

The F. tularensis protein IglE from the FPI, which is a component of the type VI-like secretion system, has been crystallized and preliminary X-ray data have been collected.
Keywords: Francisella, T6SS, lipoproteins
Abstract
Tularaemia is an uncommon but potentially dangerous zoonotic disease caused by the bacterium Francisella tularensis. As few as ten bacterial cells are sufficient to cause disease in a healthy human, making this one of the most infectious disease agents known. The virulence of this organism is dependent upon a genetic locus known as the Francisella pathogenicity island (FPI), which encodes components of a secretion system that is related to the type VI secretion system. Here, the cloning, expression, purification and preliminary X-ray diffraction statistics of the FPI-encoded protein IglE are presented. This putative lipoprotein is required for intra-macrophage growth and is thought to be a constituent of the periplasmic portion of the type VI-like protein complex that is responsible for the secretion of critical virulence factors in Francisella.
1. Introduction
Francisella tularensis is a highly virulent pathogen that occasionally infects humans. Its ability to infect a very wide range of organisms, such as mammals, birds, amphibians, fish and even invertebrates (Ellis et al., 2002 ▶), makes it an interesting model bacterium for studying the mechanisms of host–pathogen interactions. Many of the important molecules involved in the virulence of Francisella are beginning to be identified, but biochemical characterization of most of these molecules is still lacking (Meibom & Charbit, 2010 ▶). The intracellular replication and virulence of Francisella are both heavily reliant on the expression of the Francisella pathogenicity island (FPI; Nano & Schmerk, 2007 ▶; Nano et al., 2004 ▶). The FPI is comprised of 16–18 genes, depending on the Francisella strain, and a large number of these genes have been shown to be required for escape of Francisella from the phagosome and intracellular replication within the cytosol (Barker et al., 2009 ▶). A subset of the genes in the FPI (∼14 genes) encode a secretion system that is distantly related to the newly identified type VI secretion system (T6SS) that is present in 35% of all sequenced bacterial genomes. In Francisella, the FPI-encoded T6SS is thought to be responsible for the translocation of bacterial macromolecules into the cytosol of infected host cells (de Bruin et al., 2007 ▶). The molecular details of how the FPI proteins function, specifically the secretion system, are for the most part still unknown. Structural biology has been employed to characterize IglC from the FPI and other T6SS components (Hcp and VgrG from Pseudomonas aeruginosa and Escherichia coli, respectively) which have counterparts in the Francisella secretion system. As the FPI and its secretion-system components are critical to the virulence of Francisella, we have undertaken structural analysis of FPI components in order to characterize secretion in Francisella at the molecular level. Here, we present the cloning, expression, purification and preliminary X-ray diffraction analysis of one component of the FPI, IglE. This putative lipoprotein (14 469.5 Da; 125 amino acids) is required for secretion and intramacrophage growth and is believed to be a component of the Francisella T6SS-like secretion system (F. Nano, unpublished work).
2. Materials and methods
2.1. Cloning, expression and purification of IglE from F. tularensis subsp. novicida
The gene fragment encoding the soluble domain distal to the putative palmitoylation site of IglE (Ftn_1311; NCBI GeneID 4548438) was PCR-amplified from genomic DNA of F. tularensis subsp. novicida U112 using the following oligonucleotide primers: 5′-CAT ATC CATATG GAT GGT TTG TAT ATC AAC AAC-3′ and 5′-GGT GGT CTCGAG TTA ATC TTT TTC TAT GCT GCT ATC-3′. The 306 base-pair PCR-amplified gene fragment encoding the soluble domain of IglE was obtained by standard PCR methods using Phusion High-Fidelity DNA Polymerase (New England Biolabs). The product was digested with NheI and XhoI restriction endonucleases and ligated into similarly digested pET-28a(+) (Novagen) using standard cloning procedures. The resultant plasmid encodes a polypeptide consisting of residues 25–126 of IglE preceded by the residues MGSSHHHHHHSSGLVPRGSHM: an N-terminal six-histidine tag followed by a thrombin protease cleavage site.
