

Abstract
Background
Mutations in transcription factor NKX2.5 cause congenital heart disease (CHD). We identified a CHD family with atrial septal defects (ASDs), atrioventricular block, ventricular noncompaction, syncope and sudden death. Our objective is to identify the disease-causing mutation in the CHD family.
Methods
Direct DNA sequence analysis was used to identify the CHD mutation. The functional effects of the mutation were characterized by a luciferase reporter assay and immunostaining.
Results
A novel, de novo 2-bp insertion (c.512insGC) was identified in exon 2 of NKX2.5. Mutation c.512insGC co-segregates with CHD in the family, and is not present in 200 controls. Functional studies indicate that the c.512insGC mutation impedes nuclear localization of NKX2.5 and causes a total loss of transactivation activity of NKX2.5. Furthermore, no NKX2.5 mutation was identified in 125 sporadic Chinese CHD patients.
Conclusions
(1) NKX2.5 mutation c.512insGC is associated with ASDs, syncope and sudden death. It is the second de novo mutation identified in NKX2.5. (2) NKX2.5 mutations are rare in sporadic CHD patients. (3) This study for the first time identifies association between a NKX2.5 mutation and ventricular noncompaction. Our results significantly expand the phenotypic spectrum of NKX2.5 mutations.
Keywords: Transcriptional factor NKX2.5, Mutation, Ventricular noncompaction, Atrial septal defect (ASD), Syncope, Sudden death
1. Introduction
Congenital heart disease (CHD) is one of the most common birth defects resulting from abnormal development of the heart. Atrial septal defects (ASD) are one of the most common congenital heart defects due to malformation of the septum between the two atria, and account for 10% of the CHD cases [1, 2]. Some ASDs are inherited in families and caused by genetic mutations. Several genes have been reported for ASDs, including NKX2.5 (ASD1) [3], GATA4 (ASD2) [3], MYH6 (ASD3) [4], TBX20 (ASD4) [5], ACTC1 (ASD5) [6], TLL1 (ASD6) [7], and TBX5 [8, 9]. Among these ASD genes, mutations were more frequently identified in NKX2.5, which encodes a 324–amino acid transcriptional factor required for cardiac morphogenesis and development [10-15]. The NKX-2.5 protein contains a tinman domain (TN, amino acids 10-21) at the N-terminus, a homeodomain (HD, amino acids 138-197) in the middle, and a NK2-specific domain (nucleotide kinase, NK2, amino acids 212-234) near the C-terminus. The HD domain is necessary for DNA-binding, transactivation, protein-protein interaction and dimerization [16-18], whereas the NK2 domain may have a function in masking transactivation [19, 20].
Stallmeyer et al recently summarized reported NKX2.5 mutations [21], which include missense, nonsense, and frame-shift mutations. Phenotypic heterogeneity is a common finding in the NKX2.5 mutation carriers. In addition to ASDs, NKX2.5 mutations were found to be associated with ventricular septal defects (VSDs), tetralogy of Fallot (TOF), atrioventriciular block (AVB), Ebstein’s anomaly, atrial fibrillation (AF) , and mitral valve prolapse either in the context of ASDs or in the absence of ASDs [22] [23]. Here, we report the identification of a novel de novo NKX2.5 mutation in a family with ASD, AVB, syncope, and sudden death as well as ventricular noncompaction. To the best of our knowledge, this is the first time that a NKX2.5 mutation was found to be associated with ventricular noncompaction.
We also screened NKX2.5 mutations in 125 Chinese Han patients with CHD, but no mutation was identified, suggesting that NKX2.5 mutations are rare in sporadic CHD cases.
2. Materials and methods
2.1. Study subjects
A three generation American family affected with CHD was identified and clinically characterized (Fig. 1). This study was approved by the Cleveland Clinic Institutional Review Board on Human Subject Research and the Ethics Committee of College of Life Science and Technology, Huazhong University of Science and Technology and conformed to the guidelines set forth by the Declaration of Helsinki. Written consent was obtained from the study subjects.
Fig. 1.
