

Abstract
We report an approach to the fabrication of superhydrophobic thin films that is based on the ‘reactive’ layer-by-layer assembly of azlactone-containing polymer multilayers. We demonstrate that films fabricated from alternating layers of the azlactone functionalized polymer poly(2-vinyl-4,4-dimethylazlactone) (PVDMA) and poly(ethyleneimine) (PEI) exhibit micro- and nanoscale surface features that result in water contact angles in excess of 150º. Our results reveal that the formation of these surface features is (i) dependent upon film thickness (i.e., the number of layers of PEI and PVDMA deposited) and (ii) that it is influenced strongly by the presence (or absence) of cyclic azlactone-functionalized oligomers that can form upon storage of the 2-vinyl-4,4-dimethylazlactone (VDMA) used to synthesize PVDMA. For example, films fabricated using polymers synthesized in the presence of these oligomers exhibited rough, textured surfaces and superhydrophobic behavior (i.e., advancing contact angles in excess of 150º). In contrast, films fabricated from PVDMA polymerized in the absence of this oligomer (e.g., using freshly distilled monomer) were smooth and only moderately hydrophobic (i.e., advancing contact angles of ~75º). The addition of authentic, independently synthesized oligomer to samples of distilled VDMA at specified and controlled concentrations permitted reproducible fabrication of superhydrophobic thin films on the surfaces of a variety of different substrates. The surfaces of these films were demonstrated to be superhydrophobic immediately after fabrication, but they became hydrophilic after exposure to water for six days. Additional experiments demonstrated that it was possible to stabilize and prolong the superhydrophobic properties of these films (e.g., advancing contact angles in excess of 150° even after complete submersion in water for at least six weeks) by exploiting the reactivity of residual azlactones to functionalize the surfaces of the films using hydrophobic amines (e.g., aliphatic or semi-fluorinated aliphatic amines). Our results demonstrate a straightforward and substrate-independent approach to the design of superhydrophobic and reactive polymer-based coatings of potential use in a broad range of fundamental and applied contexts.
Introduction
The term ‘superhydrophobic’ is generally used to describe surfaces and interfaces that exhibit advancing water contact angles in excess of 150° with low contact angle hysteresis.1–5 These properties generally cause water droplets to bead up and roll off of (rather than adhere to or spread on) the surface of a material and, thus, result in surfaces and interfaces that can completely resist wetting by aqueous media. Methods for the fabrication or functionalization of superhydrophobic surfaces are of significant interest because of the potential utility of these non-wetting materials in a broad range of consumer, industrial, and medically-oriented contexts (e.g., the design of ‘self-cleaning’ surfaces and textiles, new non-fouling surfaces, and membranes for oil/water separation, etc.). 1–5
The non-wetting behavior of superhydrophobic surfaces is generally understood to arise from a combination of two physicochemical factors: (i) the presence (and type) of micro/nanoscale surface roughness and (ii) the presence of surface functionality that is hydrophobic in nature (e.g., surfaces presenting fluorinated alkyl groups, etc).1 As a result, many approaches to the fabrication of superhydrophobic materials make use of methods for the introduction of hierarchical micro- and nanostructure to hydrophobic surfaces or methods for the deposition of hydrophobic molecules on surfaces that already display micro/nanostructured features. Methods such as chemical vapor deposition, various approaches to lithography, the deposition or dip-coating of nanoparticles, electrospinning, and layer-by-layer assembly have been used to fabricate superhydrophobic coatings on a variety of different types of surfaces.1–5 While each of these approaches has potential advantages with respect to the needs or constraints of a given application, several of these methods require the use of complex and expensive instrumentation or require different types of surface treatments that can involve the exposure of substrates to high temperatures (thereby limiting the types of substrates to which these methods can be applied). Straightforward and substrate-independent methods for the fabrication of superhydrophobic coatings under mild conditions (e.g., room temperature) could provide opportunities for the assembly of superhydrophobic surfaces on a broader range of substrates. Here, we report a solution-based, layer-by-layer approach to the fabrication of reactive, covalently crosslinked, and superhydrophobic thin films. These thin films possess both micro- and nanoscale surface features and chemically reactive groups that can be functionalized under mild conditions to produce films that retain their superhydrophobic properties for prolonged periods.
