactivation
Model building and simulations were conducted on the NEURON software platform. A new 16-state sodium channel Markov model is shown in Fig. 6B. This model is a revision of the 13-state model of Raman & Bean (2001), which accommodates closed states C1 to C5, open state O, fast inactivation states I1 to I6, and open-blocked state OB responsible for resurgent current. The new model deletes state OB (since channels in N2A cells lack resurgent current) and adds long-term inactivation states L3 to L6 (L1 and L2 are omitted, as they are predicted to have virtually zero percent occupancy at any voltage or time). To model a sodium channel subject to long-term inactivation (comparable to association with FHF2A), kinetic parameters were set as follows: α= 353.91exp(v/13.99) mV ms−1; β= 1.272exp(−v/13.99) mV ms−1; n1= 5.422; n2= 3.279; n3= 1.83; n4= 0.738; γ= 150 ms−1; δ= 40 ms−1; Con= 0.025 ms−1; Oon= 0.75 ms−1; a= (Oon/Con)0.25; Coff= 0.5 ms−1; Ooff= 0.002 ms−1; b= (Ooff/Coff)0.25; Lon= 0.001 ms−1; Loff= 0.15 ms−1; c= 20; d= 0.075. Of particular importance in setting parameter values, rate of transition from open (O) to long-term inactivated (L6) state (Lon×c2= 0.4 ms−1) was set to approximately half the rate of transition from O to fast-inactivated state I6 (Oon= 0.75 ms−1), thereby driving one-third of channels into long-term inactivation following steep depolarization. This revised sodium channel model will be publicly available at ModelDB (http://senselab.med.yale.edu/modeldb). For neuronal excitability simulations, the previously described multicompartment model of the cerebellar granule cell (Diwakar et al. 2009) was modified by concentrating sodium channels into the hillock and axon initial segment, consistent with immunofluorescence data (Diwakar et al. 2009). Modelled sodium channels in the wild-type cell lack long-term inactivation (Lon= 0), while F2A(2–21) peptide injection was simulated by enabling long-term inactivation (Lon= 0.001 ms−1). Somatic membrane voltage was monitored during simulation of 12 pA current injection for 200 ms. These granule cell simulations also added back the resurgent sodium current component generated by O ⇌ OB, with k(O→OB)= 1.75 ms−1 and k(OB→O)= 0.0201exp(−v/25) mV ms−1, as previously described (Diwakar et al. 2009).
Figure 6. A long-term inactivation model and computer simulation.
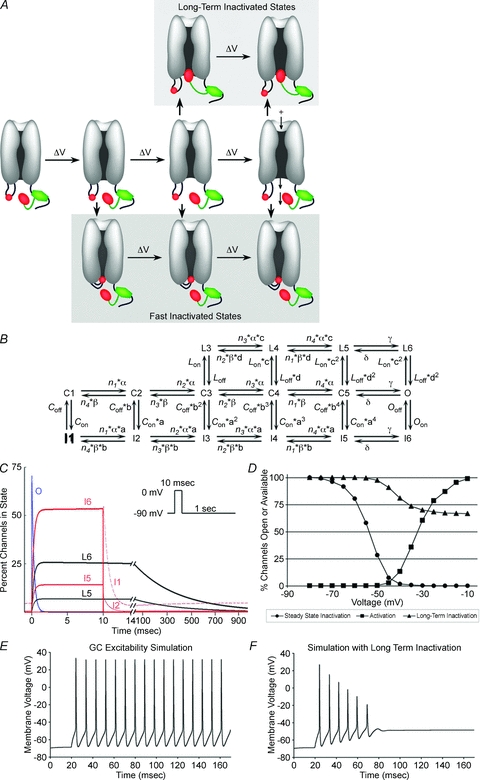
A, schematic diagram of voltage-dependent sodium channel transitions. The core domain of FHF (green) is shown tethered to the channel cytoplasmic tail. The channel's intrinsic inactivation particle in the short cytoplasmic loop (small red oval) and the larger long-term inactivation particle at N-terminus of tethered A-type FHF (larger red oval) compete for access to inactivate the channel at more depolarized voltage-driven transitions near the open state, making long-term inactivation use dependent. This illustration depicts the two particles competing for docking within the pore. Alternatively, the FHF particle may bind at a channel regulatory site elsewhere that is allosterically obscured by prior fast inactivation (not shown). B, 16-state fast kinetic model of sodium channel bearing tethered A-type FHF. This model adds four states (L3–L6) corresponding to docking of the FHF-derived particle to a previously described Markov model for activation and fast inactivation (Raman & Bean, 2001). Channels in the open state (O) or in closed states approaching the open state (C3–C5) can transit to either fast-inactivated (I) states or long-term inactivated (L) states. C, simulation of channel state occupancy during 10 ms 0 mV depolarization and 2 s −90 mV recovery. Open state (O, blue), fast-inactivation states (I1–I6, red), and long-term inactivation states (L5, L6, black) are shown. Lon= 0.001 ms−1. Depolarization drives one-third of channels into long-term inactivation. See Methods for all rate constant values. D, voltage dependence of steady-state inactivation, long-term inactivation, and activation in the simulation of channels with A-type FHF present. V1/2 long-term inactivation in simulations approximates that in recordings of Nav1.6TTXr cells with FHF2A. E, current clamp simulation of cerebellar granule neuron during 12 pA current injection (Lon= 0). Neuron fires repetitively. F, simulation of granule neuron with sodium channel long-term inactivation added (Lon= 0.001 ms−1). During 12 pA current injection, spikes decrease in amplitude until failure, similar to empirical data in Fig. 5D.
Results
A-type FHFs induce long-term inactivation of several sodium channels
FHF modulation of sodium channel physiology was studied by whole cell voltage clamp in Neuro2A (N2A) cells stably expressing an engineered tetrodotoxin (TTX)-resistant variant of Nav1.6 (Rush et al. 2006) and transiently transfected with a bicistronic vector to express both a specific FHF and green fluorescent protein. The use of a common cell line to study all FHFs allowed for reliable functional comparisons across the FHF family, and the bicistronic vector obviated the need for FHF protein modifications that could potentially affect channel modulation. A two-pulse depolarization protocol was used to monitor the recovery of channels from inactivation in fluorescent cells. In the absence of FHF, virtually all channels recovered from fast inactivation within 20 ms (Fig. 1A), whereas FHF2A drove approximately one-third of the channels into a longer-term inactivation state (Fig. 1B). Repetitive 16 ms depolarization cycles separated by 40 ms recovery periods resulted in accumulation of channels in the long-term inactivated state (Fig. 1C and Table 1). Such long-term inactivated channels exhibited slow recovery with time constants of 371 ms at −90 mV and 153 ms at −110 mV (Fig. 1D and Table 1).
Figure 1. A-type FHFs induce long-term inactivation of Nav1.6.