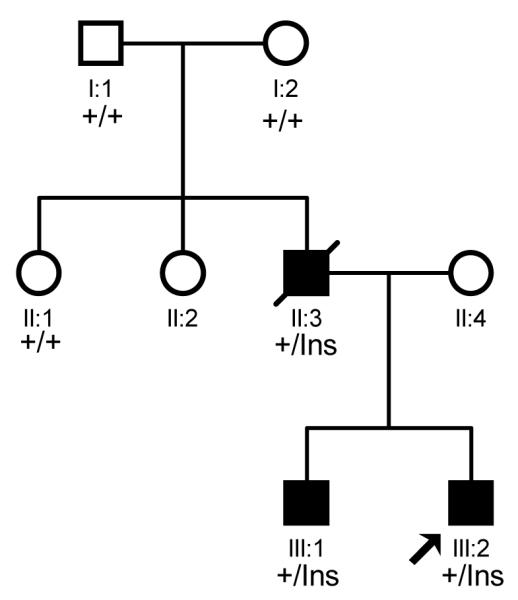
The pedigree structure of a family affected with CHD. The affected individuals are indicated by filled squares (males), and normal individuals are shown with empty squares (males) or circles (females). The deceased individual with sudden death is shown with “/”, and the proband is indicated by an arrow. +, wild type NKX2.5 allele; Ins, mutant NKX2.5 with the 2 bp insertion.
Diagnosis of CHD was made based on standard criteria, and performed by a panel of cardiologists using color doppler echocardiography. Genomic DNA was isolated from peripheral blood leukocytes using standard protocols with the Wizard Genomic DNA Purification Kit (Promega Corp. Madison, WI).
In the CHD family, there are two brothers affected with CHD, a patient with sudden death, and 5 normal family members. The proband, individual III:2 in Fig. 1, was found to be affected with ASDs and ventricular noncompaction by echocardiography. The proband also had first degree AVB and syncope. The proband’s brother was affected with ASDs and first degree AVB. The brothers’ mother was normal, but their father (individual II:3 in Fig. 1) died suddenly at the age of 29 years. Interestingly, the two parents for individual II:3 were not affected with CHD.
2.2. Mutational analysis
Mutational analysis was carried out using direct DNA sequencing analysis. The two exons and exon-intron boundaries of NKX2.5 were amplified by polymerase chain reactions (PCR) and sequenced directly as previously described to identify a mutation [1].
2.3. Modeling of the three-dimensional structure of the NKX2.5 HD domain by SWISS-model
The amino acid sequences of wild type or mutant NKX2.5 protein were sent to the SWISS-Model service, which returned the modeled three dimensional structure of the HD domain. The structure was aligned and viewed using SWISS-Model and Protein Deep View 4.0 (http://swissmodel.expasy.org/).
2.4. Expression constructs for wild type and mutant NKX2.5 protein
The full length cDNA for the human NKX2.5 gene was obtained by reverse transcription polymerase chain reactions (RT-PCR) using cardiac RNA samples. The NKX2.5 cDNA was cut using Not I and BamH I restriction enzymes and sub-cloned into the pcDNA3.1 vector, resulting in a mammalian expression construct for NKX2.5, pWT-NKX2.5. The c.512insGC insertion mutation and the pE21E variant (rs2277923) was introduced into pWT-NKX2.5 using a PCR-based mutagenesis method [24], resulting in pNKX2.5-512insGC, and pNKX2.5-E21. All expression constructs were sequenced and their identity was verified.
2.5. Cell culture and transfection
HeLa cells were cultured in Dulbecco’s Modified Eagle Medium (D-MEM, Gibco BRL) with 10% new born serum (Gibco BRL) as described previously [9].
Cells were plated onto 6-well or 12-well plates for 20 h before transfection and grown to a confluence of 50-70%. Transfection was then performed using Lipofectamine 2000 (Invitrogen) and Opti-OMEM (Gibco BRL) using the standard protocols by manufacturers.
2.6. Cellular localization of NKX2.5 by immunostaining
A goat anti-NKX2.5 antibody was purchased from Santa Cruz. A rhodamine-labeled donkey anti-goat IgG was purchased from PTG lab. Hochest33258 was from Beyotime.