Past studies demonstrating methods for the layer-by-layer assembly6 of thin polymer films and composites have focused largely on the alternate and repetitive adsorption of oppositely charged polyelectrolytes on surfaces.7–11 These methods typically make use of aqueous solutions of polymer and result in films (‘polyelectrolyte multilayers’, or PEMs) that are assembled through multivalent electrostatic interactions. These methods are particularly useful for the fabrication of polymer-based surface coatings because they (i) permit incorporation of a broad range of materials, including components that can impart surface roughness (e.g., nanoparticles, carbon nanotubes, etc.), (ii) permit molecular-level and nanometer-scale control over film thickness and chemical composition, and (iii) can be deposited on a range of different types of materials, including substrates with complex or irregular geometries. 7–11 Layer-by-layer approaches have consequently been used to fabricate polymer thin films and composites of interest in a broad range of applications.7–11 Of particular relevance to the work reported here, several past studies have demonstrated layer-by-layer approaches to the fabrication of superhydrophobic surfaces,12–24 for example, by incorporating nanoparticles 12,13,19,21,23,24 or carbon nanotubes22 as structural elements of the films during assembly. While these past studies demonstrate the potential of layer-by-layer methods for the fabrication of superhydrophobic thin films, we note again that these polyelectrolyte-based approaches are generally confined to use with aqueous solutions and, thus, may not be useful for the fabrication of films on water-soluble substrates or substrates that react with (or are degraded by) water. In addition, many, but not all,15 PEM-based approaches to the design of superhydrophobic coatings have required several different surface treatments, including procedures such as chemical vapor deposition, to deposit non-water soluble, hydrophobic small molecules (e.g., aliphatic or fluorinated silanes) on the films to render the surfaces hydrophobic.12,13,18,19,21 Finally, we note that PEMs are assembled via weak electrostatic interactions that can be disrupted under certain environmental conditions. The work reported here makes use of an organic solvent-based approach to the layer-by-layer assembly of chemically crosslinked thin films that can be rapidly functionalized post-fabrication by treatment with organic solutions of hydrophobic primary amines.
We have described in several recent reports a ‘reactive’ layer-by-layer approach25 to the assembly of covalently crosslinked multilayered films that exploits the versatile reactivity of azlactone-functionalized polymers.26–30 Polymers bearing azlactone functionality, such as poly(2-vinyl-4,4-dimethylazlactone) (PVDMA, Eq. 1), are particularly useful for this approach because they react rapidly with primary amine-functionalized nucleophiles (Eq. 1).31 We have demonstrated that PVDMA can be deposited alternately on surfaces or interfaces with primary amine-functionalized polymers, such as branched poly(ethyleneimine) (PEI), to assemble covalently crosslinked polymer multilayers.26–30 The results of these past studies establish the layer-by-layer nature of the growth of these crosslinked multilayers on a broad range of surfaces and interfaces (e.g., on planar surfaces,26,27 on topologically complex substrates,26,29 and at interfaces formed between two immiscible liquids28), and that it is possible to assemble these multilayers using a broad range of organic solvents and optimized fabrication procedures.26–30 These past studies also reveal that the resulting polymer multilayers contain residual, unreacted azlactone functionality that is accessible for further reaction post-fabrication. These films thus provide a general and facile approach to the modification of surfaces with a broad range of chemical functionality.
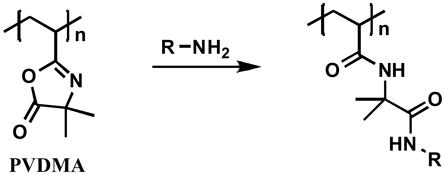 |
Eq. 1 |
The work reported here builds upon observations that arose during recent efforts in our laboratory to fabricate, characterize, and isolate freestanding PEI/PVDMA films. In a recent study, we reported that the layer-by-layer deposition of ~50 to 100 layer pairs (or ‘bilayers’) of PEI and PVDMA resulted in optically opaque films having a range of different micro- and nanoscale surface features (e.g., as characterized by scanning electron microscopy, SEM).30 Here, we describe an investigation into the origin of these observations and the development of methods that can be used to control surface roughness and fabricate films with specific surface properties (e.g., hydrophobic, hydrophilic, and superhydrophobic behavior, and films that transition in a time-dependent manner from superhydrophobic to hydrophilic upon prolonged exposure to aqueous media). We demonstrate that the roughness of these films (and thus their superhydrophobic properties) arises from the presence of cyclic azlactone-functionalized oligomers in the monomer used to synthesize PVDMA, and we report methods for the synthesis and intentional addition of these oligomers that can be used to fabricate surfaces with reproducible surface features and contact angles. Finally, we demonstrate that the superhydrophobic properties of these films are also influenced by the chemical functionality at the surface of the film (e.g., as dictated by the choice of the last polymer deposited during layer-by-layer fabrication or by post-fabrication treatment of residual azlactone groups with amine-based nucleophiles). Post-fabrication treatment with aliphatic or semi-fluorinated aliphatic amines yields films that are stable and remain superhydrophobic even after complete submersion in water for at least six weeks.