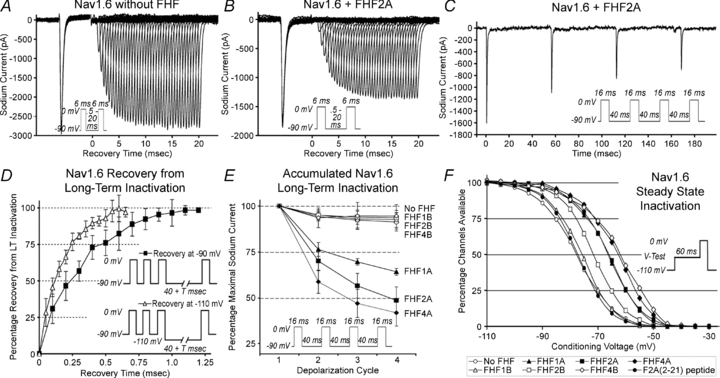
A and B, FHF2A blocks recovery of sodium channels. TTX-resistant sodium currents in N2A-Nav1.6TTXr cells with or without transiently expressed FHF2A were recorded during two 6 ms depolarizations with interpulse −90 mV interval varied between 0.5 and 20 ms (see inset). All current traces are shown. Virtually all channels recover in absence of FHF (A), while one-third of channels in FHF2A-expressing cells remain inactivated in this time frame (B). C, cycle-dependent accumulation of FHF2A-induced long-term inactivation. N2A-Nav1.6TTXr cell expressing FHF2A was depolarized four times (16 ms each) with 40 ms −90 mV recovery intervals (inset), generating smaller current peaks for each cycle. D, long-term recovery of Nav1.6 in FHF2A-expressing cells. Cells were depolarized three times to drive a substantial fraction of channels into long-term inactivation, and long-term recovery was monitored at −90 mV or −110 mV (see insets). The graph isolates the long-term inactivated channel fraction by subtracting out the fast-inactivated channels that had recovered within the first 40 ms. Channels recovered from long-term inactivation faster at the more hyperpolarized voltage. E, all A-type FHFs induce long-term inactivation. Cells were depolarized four times (16 ms each), with 40 ms −90 mV recovery intervals. Graph summarizes data for accumulation of long-term inactivation in cells expressing A- and B- isoforms of FHF1, 2, 4. Only A-type FHFs induce long-term inactivation. F, FHF-induced steady-state inactivation of Nav1.6. N2A-Nav1.6TTXr cells expressing different FHFs were tested for available sodium channels after 60 ms depolarization to test voltages (see inset). Averaged data for all recorded cells are plotted (see Suppl. Table 1 for standard deviations). FHF4B and all A-type FHFs induce large depolarizing shift in voltage dependence of inactivation, and FHF2B induces a smaller shift. Injection of F2A(2–21) peptide into untransfected cells had no effect on steady-state inactivation. Also see supporting data in Suppl. Table 1 and Suppl. Fig. 1.
Table 1.
FHF protein and peptide modulation of Nav1.6 currents
| No FHF | FHF1A | FHF1B | FHF2A | FHF2B | FHF4A | FHF4B | F2A(2–21) 1 mm peptide | ||
|---|---|---|---|---|---|---|---|---|---|
| Long term inactivation | % Available 40 ms after depolarization | ||||||||
| Cycle 1 | 94.3 ± 2.4 | 76.4 ± 3.8 | 95.2 ± 2.8 | 70.2 ± 5.2 | 95.1 ± 2.2 | 58.9 ± 6.3 | 93.6 ± 5.3 | 62.0 ± 8.8 | |
| Cycle 2 | 92.4 ± 4.8 | 69.7 ± 1.8 | 93.0 ± 4.8 | 56.7 ± 6.7 | 94.0 ± 3.8 | 47.1 ± 7.7 | 94.6 ± 7.3 | 48.1 ± 9.8 | |
| Cycle 3 | 91.4 ± 4.9 | 64.3 ± 1.8 | 92.6 ± 5.0 | 48.8 ± 7.3 | 93.6 ± 2.8 | 42.1 ± 7.2 | 94.6 ± 6.2 | 40.5 ± 9.4 | |
| No. of cells | n= 11 | n= 5 | n= 6 | n= 9 | n= 10 | n= 5 | n= 11 | n= 11 | |
| t test P value | 3 × 10−10 | 8 × 10−10 | 4 × 10−8 | 4 × 10−11 | |||||
| Tau recovery (ms) | |||||||||
| at −90 mV | 131 ± 41 | 371 ± 107 | 206 ± 67 | 323 ± 14 | |||||
| No. of cells | n= 4 | n= 9 | n= 5 | n= 4 | |||||
| at −110 mV | N.A. | 153 ± 33 | N.A. | N.A. | |||||
| No. of cells | n=5 | ||||||||
| V1/2 (mV) | −46.6 ± 2.7 | −46.7 ± 5.3 | −47.4 ± 2.1 | −42.7 ± 5.0 | |||||
| k factor (mV) | −4.0 ± 0.2 | −4.3 ± 0.4 | −3.3 ± 0.4 | −6.8 ± 1.1 | |||||
| No. of cells | n= 4 | n= 6 | n= 4 | n= 6 | |||||
| Steady state inactivation | V1/2 (mV) | −79.1 ± 3.7 | −66.4 ± 2.8 | −78.0 ± 2.6 | −66.3 ± 4.3 | −71.5 ± 2.2 | −63.3 ± 3.3 | −62.3 ± 4.3 | −77.1 ± 3.2 |
| k factor (mV) | −5.5 ± 0.5 | −5.0 ± 0.2 | −5.6 ± 0.3 | −5.6 ± 0.3 | −5.5 ± 0.5 | −6.1 ± 0.8 | −6.8 ± 1.4 | −5.3 ± 0.5 | |
| No. of cells | n= 8 | n= 5 | n= 5 | n= 8 | n= 8 | n= 5 | n= 8 | n= 7 | |
| t test P value | 3 × 10−5 | 2 × 10−5 | 3 × 10−4 | 2 × 10−5 | 1 × 10−6 | ||||
| Activation | V1/2 (mV) | −35.0 ± 3.3 | −40.0 ± 3.9 | −38.3 ± 1.3 | −39.1 ± 7.0 | −36.6 ± 3.1 | −37.6 ± 4.6 | −36.1 ± 3.7 | −34.4 ± 6.3 |
| No. of cells | n= 7 | n= 5 | n= 4 | n= 7 | n= 6 | n= 5 | n=6 | n=7 |
TTX-resistant sodium currents in N2A-Nav1.6TTXr cells were analysed after transient transfection with FHF expression plasmids or perfusion with FHF2A-derived N-terminal peptide F2A(2–21). Accumulating long-term inactivation was assayed using four 16 ms 0 mV depolarizations separated by 40 ms −90 mV recovery intervals. Recovery was assayed by varying the duration of a −90 mV recovery interval following three prior depolarization pulses. Voltage dependences for activation, steady state inactivation, and long-term inactivation were analysed using protocols described in Methods. Values shown as means ± standard deviation. Statistical significance by t tests were determined by comparing values for a given test group to those obtained in absence of FHF plasmid or peptide; highly significant values are highlighted in italic. V1/2, voltage of half-maximal effect; k, slope factor; N.A, not analysed. Also see Suppl. Fig. 2 showing lack of TTX-resistant current in parental N2A cells.