Expression plasmid pWT-NKX2.5 and pNKX2.5-512insGC were transfected into Hela cells. After 36 h, cells were washed by PBS for 3 times, and fixed in 4% paraformaldehyde for 30 min at room temperature. A goat polyclonal anti-NKX2.5 antibody (SantCruz, diluted 1:500) was diluted in TBS with 0.05% Tween 20 (Biosharp), and incubated with fixed cells for 1 h at room temperature. The cells were then washed three times with PBS for 5 min each time, incubated with a secondary antibody, rhodamine-labeled donkey anti-goat IgG (PTG lab, diluted 1:100) for 1 h at room temperature, and washed by PBS as above. The cells were counterstained with Hochest33258 (Beyotime, 2μg/ml, staining for the nucleus) for 15 min, washed by PBS as above, and analyzed with an Olympus TH4-200 fluorescence microscope (Olympus).
2.7. Dual luciferase assays
Transcriptional activity of NKX2.5 was assayed using an ANF(−638)-luciferase reporter construct (ANF-Luc) which contains NK2-binding sites as described previously [25]. The ANF(−638)-luciferase reporter contains the ANF promoter (from −638 to +1 bp transcriptional start site) inserted into the pGL3-Luciferase vector and fused to the luciferase gene. Cells were co-transfected with 1,000 ng of ANF-Luc, 300 ng of pWT-NKX2.5 or derivatives (empty pcDNA3.1 as negative control), and 50 ng of renilla as internal control. Cells were lysed 36 h after transfection. The activity of firefly and renilla luciferase was measured using a Glomax 20/20 Luminometer (Promega). The transcriptional activation was calculated by the firefly luciferase activity divided by the renilla luciferase activity. Each experiment was carried out in triplicate. All experiments were performed three times independently.
The data was presented as mean ±SD and analyzed by a student’s t-test. A P value of <0.05 was considered to be statistically significant.
3. Results
3.1. Identification of a novel de novo NKX2.5 mutation associated with CHD
A three generation CHD family was identified and studied (Fig. 1). Because NKX2.5 is the most common gene for ASDs, we sequenced its two exons and exon-intron boundaries in family members III:1, III:2, II:1, II:3, I:1, and I:2 to identify a mutation. A heterozygous 2 bp insertion (GC) at c.512 from the translation start site in exon 2 (c.512InsGC p.170X) was found in all three patients, III:1, III:2, and II:3 (Fig. 2A). Other family members did not carry this mutation. More than 200 normal control individuals did not carry the c.512InsGC insertion, either. The c.512InsGC mutation is located at codon 170 and is predicted to lead to a frame-shift and result in a truncated protein with 175 amino acid residues (170 NKX2.5 amino acid residues plus 5 additional amino acids) (Fig. 2B, 2C). These results provide strong genetic evidence that c.512InsGC is a pathogenic mutation for ASDs, ventricular noncompaction, syncope and sudden death.
Fig. 2.
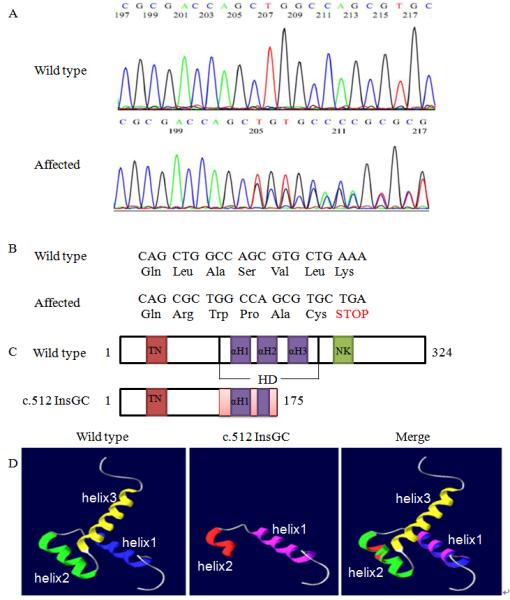
Identification of a novel NKX2.5 mutation, c.512InsGC, in the family with CHD. A: Sequencing results from one normal family member (WT) and one patient (c.512InsGC). B: Schematic diagram showing the frame-shift effect caused by the c.512InsGC mutation. C: Structure of the NKX2.5 protein and the effect of the c.512InsGC mutation. NKX2.5 contains 324 amino acids with a TN domain (amino acids 10-21), a HD with three α-helices (139-197) and a NK2-SD domain (210-233). HD αH1, 147-159; αH2, 165-175; αH3, 179-195. D: The three-dimensional structure of the NKX2.5 HD domain. For wild type NKX2.5, the three α-helices are indicated by different colors, blue, green, and yellow, respectively. For mutant NKX2.5 generated by c.512insGC, the pink color indicates α-helix 1 and red shows α-helix 2. The merged diagram indicates that the mutant NKX2.5 lost the second half of the α-helix 2 and entire α-helix 3.