Materials and Methods
Materials
Branched poly(ethyleneimine) (PEI, MW = 25,000), reagent grade acetone, DMSO, 2,2′-azoisobutyronitrile (AIBN), trifluoroaceticacid (TFA), and 4,4,5,5,6,6,7,7,8,8,9,9,10,10,11, 11,11-heptadecafluoroundecylamine (HDFA) were purchased from Sigma Aldrich (Milwaukee, WI). Reagent grade THF and glass microscope slides were purchased from Fischer Scientific (Pittsburgh, PA). The monomer 2-vinyl-4,4-dimethylazlactone (VDMA) was a kind gift from Dr. Steven M. Heilmann (3M Corporation, Minneapolis, MN). Decylamine was purchased from TCI America (Portland, OR). All materials were used as received without further purification unless noted otherwise. Compressed air used to dry films and coated substrates was filtered through a 0.4 μm membrane syringe filter.
General Considerations
Gel permeation chromatography (GPC) was performed using a GPCmax-VE2001 Solvent/Sample module (Viscotek Corp., Houston, TX) and two PlusPore Organic GPC Columns (Polymer Laboratories, Amherst, MA) equilibrated to 40 °C. THF was used as the eluent at a flow rate of 1.0 mL/min. Data were collected using the refractive index detector of a Viscotek TDA 302 triple detector array and processed using the OmniSEC 4.5 software package. Molecular weights and polydispersities are reported relative to monodisperse polystyrene standards. Digital images were acquired using a Nikon Coolpix 4300 digital camera. Scanning electron micrographs were acquired on a LEO DSM 1530 scanning electron microscope at an accelerating voltage of 3 kV. Samples were coated with a thin layer of gold using a sputterer (30 s at 45 mA, 50 mTorr) prior to imaging. LC-MS data were obtained using a Shimadzu LCMS-2010 equipped with two LC-10ADvp pumps, an SCL-10Avp controller, an SIL-10ADvp autoinjector, an SPD-M10Avp UV/vis diode array detector, and a single quadrupole analyzer (for ESI). A Supelco 15 cm × 2.1 mm C18 wide-pore column was used for all LC-MS work. Standard RP-HPLC conditions for LC-MS were as follows: flow rate = 200 μL/min; mobile phase A = 0.4% formic acid in water; mobile phase B = 0.2% formic acid in acetonitrile. Static and dynamic water contact angles were measured using a Dataphysics OCA 15 Plus instrument and ImageJ (NIH). Glass substrates were cleaned with acetone, ethanol, methanol, and deionized water and dried under a stream of compressed air prior to the fabrication of multilayered films.