FHFs are encoded by four genes, each giving rise to multiple isoforms differing in their N-terminal sequences through alternative promoter usage and splicing (Goldfarb, 2005). Among six FHFs (1A, 1B, 2A, 2B, 4A, 4B) assayed for modulation of Nav1.6, only the three A-type FHFs induced accumulating long-term inactivation (Fig. 1E and Table 1). The observed variability in the accumulation of long-term inactivation induced by A-type FHFs may be explained in part by different rates of recovery (Table 1). All FHFs were also tested for modulation of voltage-dependent steady-state inactivation of Nav1.6. FHF1A, FHF2A, FHF4A and FHF4B each induced 13–16 mV depolarizing shifts in the V1/2 of inactivation (Fig. 1F and Table 1), while FHF2B induced a lesser shift and FHF1B had no demonstrable effect. These data show that induction of long-term inactivation and shift in steady-state inactivation are not tightly correlated across the spectrum of tested FHFs.
The ability of A-type FHFs to induce long-term inactivation extends to other sodium channels in addition to Nav1.6. N2A cells were transiently transfected with expression vectors for the cardiac channel Nav1.5 (naturally TTX-resistant) and a GFP expression vector with or without FHF2A. In the presence of FHF2A, Nav1.5 channels in fluorescent cells assayed in the presence of TTX experienced accumulating long-term inactivation (Suppl. Table 1). Additional assays were conducted in the absence of TTX to analyse the behaviour of endogenous sodium channels in N2A cells, which express Nav1.1, Nav1.2, Nav1.3 and Nav1.7, but not Nav1.6. (Lou et al. 2005). Both FHF1A and FHF2A induced robust accumulating long-term inactivation of this endogenous mixture of channels as well (Suppl. Table 1).
FHF-induced long-term inactivation and intrinsic fast inactivation are mutually exclusive channel states
Several lines of experiments described here defined long-term inactivation as an FHF-induced channel state distinct from fast inactivation. To determine the voltage dependence of long-term inactivation, cells expressing Nav1.6 and FHF2A were clamped at different test voltages for 16 ms, returned to −90 mV for 40 ms to allow for fast channel recovery, and depolarized to 0 mV to assess available channels. V1/2 for long-term channel inactivation (–46.7 mV) was far more positive than V1/2 for steady-state inactivation (−66.3 mV), though still negative to V1/2 for activation (−39.1 mV) (Fig. 2A and Table 1). This result suggested that at more negative voltages, most steady-state inactivation would represent fast inactivation. To test this prediction, the recovery of Nav1.6 from inactivation in a representative cell expressing FHF2A was monitored after depolarization to either 0 mV or −55 mV. While approximately one-third of channels depolarized to 0 mV had undergone long-term inactivation, 90% of channels recovered rapidly following inactivation at −55 mV (Fig. 2B). In another experiment, the time dependence for onset of long-term inactivation was assessed by varying the duration of each depolarization cycle through the range of 2–16 ms. The degree of long-term inactivation was found to be near maximal after only 2 ms depolarization per cycle (Fig. 2C). Accumulation of channels into long-term inactivation was more determined by the number of depolarization cycles than by the duration of depolarization.
Figure 2. Long-term and fast inactivation are distinct and competing channel states.
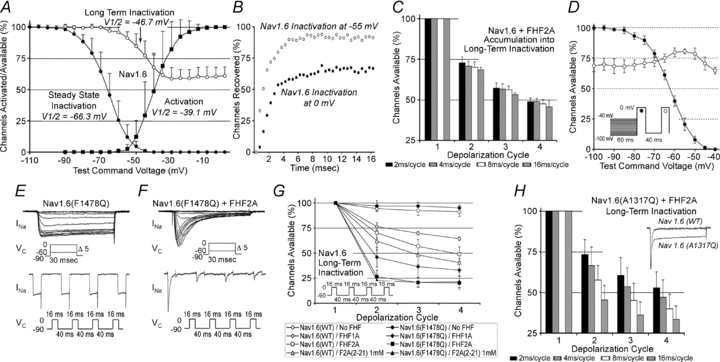
A, long-term inactivation and steady-state inactivation have different voltage thresholds. N2A-Nav1.6TTXr cells expressing FHF2A were assayed for voltage dependence of activation (n= 7), steady-state inactivation (n= 8) and long-term inactivation (n= 6). Average V1/2 for long-term inactivation (−46.7 ± 5.3 mV) is close to that for activation (−39.1 ± 7.0 mV) and far more positive than that for steady-state inactivation (−66.3 ± 4.3 mV). Also see supporting data in Table 1. B, virtually all steady-state inactivation at −55 mV is fast inactivation. An N2A-Nav1.6TTXr cell expressing FHF2A was depolarized to either 0 mV or −55 mV followed by recovery (–90 mV) for 0.5–20 ms before measuring current induced by a second depolarization to 0 mV. Long-term inactivation is induced at 0 mV, but steady-state inactivation at −55 mV is almost exclusively fast inactivation. C, effect of depolarization interval duration on long-term inactivation. N2A-Nav1.6TTXr cells expressing FHF2A (n= 8) were each subjected to four 0 mV depolarization cycles of varying duration (2–16 ms) spaced by 40 ms −90 mV recovery periods, and available sodium current for each cycle was expressed as a percentage of first-cycle current. Long-term inactivation is driven predominantly by the number of depolarization cycles, and not by cycle duration. D, voltages favouring Nav1.6 fast inactivation inhibit subsequent entry into long-term inactivation. N2A-Nav1.6TTXr cells expressing FHF2A (n= 4) were clamped to test voltages for 60 ms, followed by two depolarizations to 0 mV separated by a 40 ms recovery period at −90 mV (inset). The current induced in the first 0 mV depolarization (filled circles) reports degree of steady state inactivation, while the second 0 mV depolarization (open circles) reports long-term inactivation. The −50 to −60 mV range, which preferentially induces fast inactivation (panels A and B), partially protects against long-term inactivation upon further depolarization to 0 mV. E and F, FHF2A-induced long-term inactivation of channels lacking intrinsic fast inactivation. N2A cells were transiently transfected with vectors expressing Nav1.6(F1478Q)TTXr and GFP without (E) or with FHF2A (F). In absence of FHF2A, Nav1.6(F1478Q)TTXr opens without inactivation (note increased tail current spike upon repolarizations), while FHF2A restores inactivation that is long term. See supporting data in Suppl. Table 2. G, A-type FHFs induce enhanced long-term inactivation of Nav1.6(F1478Q)TTXr. Accumulating long term inactivation through four 0 mV 16 ms depolarization cycles and −90 mV 40 ms recovery intervals was measured for Nav1.6TTXr and Nav1.6(F1478Q)TTXr upon transfection of FHF1A, FHF2A or cell perfusion with F2A(2–21) peptide (see Table 1 and Suppl. Table 2 for number of cells analysed). H, FHF2A induces time-dependent accumulation into long-term inactivation of Nav1.6(A1317Q) channels. FHF2A-induced long-term inactivation of Nav1.6(A1317Q)TTXr channels was measured after four 0 mV depolarization cycles of 2–16 ms separated by 40 ms −90 mV recovery intervals (n= 4). In each depolarization cycle, mutant channels continue to enter the long-term inactivated state during the 16 ms depolarization interval. Scaled sodium current traces for Nav1.6TTXr and Nav1.6(A1317Q)TTXr upon depolarization (inset); mutant channel shows enhanced persistent current.