We subscribed the amino acid sequences of wild type NKX2.5 and mutant NKX2.5 generated by c.512insGC to online Swissmodel Service for prediction of the three-dimension structure. Compared with wild type NKX2.5, the mutant NKX2.5 protein lost the second half of α-helix 2 and entire α-helix 3 in the HD domain (Fig. 2D).
Interestingly, family member II:3 carried the c.512insGC insertion, but his two parents did not carry the mutation. The paternal relationship was confirmed by genotyping and linkage analysis of 6 highly polymorphic microsatellite markers (data not shown). These results indicate that the c.512insGC insertion is a de novo mutation.
3.2. NKX2.5 mutations are rare in sporadic patients with CHD
A total of 125 patients with sporadic CHD were also investigated for mutations in NKX2.5. The clinical characteristics of the study population are listed in Table 1. Thirty-nine patients had ASDs, and 64 patients had VSDs. No mutation was identified.
Table 1.
Characteristics of the study population of 125 patients with sporadic CHD.
| Type of congenital heart disease | Number of patients (n) | % |
|---|---|---|
| ASDs | 17 | 13.6 |
| VSDs | 51 | 40.8 |
| PDA | 10 | 8.0 |
| TOF | 7 | 5.6 |
| ASDs, CAD | 1 | 0.8 |
| ASDs, MI | 1 | 0.8 |
| ASDs, PFO | 1 | 0.8 |
| ASDs, SMTVS | 1 | 0.8 |
| ASDs, Ebstein anomaly | 1 | 0.8 |
| ASDs, LVOTS | 1 | 0.8 |
| ASDs, PDA, TGA | 2 | 1.6 |
| ASDs, PS | 1 | 0.8 |
| ASDs, RHD | 1 | 0.8 |
| ASDs, TOF | 1 | 0.8 |
| ASDs, TVR, CT | 1 | 0.8 |
| ASDs, VSDs | 7 | 5.6 |
| ASDs, VSDs, PDA | 1 | 0.8 |
| AVSDs | 1 | 0.8 |
| PDA, CAD, AI | 1 | 0.8 |
| PDA, Eisenmengor syndrome | 1 | 0.8 |
| PDA, TOF | 1 | 0.8 |
| PDA, TOF, PA | 1 | 0.8 |
| TOF, AVB | 1 | 0.8 |
| VSDs, AI | 4 | 0.8 |
| VSDs, ECD, TGA, PA | 1 | 0.8 |
| VSDs, PDA | 1 | 0.8 |
| VSDs, PFO | 3 | 2.4 |
| VSDs, PFO, PA | 1 | 0.8 |
| VSDs, PS, MVR | 1 | 0.8 |
| VSDs, RVOTS | 2 | 1.6 |
| ASD,PDA | 1 | 0.8 |
| Total | 125 | 100 |
ASD, atrial septal defect; VSD, ventricular septal defect; PDA, patent ductus arteriosus; TOF, tetralogy of Fallot; AVSD, atrioventricular septal defect. CAD, coronary artery disease; MI, mitral insufficiency; PFO, patent foramen ovale; SMTVS, sever mitral valve stenosis; LVOTS, left ventricular outflow tract stenosis; TGA, transposition of the great arteries; PS, pulmonary stenosis; RHD, rheumatic heart disease; TVR, tricuspid valve reggitation; CT, cor triatriatum; AI, aortic insufficiency; ECD, Endocardial cushion defect; PA, pulmonary atresia; MVR, RVOTC, right ventricular outflow tract stenosis.