Recombinant IglE was produced in E. coli BL21 Star (DE3) (Invitrogen) in 4 l cultures of 2× YT broth containing 50 µg ml−1 kanamycin (Sigma). Cells were harvested by centrifugation and the cell paste was stored at 253 K prior to chemical lysis. The frozen cell paste was resuspended in 30 ml sucrose solution (25% sucrose, 20 mM Tris–HCl pH 8.0) and treated with 10 mg lysozyme for 10 min. 60 ml deoxycholate solution (1% deoxycholate, 1% Triton X-100, 100 mM NaCl, 20 mM Tris–HCl pH 7.5) was then added to the solution mixture and stirred for another 10 min. Finally, genomic DNA was digested by adding 0.5 mg DNase (Sigma) and MgCl2 to a final concentration of 5 mM and incubating at room temperature for 5 min. The cell lysate was clarified by centrifugation at 27 000g for 45 min. The clarified lysate was applied onto an immobilized metal-affinity resin column (Sigma His-Select) charged with Ni2+ and equilibrated in binding buffer (0.5 M NaCl, 20 mM Tris–HCl pH 8.0). Bound protein was then eluted using a linear gradient to 100% elution buffer (0.5 M imidazole, 0.5 M NaCl, 20 mM Tris–HCl pH 8.0). Fractions were analyzed for protein content and purity by SDS–PAGE and appropriate fractions were pooled and concentrated in a stirred ultrafiltration unit (Amicon) using a 10 kDa molecular-weight cutoff membrane (Filtron). The concentrated protein was then buffer-exchanged using gel filtration on a PD10 column (GE Biosciences) which was pre-equilibrated in thrombin cleavage buffer (20 mM Tris–HCl pH 8.0, 150 mM NaCl, 5 mM CaCl2). The protein was treated with thrombin (1 U per 5 mg protein) overnight at 291 K and the completeness of the cleavage was checked by SDS–PAGE. Purified thrombin-cleaved protein with the extra residues GSHM was then separated from the free histidine tag and soluble aggregate by size-exclusion chromatography using a Sephacryl S-200 column (GE Biosciences) in 20 mM Tris–HCl pH 8.0. The concentration of IglE was determined by absorbance at 280 nm using the calculated molar extinction coefficient of 5960 M −1 cm−1 (Gasteiger et al., 2003 ▶).
2.2. Crystallization and X-ray data collection
IglE at 25 mg ml−1 was crystallized using the hanging-drop vapour-diffusion method by mixing 1 µl protein solution (20 mM Tris–HCl pH 8.0) and 1 µl well solution [20 mM CdCl2, 100 mM sodium acetate pH 4.4, 30%(v/v) PEG 400] and suspending the drop over 0.5 ml well solution. Crystals were fully formed after one night at 291 K. Crystals were cryocooled directly in a nitrogen stream at 113 K without the need for additional cryoprotectant. Diffraction experiments were carried out on a ‘home-beam’ comprising a MicroMax-002 X-ray source equipped with Osmic Blue Optics, an Oxford Cryosystems Cryostream 700 and an R-AXIS IV++ area detector. Diffraction data were processed using iMOSFLM and SCALA (Leslie, 2006 ▶; Evans, 2006 ▶).
3. Results and discussion
A soluble fragment of IglE, the smallest protein encoded by the FPI, was successfully cloned, overproduced in E. coli and purified to greater than 95% homogeneity as judged by SDS–PAGE analysis. Initial crystallization conditions were obtained by the use of various sparse-matrix screens. Optimization of the initial condition, E12 from Crystal Screen 2 [Hampton Research; 100 mM CdCl2, 0.1 M sodium acetate pH 4.6, 30%(v/v) PEG 400], resulted in the growth of diffraction-quality crystals that started to form almost instantly and grew to full size overnight (Fig. 1 ▶). The final condition was 20 mM CdCl2, 0.1 M sodium acetate pH 4.4, 30%(v/v) PEG 400. The crystals diffracted very well, allowing the collection of a complete diffraction data set to a resolution of 1.90 Å (Fig. 2 ▶). The space group of the crystals proved to be P21212, as determined by POINTLESS (Evans, 2006 ▶), and the data were reduced with an overall R merge of 0.056 (see Table 1 ▶ for complete data statistics). Analysis of the content of the asymmetric unit indicated that it contains two 12 kDa molecules with a Matthews coefficient and solvent content of 1.95 Å3 Da−1 and 37%, respectively (Matthews, 1968 ▶). Using MOLREP, a pseudo-translation was detected between the two monomers in the asymmetric unit (Vagin & Teplyakov, 2010 ▶). The primary structure of IglE contains no methionine residues, making selenomethionine labelling impossible and thus necessitating an alternative approach to phasing that includes the use of halide soaks and/or the production of methionine-substitution mutations.