The above data showed that FHF2A-dependent long-term inactivation occurs only at greater voltage-driven channel transitions than are needed for fast inactivation, and further showed that the fraction of channels driven into long-term inactivation is restricted by a rapid process. A series of experiments were conducted to further test the hypothesis that fast inactivation blocks channel entry into the long-term inactivated state. First, Nav1.6 channels were preferentially driven into fast inactivation and tested for whether they were protected against long-term inactivation upon subsequent steep depolarization. As shown in the inset of Fig. 2D, cells expressing Nav1.6 and FHF2A were clamped to variable test voltages for 60 ms, followed by two 16 ms depolarizations to 0 mV separated by a 40 ms recovery period at −90 mV. The current induced in the first 0 mV depolarization reported fraction of channels having undergone of steady state inactivation, while the second 0 mV depolarization reported fraction of channels having undergone long-term inactivation (Fig. 2D). Test voltages in the range of −50 to −60 mV, which drove 60–90% of the channels into steady state inactivation, also partially protected against subsequent 0 mV-induced long-term inactivation (Fig. 2D). We suspect that the inability to fully block long-term inactivation with this protocol reflects some overlap in the voltage ranges of Boltzmann curves for voltage dependence of fast and long-term inactivation (Fig. 2A), thereby preventing selection of a test voltage that exclusively and efficiently induces fast inactivation. More depolarizing test voltages (−40 to −45 mV) failed to inhibit long-term inactivation (Fig. 2D), as these voltages were themselves capable of inducing the long-term inactivated state (Fig. 2A).
In a second experiment, long-term inactivation was assayed in mutant channels with impaired fast inactivation. For several sodium channel isoforms, a phenylalanine residue in the so-called IFM particle within the DIII/DIV cytoplasmic loop is essential for fast inactivation (West et al. 1992). The corresponding F1478Q substitution engineered into Nav1.6TTXr successfully suppressed channel fast inactivation at any test voltage in transfected N2A cells (Fig. 2E). In the presence of FHF2A, inactivation of Nav1.6(F1478Q) was restored, with most channels driven into long-term inactivation by a single 16 ms depolarization cycle (Fig. 2F and G and Suppl. Table 2). FHF1A could also induce robust long-term inactivation of Nav1.6(F1478Q) (Fig. 2G and Suppl. Table 2). Because Nav1.6(F1478Q) is defective for fast inactivation, the voltage dependence of FHF2A-induced Nav1.6(F1478Q) steady state inactivation was depolarized near to the value of V1/2 for long-term inactivation (Suppl. Table 2).
As an alternative to disruption of the IFM particle, a mutation was engineered in the docking site within the channel pore for this particle. As predicted from prior studies on Nav1.2 (Smith & Goldin, 1997), the corresponding A1317Q mutation in Nav1.6TTXr rendered fast inactivation leaky, yielding far greater persistent current attributable to unstable docking of the IFM particle (Fig. 2H inset). Whereas 2 ms depolarization cycles induced FHF2A-dependent Nav1.6(A1317Q) of magnitude similar to wild-type channel (Fig. 2D and H), the extent of Nav1.6(A1317Q) long-term inactivation increased substantially as the duration of depolarization cycles were lengthened (Fig. 2H). This finding suggests that fast-inactivated Nav1.6(A1317Q) channels remained sensitive to long-term inactivation in an ongoing manner as its weakly bound IFM particle dissociated.
The above experiments demonstrate that sodium channel fast inactivation and long-term inactivation are distinct channel states, and that channel fast inactivation inhibits entry into long-term inactivation.
Long-term inactivation requires FHF2A channel binding and N-terminal cytoplasmic effector domains
To better understand the molecular mechanism of sodium channel long-term inactivation, mutant versions of FHF2A were tested for their ability to modulate Nav1.6. The 62–66 residue N-terminal extensions of FHF1A, 2A, and 4A share substantial sequence similarity, including 94% identity for residues 1–18 and a cluster of basic residues close to the FHF core (Fig. 3A). FHF2A variants bearing mutations in the N-terminal regions or in core residues known to be necessary for association with channel carboxy-terminal tails (Goetz et al. 2009) were assayed for their ability to induce Nav1.6 long-term inactivation. While mutation of seven consecutive core-proximal basic residues to glutamines (7Q mutant) (Fig. 3A) did not interfere with induction of long-term inactivation, mutations near the N-terminus had significant effects (Fig. 3B). FHF2A mutants bearing five lysine/arginine-to-glutamine substitutions between residues 13–21 (5Q mutant) or bearing two alanine substitutions at leucine-9 and isoleucine-10 (LI/AA mutant) (Fig. 3A) were defective for induction of long-term inactivation (Fig. 3B, Suppl. Table 3), while their ability to induce a depolarizing shift in V1/2 steady-state (fast) inactivation remained intact (Fig. 3C, Suppl. Table 3). FHF2A(I5A) (Fig. 3B) retained competence to induce long-term inactivation, albeit to a somewhat reduced extent and with a faster rate of recovery (Fig. 3C, Suppl. Table 3). The core octamutant version of FHF2A(8xMT) (Goetz et al. 2009) was incapable of modulating fast inactivation or of inducing long-term inactivation (Fig. 3B, Suppl. Table 3). This survey demonstrated that long-term inactivation requires the channel-tethering domain in the FHF core and an N-terminal effector domain.