A nonsynonymous single nucleotide polymorphism (SNP) that changes nucleotide A to G at codon E21 (c.63A>G pE21E, rs2277923) was identified in NKK2.5 during the sequencing analysis. The frequency of the minor allele A of SNP rs2277923 was 46.1% in 125 patients and 36.7% in 105 normal controls. A Chi-square test (2 × 2 contingency table) found that the two frequencies were marginally significant (P=0.04, odds ratio of 1.48 with 95% confidence interval of 1.01-2.14).
3.3. Functional characterization of NKX2.5 mutation c.512insGC
To identify the molecular mechanism by which the c.512insGC insertion causes CHD, we functionally characterized the effects of the c.512insGC insertion on nuclear localization and transcriptional activity of NKX2.5. Wild type and mutant NKX2.5 expression plasmids were transfected into HeLa cells, and immunofluorescence staining with an anti-NKX2.5 antibody was used to characterize cellular localization of NKX2.5. As expected, wild type NKX2.5 is localized into nuclei, however, the mutant NKX2.5 protein generated by the c.512inGC insertion was distributed in both cytoplasm and nuclei (Fig. 3).
Fig. 3.

Cellular localization of wild type NKX2.5 (WT) and mutant NKX2.5 generated by mutation c.512insGC. HeLa cells were transfected with pWT-NKX2.5 and pNKX2.5-512insGC and stained with a goat anti-NKX2.5 antibody as the primary antibody and rhodamine-labeled donkey anti-goat antibody as the secondary antibody (red signal). Nuclei were stained with Hochest 33258 (blue signal). Wild type NKX2.5 was localized completely into the nucleus, whereas the mutant NKX2.5 was localized in both nucleus and cytoplasm.
The effect of the c.512insGC insertion on the transcriptional activation activity of NKX2.5 was assessed using an ANFp-Luc reporter with dual luciferase assays. Wild type NKX2.5 can activate the transcription of the ANF promoter by about five fold compared to the empty vector pcDNA3.1, whereas mutant NKX2.5 generated by c.512insGC did not possess any transactivation activity (Fig. 4). Western blot analysis showed that both wild type and mutant NKX2.5 proteins were successfully expressed in transfected cells (data not shown).
Fig. 4.

Transcriptional activation activity of wild type NKX2.5 (WT), mutant NKX2.5 generated by mutation c.512insGC, and mutant NKX2.5 with SNP rs2277923 (63a>g). pcDNA3.1, empty vector as negative control. The data were shown as mean ± SD (n=9).
The transcriptional activity of the mutant NKX2.5 protein with SNP rs2277923 was also studied, and interestingly the SNP reduced the transactivation activity of NKX2.5 by 20% in multiple independent experiments (n=9).
4. Discussion
In this study, we identified a novel 2 bp insertion mutation, c.512InsGC p.170X, in NKX2.5 that causes ASDs, AVB, and ventricular noncompaction as well as syncope and sudden death. Multiple pieces of evidence indicate that c.512InsGC is a pathogenic mutation. First, c.512InsGC co-segregates with CHD in the family. Second, the mutation was not identified in 200 normal controls. Third, the c.512InsGC insertion is a frame-shift mutation and generates a truncated NKX2.5 protein that deletes the C-terminal 154 amino acids containing the second half of the HD domain and the entire NK2 domain. Forth, functional studies showed that contrary to wild type NKX2.5, the mutant NKX2.5 protein generated by c.512InsGC was distributed in both the cytoplasm and nuclei, and completely lost the transcription activation activity.
A novel finding from this study is that NKX2.5 mutation is associated with ventricular noncompaction. Ventricular noncompaction is sometimes referred to as “ventricular hypertrabeculation” and is a cardiomyopathy which may be caused by failed compaction of trabeculated muscle structures due to arrest of normal embryogenesis of the endocardium and myocardium [26]. Our finding is consistent with results from knockout mice with ventricular-restricted deletion of NKX2.5 [27]. These NKX2.5 knockout mice exhibited massive trabecular muscle overgrowth. To the best of our knowledge, this is the first time that ventricular noncompaction was found in a human NKX2.5 mutation carrier. These results significantly expand the phenotypic spectrum of NKX2.5 mutations. The molecular mechanism by which a NKX2.5 mutation causes ventricular noncompaction is unknown. Ventricular noncompaction is a cardiac disorder associated with mutations in sarcomere genes [28]. NKX2.5 may regulate expression of some sarcomere genes, facilitating development of ventricular noncompaction.