Figure 1.

Crystal of IglE grown in 30%(v/v) PEG 400, 70 mM CdCl2, 100 mM sodium acetate pH 4.4. Crystal dimensions are 0.3 × 0.1 × 0.1 mm.
Figure 2.

X-ray diffraction image of IglE crystals, showing diffraction to 1.90 Å resolution.
Table 1. Data-collection statistics.
| No. of crystals | 1 |
| Beamline | Home beam |
| Wavelength (Å) | 1.5418 |
| Detector | R-AXIS IV++ area detector |
| Crystal-to-detector distance (mm) | 72.0 |
| Rotation range per image (°) | 0.5 |
| Total rotation range (°) | 220 |
| Exposure time per image (s) | 60 |
| Resolution range (Å) | 19.826–1.90 (1.95–1.90) |
| Space group | P21212 |
| Unit-cell parameters (Å, °) | a = 43.66, b = 90.39, c = 47.37, α = β = γ = 90.00 |
| Mosaicity (°) | 0.89 |
| Total no. of measured intensities | 62528 (8122) |
| Unique reflections | 14748 (2156) |
| Multiplicity | 4.2 (3.8) |
| Mean I/σ(I) | 11.9 (2.4) |
| Completeness (%) | 96.7 (98.7) |
| Rmerge† | 0.063 (0.414) |
| Rmeas | 0.082 (0.540) |
| Overall B factor from Wilson plot (Å2) | 23.6 |
R
merge = 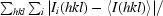
 , where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
, where I
i(hkl) is the ith observation of reflection hkl and 〈I(hkl)〉 is the weighted average intensity for all observations i of reflection hkl.
Structural studies of proteins within the FPI have already begun with the structure of IglC and progress has been made towards an understanding of the mechanism of secretion (Sun et al., 2007 ▶). However, a complete molecular model of the secretion system is still lacking and requires further attention. Here, we present the preliminary diffraction analysis of one component of the secretion system encoded by the FPI. These crystals are of sufficient quality to enable the determination of a high-resolution crystal structure of this protein. Determining the three-dimensional structure and binding partners of this particular protein will provide considerable insight into the overall model of secretion in Francisella and may shed light on type VI secretion in general.
Acknowledgments
This work was supported by a Canadian Institutes of Health Research Operating Grant (FRN 89812). ABB is a Canada Research Chair in Molecular Interactions and a Michael Smith Foundation for Health Research Career Scholar.
References
- Barker, J. R., Chong, A., Wehrly, T. D., Yu, J.-J., Rodriguez, S. A., Liu, J., Celli, J., Arulanandam, B. P. & Klose, K. E. (2009). Mol. Microbiol.74, 1459–1470. [DOI] [PMC free article] [PubMed]
- Bruin, O. M. de, Ludu, J. S. & Nano, F. E. (2007). BMC Microbiol.7, 1. [DOI] [PMC free article] [PubMed]
- Ellis, J., Oyston, P. C., Green, M. & Titball, R. W. (2002). Clin. Microbiol. Rev.15, 631–646. [DOI] [PMC free article] [PubMed]
- Evans, P. (2006). Acta Cryst. D62, 72–82. [DOI] [PubMed]
- Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R. D. & Bairoch, A. (2003). Nucleic Acids Res.31, 3784–3788. [DOI] [PMC free article] [PubMed]
- Leslie, A. G. W. (2006). Acta Cryst. D62, 48–57. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol.33, 491–497. [DOI] [PubMed]
- Meibom, K. L. & Charbit, A. (2010). Curr. Opin. Microbiol.13, 11–17. [DOI] [PubMed]
- Nano, F. E. & Schmerk, C. (2007). Ann. NY Acad. Sci.1105, 122–137. [DOI] [PubMed]
- Nano, F. E., Zhang, N., Cowley, S. C., Klose, K. E., Cheung, K. K., Roberts, M. J., Ludu, J. S., Letendre, G. W., Meierovics, A. I., Stephens, G. & Elkins, K. L. (2004). J. Bacteriol.186, 6430–6436. [DOI] [PMC free article] [PubMed]
- Sun, P., Austin, B. P., Schubot, F. D. & Waugh, D. S. (2007). Protein Sci.16, 2560–2563. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]