If the N-terminal effector region of channel-bound FHF2A is freely exposed to the cytoplasm, an antibody to the effector epitope would be predicted to block long-term inactivation without blocking FHF modulation of fast inactivation. An N-terminal acetylated peptide corresponding to residues 2–21 of FHF2A was used as immunogen to raise polyclonal antibodies, and the specificity of the affinity-purified antibodies was tested by immunoblotting. Whereas previously described antibodies against the FHF2 C-terminus (Wittmack et al. 2004) recognize FHF2B, FHF2A and all FHF2A mutant proteins (Fig. 3D, top panel), the FHF2A N-terminal antibodies only recognized FHF2A and its 5Q, 7Q and 8xMT derivatives efficiently and detected FHF2A mutants I5A and LI/AA poorly (Fig. 3D, lower panel). These N-terminal antibodies were delivered into cells expressing Nav1.6 and FHF2A by diffusion from the patch pipette. Shortly after attaining whole cell access, sodium channels could still undergo long-term inactivation (Fig. 3E, left panel). After a 30 min period to allow diffusion of antibody into cells, long-term inactivation was virtually abolished (Fig. 3E, right panel and Suppl. Table 3). FHF2A antibody perfusion did not affect the depolarizing shift in steady-state fast inactivation induced by FHF2A (Suppl. Table 3), demonstrating the specificity of the antibody's effect. As a further test of specificity, long-term inactivation induced by FHF2A(I5A) was not strongly inhibited by antibody, reflecting poorer recognition of the mutant FHF by the antibodies (Fig. 3D and Suppl. Table 3).
FHF2A residues 2–21 constitute a long-term inactivation particle
Long-term inactivation of sodium channels requires tethering and effector domains of FHF2A. We considered the possibility that the N-terminal effector region of an A-type FHF in the form of a synthetic peptide may be sufficient to induce long-term inactivation. We also speculated that the requirement for channel tethering could be obviated by high peptide concentration. Peptides were synthesized corresponding to residues 2–12, 11–21, or 2–21 of FHF2A. Peptides were N-terminally acetylated and Met-1 was omitted due to predicted posttranslational cleavage of FHF2A's N-terminal methionine in vivo (Frottin et al. 2006). When 1 mm F2A(2–21) peptide was added to patch pipettes for diffusion-driven delivery into cells, long-term inactivation of Nav1.6 was evident within 10 min of cell break-in (Fig. 4A and Table 1).
Figure 4. N-terminal FHF2A-derived synthetic peptide acts as a long-term inactivation particle.
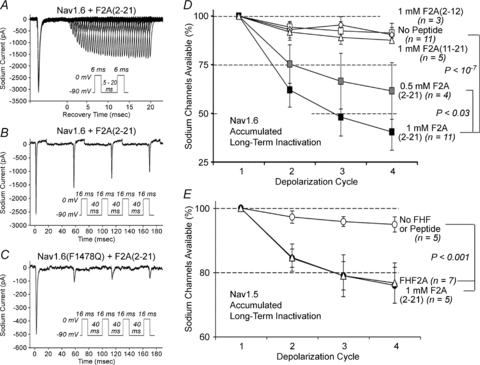
A and B, peptide-induced long-term inactivation of Nav1.6TTXr. TTX-resistant sodium current in N2A-Nav1.6TTXr cells patched with pipettes containing 1 mm peptide corresponding to FHF2A residues 2–21 (F2A(2–21)). Approximately 40% of channels underwent long-term inactivation after one 0 mV 6 ms depolarization cycle (A) and accumulated to ∼60% following three prior 16 ms depolarization cycles separated by −90 mV 40 ms recovery intervals (B). See supporting data in Table 1. C, enhanced induction of Nav1.6(F1478Q)TTXr long-term inactivation by F2A(2–21). Cell expressing Nav1.6(F1478Q)TTXr was patched with pipette containing 1 mm F2A(2–21) and sodium currents recorded during the four-pulse protocol (as in panel B). One depolarization cycle induced maximum long-term inactivation. See supporting data in Suppl. Table 2. D, dose dependence and sequence specificity of peptide-induced channel inactivation. N2A-Nav1.6TTXr cells were patched with pipettes filled with 0.5 mm F2A(2–21) (n= 4), 1 mm F2A(2–21) (n= 11), 1 mm F2A(2–12) (n= 3), or 1 mm F2A(11–21) (n= 5) and subjected to four 16 ms depolarizations spaced by 40 ms −90 mV recovery intervals. F2A(2–21) activity is dose dependent, and other peptides lack significant effect. See supporting data in Suppl. Table 4. E, peptide-induced long-term inactivation of Nav1.5. TTX-resistant sodium currents in N2A cells transiently transfected with Nav1.5 vector with or without 1 mm F2A(2–21) or with FHF2A cotransfection. Four 16 ms −30 mV depolarizations were spaced with 40 ms recoveries at −130 mV. Peptide-induced long-term inactivation was comparable to effect of FHF2A transfection.
By several criteria, the properties of long-term inactivation induced by pipette injection were very similar to those induced by expression of full-length FHF2A protein. In the presence of peptide, channels displayed a V1/2=−42.7 mV for long-term inactivation, accumulated into the long-term inactivated state following multiple cycles of depolarization, and recovered from long-term inactivation with a time constant of 323 ms at −90 mV (Fig. 4B and Table 1). Additionally, this peptide could drive most inactivation-defective Nav1.6(F1478Q) channels into long-term inactivation with a single 16 ms depolarization cycle (Figs 2G and 4C and Suppl. Table 2), analogous to full-length FHF2A. F2A(2–21) peptide acted in a dose-dependent manner, and the shorter peptides lacking either the basic residue cluster or the hydrophobic N-terminus lacked significant activity at 1 mm concentrations (Fig. 4D and Suppl. Table 4). Together with the FHF mutagenesis data (Fig. 3B), these findings show that the N-terminal region of FHF2A is a necessary and sufficient long-term channel inactivation gating particle. Additionally, F2A(2–21) peptide did not shift the voltage dependence of steady-state fast inactivation (Fig. 1F and Table 1), consistent with FHF2A mutagenesis data showing that the N-terminal region is not required for modulation of fast inactivation (Fig. 3C).
F2A(2–21) peptide was also tested for its ability to induce long-term inactivation of cardiac channel Nav1.5. Indeed, at 1 mm concentration, the peptide induced accumulating long-term inactivation of Nav1.5 to an extent indistinguishable from that driven by expression of FHF2A protein (Fig. 4E and Suppl. Table 1).