NKX2.5 mutation c.512InsGC is associated with syncope and sudden death. Two other mutations, Q170X identified in one ASD family and a double mutation Q170X/Q198X in another ASD family, were also previously reported to be associated with sudden death in families [29].
The c.512InsGC insertion arose de novo. This is the second de novo mutation identified in NKX2.5. The first de novo mutation in NKX2.5 was a one bp C insertion at codon 167, and was identified in a single patient with ASDs and AVDs [23].
We failed to identify any NKX2.5 mutation in 125 Chinese Han patients with CHD, suggesting that NKX2.5 mutations are rare in sporadic CHD cases. Our results are more consistent with several previously-reported studies [21, 30-34].
In conclusion, this study identified a novel, de novo mutation in NKX2.5 that is associated not only with ASDs, AVB, syncope, and sudden death, but also with ventricular noncompaction. The results provide insights into the molecular pathogenic mechanism of ventricular noncompaction.
Acknowledgments
We thank the patients and family members for their support of this study, and Dr. X. Yang for help. This work was supported by the China National Natural Science Foundation (NSF30670857), an NIH grant (R01 HL094498), a Hubei Province Natural Science Key Program (2008CDA047), the China National Basic Research Program (973 Program 2007CB512002), a National Natural Science Foundation grant of China (30800457) and a Key Academic Program Leader Award of Wuhan City (200951830560).
Footnotes
Competing interests All authors have declared no conflict of interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pabst S, Wollnik B, Rohmann E, Hintz Y, Glänzer K, Vetter H, Nickenig G, Grohé C. A novel stop mutation truncating critical regions of the cardiac transcription factor NKX2-5 in a large family with autosomaldominant inherited congenital heart disease. Clin Res Cardiol. 2008;97:39–42. doi: 10.1007/s00392-007-0574-0. [DOI] [PubMed] [Google Scholar]
- 2.Elliott DA, Kirk EP, Yeoh T, Chandar S. Cardiac Homeobox Gene NKX2-5 Mutations and Congenital Heart Disease. JACC. 2003;41:2072–2075. doi: 10.1016/s0735-1097(03)00420-0. [DOI] [PubMed] [Google Scholar]
- 3.Hirayama-Yamada K, Kamisago M, Akimoto Kaoru. Phenotypes With GATA4 or NKX2.5 Mutations in Familial Atrial Septal Defect. American Journal of Medical Genetics. 2005;135A:47–52. doi: 10.1002/ajmg.a.30684. [DOI] [PubMed] [Google Scholar]
- 4.Ching Y-H, Ghosh TK, Cross SJ, Packham EA. Mutation in myosin heavy chain 6 causes atrial septal defect. Nature Genet. 2005;37:423–428. doi: 10.1038/ng1526. [DOI] [PubMed] [Google Scholar]
- 5.Kirk EP, Sunde M, Costa MW, Rankin SA, Wolstein O. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet. 2007;81:280–291. doi: 10.1086/519530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Matsson H, Eason J, Bookwalter CS, Klar J, Gustavsson P. Alpha-cardiac actin mutations produce atrial septal defects. Hum Molec Genet. 2008;17:256–265. doi: 10.1093/hmg/ddm302. [DOI] [PubMed] [Google Scholar]
- 7.Stanczak P, Witecka J, Szydlo A, Gutmajster E, Lisik M. Mutations in mammalian tolloid-like 1 gene detected in adult patients with ASD. Europ J Med Genet. 2009;17:344–351. doi: 10.1038/ejhg.2008.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bruneau BG, Nemer G, Schmitt JP. A Murine Model of Holt-Oram Syndrome Defines Roles of the T-Box Transcription Factor Tbx5 in Cardiogenesis and Disease. Cell. 2001;106:709–721. doi: 10.1016/s0092-8674(01)00493-7. [DOI] [PubMed] [Google Scholar]