F2A(2–21) peptide-mediated long-term inactivation of sodium channels blocks repetitive neuronal firing
Sustained high-frequency neuronal firing requires efficient fast recovery of sodium channels from inactivation. For example, cerebellar granule neurons cultured for 16 days undergo reproducible inactivating inward sodium current and non-inactivating outward potassium current upon repeated voltage clamped cycles of 0 mV depolarization (Fig. 5A and Suppl. Table 5). Rapid recovery enables granule neurons to fire continuously when depolarized under current clamp (Fig. 5B and Suppl. Table 5). When 0.5 mm F2A(2–21) peptide was included in a patch pipette, inward transient currents decreased upon repeated depolarization cycles, while outward currents were unaffected (Fig. 5C and Suppl. Table 5), reflecting sodium channel long-term inactivation. Concomitantly, the peptide-injected cell could not undergo sustained firing at any amplitude of injected current (Fig. 5D and Suppl. Table 5). Peptide injection did not alter the first inward transient current nor the voltage peak amplitude of the first induced action potential (Fig. 5C and D and Suppl. Table 5). A cohort of granule neurons were recorded using pipettes filled with 0, 0.05, 0.1, or 0.5 mm F2A(2–21) peptide, and all neurons were characterized for accumulating loss of inward sodium current and for maximal spike generation. Sodium currents were inhibited in a dose-dependent manner, and there was a strong correlation between sodium channel long-term inactivation and reduction in spike frequency (Fig. 5E and Suppl. Table 5).
Figure 5. F2A(2–21) peptide modulation of sodium current and excitability of mouse cerebellar granule neurons.
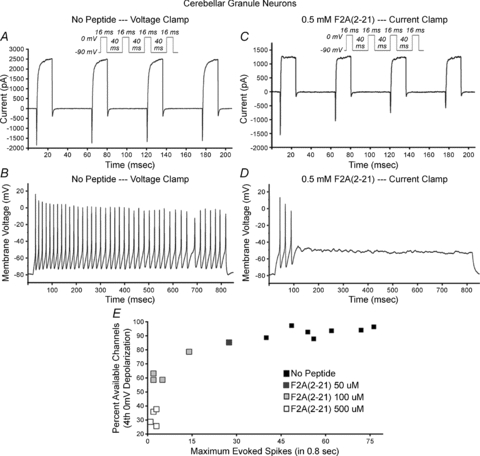
Cultured granule neurons (16 days in vitro) were patched with pipette solution with or without F2A(2–21) peptide. A cell patched without peptide (A and B) was subjected to four 16 ms 0 mV depolarizations separated by 40 ms −90 mV recovery intervals (A) and current clamped depolarization (B). Inactivating inward current and sustained outward currents were virtually reproduced through four depolarization cycles (A), and current injection yielded sustained firing (B). Cell patched with 0.5 mm F2A(2–21) (C and D) showed accumulating loss of inward current (C) and generated three spikes of reducing amplitude until failure (D). E, accumulating inactivation of inward current and loss of spike generation potential. Cells were patched with pipettes containing 0, 0.05, 0.1, or 0.5 mm F2A(2–21) peptide. The graph plots, for each cell, percentage accumulated inactivation of inward current vs. maximum spikes generated. Inward current long-term inactivation is peptide dose dependent, and long-term inactivation correlates with inhibition of spiking.
A revised transition state model for sodium channels
The empirical data presented in this paper suggest an expanded transition state model for voltage-gated sodium channels (Fig. 6A and B). By analogy to the transitions documented for voltage-gated potassium channels (Long et al. 2005; Pathak et al. 2007), rising voltage allows for widening of the sodium channel's cytoplasmic face, reaching a final conducting open conformation (Fig. 6A). Lesser transitions attained at more negative potentials render the channel competent for inactivation, even without channel opening. The intrinsic fast inactivation particle in the DIII/DIV loop can gain access to the channel pore's cytoplasmic face at more negative potential than can the FHF-associated long-term inactivation particle, and docking of the fast-inactivation particle blocks access of the FHF particle (Fig. 6A). The model reflects several empirical observations: (1) greater depolarization is required to achieve long-term inactivation than is needed for fast inactivation (Fig. 2A and B), (2) somewhat greater depolarization is needed to achieve channel opening than is required for long-term inactivation (Fig. 2A), suggesting potential entrance into long-term inactivation from a closed state as well as the open state (Fig. 6A), (3) fast inactivation blocks entrance into long-term inactivation (Fig. 2C–H), and (4) long-term inactivation is mediated by an A-type FHF N-terminal particle that is positioned near the channel by FHF core domain interaction with the channel C-terminal tail (Figs 3, 4). The schematic diagram in Fig. 6A presents one potential model for particle competition, in which the FHF particle binding site lies deep within the cytoplasmic face of the pore and is occluded by fast inactivation. Alternatively, the FHF particle may bind to a regulatory site lying outside the pore that is allosterically obscured by prior fast inactivation.
Independent of precise knowledge of the underlying structural mechanism, the kinetics of FHF-induced sodium channel inactivation can be modelled computationally. The previously described 12-state fast kinetic model for sodium channels posited a series of voltage-dependent rates for transitions through closed states C1 through C5, a final voltage-independent equilibrium between C5 and open state (O), and voltage-independent rate constants for transitions between C1–C5 and O to fast-inactivated states I1 through I6, respectively (Fig. 6B) (Raman & Bean, 2001). Long-term inactivation can be modelled as alternative, mutually exclusive voltage-independent transitions from C3–C5 and O to states L3–L6, respectively, in which the FHF inactivation particle has blocked conduction (Fig. 6B). Appropriate selection of rate constants for state transitions into this 16-state model (see Methods) yields voltage-clamp simulations that approximate the recorded behaviour of Nav1.6 in the presence of FHF2A (Fig. 6C and D). In a simulated 10 ms depolarization to 0 mV, most channels briefly opened followed by entry of approximately one-third of channels into long-term inactivated states L5 and L6, with the remaining channels undergoing fast inactivation (I states). Upon repolarization, the fast-inactivated states I5 and I6 rapidly recovered, while those in L5 and L6 recovered with a time constant on the order of 400 ms (Fig. 6C). Simulations to assay voltage dependence of steady-state inactivation, long-term inactivation, and activation yielded a V1/2 long-term inactivation substantially higher than V1/2 for steady-state (fast) inactivation (Fig. 6D), in good accord with the recorded data (Fig. 2A and Table 1).
We modified a previously described computer model of the cerebellar granule cell (D’Angelo et al. 2001; Diwakar et al. 2009) by incorporating sodium channel long-term inactivation states into the model. In a current injection simulation of the granule cell lacking A-type FHF peptide (i.e. transition rates to L states = 0), the neuron could fire continuously at high frequency (Fig. 6E). By contrast, upon modelling the neuron in the presence of A-type FHF peptide, current injection induced a few spikes of declining amplitude followed by failure (Fig. 6F), comparable to our empirical recordings (Fig. 5D and E). The ability of long-term sodium channel inactivation to impair repetitive firing in the model provides evidence that experimental peptide injection into cultured granule cells restricts neuronal excitability specifically through its action on sodium channels.
Discussion
A-type FHFs associate with voltage-gated sodium channels and provide a long-term use-dependent inactivation particle. The FHF particle can only function at substantial voltage-dependent channel transitions approaching the open state, with ensuing competition between FHF and the channel-intrinsic particles dictating the fraction of channels driven into long-term inactivation. Although the competing particles exert different effects on a sodium channel, they possess curious similarities. Each particle bears and relies upon a cluster of hydrophobic residues flanked by an adjacent cluster of basic residues (West et al. 1992; Eaholtz et al. 1999) (and Figs 3 and 4). While the channel fast inactivation particle is ∼8 residues in length, the larger FHF particle (∼20 residues) may provide a greater surface for channel interaction underlying this particle's greater stability upon binding.