- 9.Fan C, Liu M, Wang Q. Functional Analysis of TBX5 Missense Mutations Associated with Holt-Oram Syndrome. J Biol Chem. 2003;278:8780–8785. doi: 10.1074/jbc.M208120200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toko H, Zhu W, Takimoto E. Csx/Nkx2-5 Is Required for Homeostasis and Survival of Cardiac Myocytes in the Adult Heart. The Journal of Biological Chemistry. 2002;277:24735–24743. doi: 10.1074/jbc.M107669200. [DOI] [PubMed] [Google Scholar]
- 11.Prall OW, Menon MK, Solloway MJ. An Nkx2-5/Bmp2/Smad1 negative feedback loop controls second heart field progenitor specification and proliferation. Cell. 2007;128:947–959. doi: 10.1016/j.cell.2007.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joziasse rC, Smagt JJvd, Smith K. Genes in congenital heart disease: atrioventricular valve formation. Basic Res Cardiol. 2008;103:216–227. doi: 10.1007/s00395-008-0713-4. [DOI] [PubMed] [Google Scholar]
- 13.Riazi1 AM, Takeuchi JK, Hornberger LK. NKX2-5 Regulates the Expression of b-Catenin and GATA4 in Ventricular Myocytes. PLoS ONE. 2009;4:e5698. doi: 10.1371/journal.pone.0005698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tu C-T, Yang T-C, Tsai H-J. Nkx2.7 and Nkx2.5 Function Redundantly and Are Required for Cardiac Morphogenesis of Zebrafish Embryos. PLoS ONE. 2009;4:e4249. doi: 10.1371/journal.pone.0004249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ueyama T, Kasahara H, Ishiwata T. Myocardin Expression Is Regulated by Nkx2.5, and Its Function Is Required for Cardiomyogenesis. MOLECULAR AND CELLULAR BIOLOGY. 2003;23:9222–9232. doi: 10.1128/MCB.23.24.9222-9232.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasahara H, Usheva A, Ueyama T. Characterization of Homo- and Heterodimerization of Cardiac Csx/Nkx2.5 Homeoprotein. THE JOURNAL OF BIOLOGICAL CHEMISTRY. 2001;276:4570–4580. doi: 10.1074/jbc.M004995200. [DOI] [PubMed] [Google Scholar]
- 17.Lee Y, Shioi T, Kasahara H. The Cardiac Tissue-Restricted Homeobox Protein Csx/Nkx2.5 Physically Associates with the Zinc Finger Protein GATA4 and Cooperatively Activates Atrial Natriuretic Factor Gene Expression. Mol Cell Biol. 1998;18:3120–3129. doi: 10.1128/mcb.18.6.3120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hiroi Y, Kudoh S, Monzen K. Tbx5 associates with Nkx2-5 and synergistically promotes cardiomyocyte differentiation. Nature Genetics. 2001;28:276–280. doi: 10.1038/90123. [DOI] [PubMed] [Google Scholar]
- 19.Watada H, Mirmira RG, Kalamaras J. Intramolecular control of transcriptional activity by the NK2-specific domain in NK-2 homeodomain proteins. PNAS. 2000;97:9443–9448. doi: 10.1073/pnas.97.17.9443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bartlett H, Veenstra GJC, Weeks DL. Examining the Cardiac NK-2 Genes in Early Heart Development. Pediatr Cardiol. 2010;31:335–341. doi: 10.1007/s00246-009-9605-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stallmeyer B, Fenge H, Nowak-Göttl U. Mutational spectrum in the cardiac transcription factor gene NKX2.5 (CSX) associated with congenital heart disease. Clinical Genetics. 2010 doi: 10.1111/j.1399-0004.2010.01422.x. [DOI] [PubMed] [Google Scholar]