Several lines of evidence have shown that the fast inactivation particle and the A-type FHF long-term inactivation particle compete for inactivation of sodium channels: (1) whereas some channels rapidly enter long-term inactivation upon steep depolarization, further entry is inhibited coincident with onset of fast inactivation, (2) weaker depolarization to voltages that favour fast inactivation reduce entry into long-term inactivation upon subsequent further depolarization, (3) by contrast, Nav1.6(A1317Q) channels with leaky fast inactivation continue to accumulate into long-term inactivation during a depolarization phase, and (4) almost all fast-inactivation-defective Nav1.6(F1478Q) channels rapidly enter long-term inactivation in a single depolarization cycle. Competition between inactivation particles may reflect FHF long-term inactivation particle binding deep within the widening channel pore, so as to be inhibited by prior binding of the intrinsic fast inactivation particle (Fig. 6A). Alternatively, fast particle docking may prevent conformational changes that expose an FHF particle's docking site elsewhere on the channel's cytoplasmic face. Indeed, fast inactivation particle docking has been shown to inhibit conformational changes associated with slow inactivation (Featherstone et al. 1996; Alekov et al. 2001). A related issue is whether FHF-induced long-term inactivation proceeds through a unique structural mechanism or by dramatically accelerating a slow inactivation-type structural change. Clarification of the structural basis for long-term inactivation will require additional functional scanning of the channel surface by site-specific mutagenesis.
Cell perfusion with FHF-derived peptide induces long-term sodium channel inactivation and restricts firing of action potentials in cerebellar granule cells. We speculate that endogenously expressed A-type FHFs underly spike accommodation or arrest of repetitive firing in certain neurons or neuronal subcellular compartments. Consistent with their potential roles as excitation modulators, we have detected endogenously expressed A-type FHFs at axon initial segments of some neurons in the central nervous system (K. Dover and M. Goldfarb, unpublished data). Neuronal excitability may be determined by the relative occupancy of sodium channels by A-type vs. B-type FHF isoforms. We also note that the effects of A-type FHFs on excitability are likely to be complex, as their ability to raise the voltage dependence of sodium channel steady-state inactivation is expected to favour neuronal excitability. Additionally, accumulating long-term inactivation of sodium channels mediated by A-type FHFs bears striking physiological resemblance to long-term use-dependent inactivation of dendritic sodium channels in hippocampal neurons responsible for limiting action potential back-propagation from the soma (Jung et al. 1997; Remy et al. 2009). Future experiments are warranted to determine whether dendritic action potentials in some neurons are regulated by A-type FHFs.
Long-term inactivation mediated by A-type FHF requires both its N-terminal inactivation particle and a surface on its β-trefoil core that binds to the channel cytoplasmic tail. The inactivation particle in isolation can block channels if present at sufficiently high concentration, even if another FHF is docked at the channel tail, as is the case in cerebellar granule neurons (Goldfarb et al. 2007). The superseding effect of FHF N-terminus-derived peptides on long-term inactivation suggests potentially efficacious use of peptides or small molecule mimetics for managing disorders of cellular hyperexcitability in nervous and cardiac systems.
Acknowledgments
We thank Robert Kass (Columbia University) for providing the human Nav1.5 expression vector, David Linden (Johns Hopkins University) for commentary on the manuscript and for suggesting BDNF addition to cerebellar neuron cultures, and Annie Yam Shen and Ivan Anastassov for technical assistance. This work was supported by NIH awards to M.G. (R01-NS39906 and U54-NS40173). The cover photo employed a monoclonal antibody against A-type FHFs developed and maintained by the UC Davis/NeuroMab Facility in the Dept. of Neurobiology, Physiology, and Behavior at UC Davis (supported by PHS grant U24NS50606).
Glossary
Abbreviations
- BDNF
brain-derived neurotrophic factor
- FHF
fibroblast growth factor homologous factor
Author contributions
K.D. generated all expression vectors, performed immunoblotting experiments, conducted the majority of patch clamp experiments on Neuro2A and derivative cell lines, contributed to data analysis and interpretation, and provided critiques of manuscript drafts. S.S. provided M.G. with training in computer modeling and formatted new models for submission to public online database. E.D'A. provided M.G. with training on his laboratory's cerebellar granule cell model and provided critiques of manuscript drafts. M.G. supervised the project, designed all recording protocols, conducted patch clamp experiments on Neuro2A and cerebellar granule cells, performed the majority of data analysis, generated the revised sodium channel computer model, and wrote the manuscript.
Supplement Information
References
- Alekov AK, Peter W, Mitrovic N, Lehmann-Horn F, Lerche H. Two mutations in the IV/S4-S5 segment of the human skeletal muscle Na+ channel disrupt fast and enhance slow inactivation. Neurosci Lett. 2001;306:173–176. doi: 10.1016/s0304-3940(01)01895-x. [DOI] [PubMed] [Google Scholar]
- Caterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- Courtney MJ, Akerman KE, Coffey ET. Neurotrophins protect cultured cerebellar granule neurons against the early phase of cell death by a two-component mechanism. J Neurosci. 1997;17:4201–4211. doi: 10.1523/JNEUROSCI.17-11-04201.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angelo E, Nieus T, Maffei A, Armano S, Rossi P, Taglietti V, Fontana A, Naldi G. Theta-frequency bursting and resonance in cerebellar granule cells: experimental evidence and modeling of a slow K+-dependent mechanism. J Neurosci. 2001;21:759–770. doi: 10.1523/JNEUROSCI.21-03-00759.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diwakar S, Magistretti J, Goldfarb M, Naldi G, D’Angelo E. Axonal Na+ channels ensure fast spike activation and back-propagation in cerebellar granule cells. J Neurophysiol. 2009;101:519–532. doi: 10.1152/jn.90382.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaholtz G, Colvin A, Leonard D, Taylor C, Catterall WA. Block of brain sodium channels by peptide mimetics of the isoleucine, phenylalanine, and methionine (IFM) motif from the inactivation gate. J Gen Physiol. 1999;113:279–294. doi: 10.1085/jgp.113.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaholtz G, Scheuer T, Catterall WA. Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron. 1994;12:1041–1048. doi: 10.1016/0896-6273(94)90312-3. [DOI] [PubMed] [Google Scholar]