- 22.Goldmuntz E, Geiger E, Benson DW. NKX2.5 Mutations in Patients With Tetralogy of Fallot. Circulation. 2001;104:2565–2568. doi: 10.1161/hc4601.098427. [DOI] [PubMed] [Google Scholar]
- 23.Benson D. Woodrow, Silberbach G. Michael, Kavanaugh-McHugh Ann. Mutations in the cardiac transcription factor NKX2.5 affect diverse cardiac developmental pathways. J Clin Invest. 1999;104:1567–1573. doi: 10.1172/JCI8154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Zeng JQ, Gang Wan. Construction of siRNA/miRNA expression vectors based on a one-step PCR process. BMC Biotechnology. 2009;9:53. doi: 10.1186/1472-6750-9-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan C, Chen Q, Wang Q. Functional role of transcriptional factor TBX5 in pre-mRNA splicing and Holt-Oram syndrome via association with SC35. J Biol Chem. 2009;284:25653–25663. doi: 10.1074/jbc.M109.041368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zambrano E, Marshalko SJ, Jaffe CC, Hui1 P. Isolated Noncompaction of the Ventricular Myocardium: Clinical and Molecular Aspects of a Rare Cardiomyopathy. Lab Invest. 2002;82:117–122. doi: 10.1038/labinvest.3780404. [DOI] [PubMed] [Google Scholar]
- 27.Pashmforoush M, Lu JT, Chen H, Amand TS, Kondo R. Nkx2-5 Pathways and Congenital Heart Disease. Cell. 2004;117:373–386. doi: 10.1016/s0092-8674(04)00405-2. [DOI] [PubMed] [Google Scholar]
- 28.Hoedemaekers YM, Caliskan K, Michels M, Frohn-Mulder I, van der Smagt JJ, Phefferkorn JE, Wessels MW, ten Cate FJ, Sijbrands EJG, Dooijes D, et al. The Importance of Genetic Counseling, DNA Diagnostics, and Cardiologic Family Screening in Left Ventricular Noncompaction Cardiomyopathy. Circ-Cardiovasc Gene. 2010;3(3):232–U243. doi: 10.1161/CIRCGENETICS.109.903898. [DOI] [PubMed] [Google Scholar]
- 29.Schott J-J, Benson DW, Basson CT. Congenital Heart Disease Caused by Mutations in the Transcription Factor NKX2-5. Science. 1998;281:108–111. doi: 10.1126/science.281.5373.108. [DOI] [PubMed] [Google Scholar]
- 30.Posch Maximilian G., Perrot Andreas, Schmitt K. Mutations in GATA4, NKX2.5, CRELD1, and BMP4 Are Infrequently Found in Patients With Congenital Cardiac Septal Defects. American Journal of Medical Genetics Part A. 2008;146A:251–253. doi: 10.1002/ajmg.a.32042. [DOI] [PubMed] [Google Scholar]
- 31.Esposito Giorgia, Grutter Giorgia, Drago Fabrizio, Costa Mauro W., De Santis Antonella, Bosco Giovanna, Marino Bruno, Bellacchio Emanuele, Lepri Francesca, Harvey Richard P., et al. Molecular Analysis of PRKAG2, LAMP2, and NKX2-5 Genes in a Cohort of 125 Patients With Accessory Atrioventricular Connection. Am J Med Genet Part A. 2009;149A:1574–1577. doi: 10.1002/ajmg.a.32907. [DOI] [PubMed] [Google Scholar]
- 32.Wei-min Z, Xiao-feng L, Zhong-yuan M. GATA4 and NKX2.5 gene analysis in Chinese Uygur patients with congenital heart disease. Chin Med J (Engl) 2009;122:416–419. [PubMed] [Google Scholar]
- 33.Zhang W, Li X, Shen A. Screening NXK2.5 Mutation in a Sample of 230 Han Chinese Children with Congenital Heart Diseases. Genet Test Mol Biomarkers. 2009;13:159–162. doi: 10.1089/gtmb.2008.0044. [DOI] [PubMed] [Google Scholar]
- 34.Gioli-Pereira L, Pereira AC, Mesquita SM. NKX2.5 mutations in patients with non-syndromic congenital heart disease. International Journal of Cardiology. 2010;138:261–265. doi: 10.1016/j.ijcard.2008.08.035. [DOI] [PubMed] [Google Scholar]