- Featherstone DE, Richmond JE, Ruben PC. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys J. 1996;71:3098–3109. doi: 10.1016/S0006-3495(96)79504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frottin F, Martinez A, Peynot P, Mitra S, Holz RC, Giglione C, Meinnel T. The proteomics of N-terminal methionine cleavage. Mol Cell Proteomics. 2006;5:2336–2349. doi: 10.1074/mcp.M600225-MCP200. [DOI] [PubMed] [Google Scholar]
- Goetz R, Dover K, Laezza F, Shtraizent N, Tchetchik D, Huang X, Eliseenkova AV, Xu C, Neubert TA, Ornitz DM, Goldfarb M, Mohammadi M. Crystal structure of a fibroblast growth factor homologous factor (FHF) defines a conserved surface on FHFs for binding and modulation of voltage-gated sodium channels. J Biol Chem. 2009;284:17883–17896. doi: 10.1074/jbc.M109.001842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb M. Fibroblast growth factor homologous factors: evolution, structure, and function. Cytokine Growth Factor Rev. 2005;16:215–220. doi: 10.1016/j.cytogfr.2005.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfarb M, Schoorlemmer J, Williams A, Diwakar S, Wang Q, Huang X, Giza J, Tchetchik D, Kelley K, Vega A, Matthews G, Rossi P, Ornitz DM, D’Angelo E. Fibroblast growth factor homologous factors control neuronal excitability through modulation of voltage-gated sodium channels. Neuron. 2007;55:449–463. doi: 10.1016/j.neuron.2007.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grieco TM, Malhotra JD, Chen C, Isom LL, Raman IM. Open-channel block by the cytoplasmic tail of sodium channel β4 as a mechanism for resurgent sodium current. Neuron. 2005;45:233–244. doi: 10.1016/j.neuron.2004.12.035. [DOI] [PubMed] [Google Scholar]
- Hartung H, Feldman B, Lovec H, Coulier F, Birnbaum D, Goldfarb M. Murine FGF-12 and FGF-13: expression in embryonic nervous system, connective tissue and heart. Mech Dev. 1997;64:31–39. doi: 10.1016/s0925-4773(97)00042-7. [DOI] [PubMed] [Google Scholar]
- Jung HY, Mickus T, Spruston N. Prolonged sodium channel inactivation contributes to dendritic action potential attenuation in hippocampal pyramidal neurons. J Neurosci. 1997;17:6639–6646. doi: 10.1523/JNEUROSCI.17-17-06639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laezza F, Lampert A, Kozel MA, Gerber BR, Rush AM, Nerbonne JM, Waxman SG, Dib-Hajj SD, Ornitz DM. FGF14 N-terminal splice variants differentially modulate Nav1.2 and Nav1.6-encoded sodium channels. Mol Cell Neurosci. 2009;42:90–101. doi: 10.1016/j.mcn.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CJ, Dib-Hajj SD, Renganathan M, Cummins TR, Waxman SG. Modulation of the cardiac sodium channel Nav1.5 by fibroblast growth factor homologous factor 1B. J Biol Chem. 2003;278:1029–1036. doi: 10.1074/jbc.M207074200. [DOI] [PubMed] [Google Scholar]
- Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1.2: structural basis of electromechanical coupling. Science. 2005;309:903–908. doi: 10.1126/science.1116270. [DOI] [PubMed] [Google Scholar]
- Lou JY, Laezza F, Gerber BR, Xiao M, Yamada KA, Hartmann H, Craig AM, Nerbonne JM, Ornitz DM. Fibroblast growth factor 14 is an intracellular modulator of voltage-gated sodium channels. J Physiol. 2005;569:179–193. doi: 10.1113/jphysiol.2005.097220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathak MM, Yarov-Yarovoy V, Agarwal G, Roux B, Barth P, Kohout S, Tombola F, Isacoff EY. Closing in on the resting state of the shaker K+ channel. Neuron. 2007;56:124–140. doi: 10.1016/j.neuron.2007.09.023. [DOI] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Resurgent sodium current and action potential formation in dissociated cerebellar Purkinje neurons. J Neurosci. 1997;17:4517–4526. doi: 10.1523/JNEUROSCI.17-12-04517.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Inactivation and recovery of sodium currents in cerebellar Purkinje neurons: evidence for two mechanisms. Biophys J. 2001;80:729–737. doi: 10.1016/S0006-3495(01)76052-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remy S, Csicsvari J, Beck H. Activity-dependent control of neuronal output by local and global dendritic spike attenuation. Neuron. 2009;61:906–916. doi: 10.1016/j.neuron.2009.01.032. [DOI] [PubMed] [Google Scholar]
- Rush AM, Wittmack EK, Tyrrell L, Black JA, Dib-Hajj SD, Waxman SG. Differential modulation of sodium channel Nav1.6 by two members of the fibroblast growth factor homologous factor 2 subfamily. Eur J Neurosci. 2006;23:2551–2562. doi: 10.1111/j.1460-9568.2006.04789.x. [DOI] [PubMed] [Google Scholar]
- Schoorlemmer J, Goldfarb M. Fibroblast growth factor homologous factors are intracellular signaling proteins. Curr Biol. 2001;11:793–797. doi: 10.1016/s0960-9822(01)00232-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkottai VG, Xiao M, Xu L, Wong M, Nerbonne JM, Ornitz DM, Yamada KA. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol Dis. 2009;33:81–88. doi: 10.1016/j.nbd.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smallwood PM, Munoz-Sanjuan I, Tong P, Macke JP, Hendry SH, Gilbert DJ, Copeland NG, Jenkins NA, Nathans J. Fibroblast growth factor (FGF) homologous factors: new members of the FGF family implicated in nervous system development. Proc Natl Acad Sci U S A. 1996;93:9850–9857. doi: 10.1073/pnas.93.18.9850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MR, Goldin AL. Interaction between the sodium channel inactivation linker and domain III S4-S5. Biophys J. 1997;73:1885–1895. doi: 10.1016/S0006-3495(97)78219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker K, Fadool DA. Neurotrophin modulation of voltage-gated potassium channels in rat through TrkB receptors is time and sensory experience dependent. J Physiol. 2002;542:413–429. doi: 10.1113/jphysiol.2002.017376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulbricht W. Sodium channel inactivation: molecular determinants and modulation. Physiol Rev. 2005;85:1271–1301. doi: 10.1152/physrev.00024.2004. [DOI] [PubMed] [Google Scholar]
- Wang Q, McEwen DG, Ornitz DM. Subcellular and developmental expression of alternatively spliced forms of fibroblast growth factor 14. Mech Dev. 2000;90:283–287. doi: 10.1016/s0925-4773(99)00241-5. [DOI] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proc Natl Acad Sci U S A. 1992;89:10905–10909. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittmack E, Rush AM, Craner MJ, Goldfarb M, Waxman SG, Dib-Hajj SD. Fibroblast growth factor homologous factor 2B: Association with Nav1.6 and selective colocalization at nodes of Ranvier of dorsal root axons. J Neurosci. 2004;24:6765–6775. doi: 10.1523/JNEUROSCI.1628-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.