Synthesis of Poly(2-vinyl-4,4-dimethylazlactone) (PVDMA)
VDMA used for the polymerization of PVDMA was either distilled and stored with triethylamine (TEA) prior to polymerization (clear liquid) or was used without distillation (viscous, yellow liquid) (see text). The amounts of each reagent used for the polymerization of distilled or non-distilled monomer and the resulting polymer characteristics are noted below. VDMA was passed through a phenolic inhibitor removal resin followed by passage through a short plug of silica gel prior to polymerization. AIBN was weighed into a 25 mL round-bottomed flask equipped with a stir bar. Ethyl acetate was added and the solution was stirred to dissolve the AIBN. VDMA was added to the flask, the flask was capped with a septum, and the solution was purged with N2 for 10 minutes. The solution was stirred under N2 at 60 °C for 24 hours, after which time the viscous reaction mixture was cooled to room temperature and CH2Cl2 (5 mL) was added to the flask. The polymer was precipitated into hexanes to yield a white solid. The polymer was filtered and washed with hexanes, then redissolved in CH2Cl2 and precipitated once more in hexanes. PVDMA was isolated as a white solid. Polymerization using distilled VDMA: AIBN (11.8 mg, 0.0719 mmol), VDMA (1.0 g, 7.19 mmol), ethyl acetate (3 mL). 1H-NMR (300 MHz, CDCl3): δ = 1.37 (br s, (-CH3)2), 1.62–2.1 (br m, -CH2CH-), 2.69 (br s, -CH2CH-). FT-IR (ATR, cm−1): 2980–2900 (C-H), 1820 (lactone C=O), 1672 (C=N). Mn: 55,132; PDI = 3.6. Polymerization using undistilled VDMA: AIBN (27.2 mg, 0.166 mmol), VDMA (2.4 g, 17.4 mmol), ethyl acetate (6 mL). 1H-NMR (300 MHz, CDCl 3): δ = 1.37 (br s, (-CH3)2), 1.62–2.1 (br m, -CH2CH-), 2.69 (br s, -CH2CH-). FT-IR (ATR, cm−1): 2980–2900 (C-H), 1820 (lactone C=O), 1672 (C=N). Mn: 3,705; PDI = 2.4. For PVDMA polymerized in the presence of synthesized oligomer (synthesized as described below), distilled VDMA was used and oligo(VDMA) was added to the polymerization mixture at 7% (w/w) relative to VDMA. The polymerization was then carried out in analogy to the procedures described above. AIBN (11.8 mg, 0.0719 mmol), VDMA (1.0095 g, 7.19 mmol), oligo(VDMA) (71.5 mg, 0.504 mmol), ethyl acetate (3 mL). 1H-NMR (300 MHz, CDCl3): δ = 1.37 (br s, (-CH3)2), 1.62–2.1 (br m, -CH2CH-), 2.69 (br s, -CH2CH-). FT-IR (ATR, cm−1): 2980–2900 (C-H), 1820 (lactone C=O), 1672 (C=N). Mn: 20,724; PDI = 2.4.
Synthesis of Oligo(2-vinyl-4,4-dimethylazlactone)
Samples of oligo(VDMA) were synthesized in analogy to methods reported previously by Heilmann.32 Briefly, VDMA (1.0061 g, 7.2 mmol) was weighed into a 10 mL round-bottomed flask equipped with a stir bar. Ethyl acetate (1.7 mL) was added and stirred to dissolve the VDMA. TFA (37.5 μL, 0.505 mmol) was added, the flask was capped with a septum, and the solution was stirred at 65 °C for 24 hours. The reaction mixture turned yellow within 10 mins of stirring. After 24 hours, the dark orange mixture was precipitated into ~20 volumes of heptane and filtered to yield a yellow solid. The solid was redissolved in acetone (~1 mL) and precipitated a second time in heptane to give a yellow solid in 88% yield. 1H-NMR (300 MHz, CDCl3): δ = 1.2–1.8 (br m), 1.8–3.05 (br m), 3.57 (br s), 3.86 (br s). FT-IR (ATR, cm−1): 2974–2854 (C-H), 1816 (C=O, azlactone), 1735 (C=O, carboxylic acid of hydrolyzed azlactone), 1672 (C=O, amide of hydrolyzed azlactone). Mn: 806; PDI = 1.09.
Hydrolysis of Oligo(2-vinyl-4,4-dimethylazlactone)
Samples of oligo(VDMA) used for characterization by LC-MS were hydrolyzed prior to characterization using a protocol similar to that reported previously by Heilmann.32 Oligo(VDMA) (50.9 mg, 0.366 mmol) was weighed into a 4 dram vial equipped with a stir bar and dissolved in THF (0.5 mL). H2O (100 μL) and TFA (9 μL) were added to the vial, the vial was capped, and the solution was stirred at room temperature for 8 days. The mixture was precipitated into diethyl ether and filtered to produce a light yellow solid in 57% yield. FT-IR (ATR, cm−1): 2974–2854 (C-H), 1735 (C=O, hydrolyzed azlactone), 1672 (C=N, azlactone).
Fabrication of Multilayered Films
Solutions of PEI or PVDMA used for the fabrication of multilayered films were prepared in acetone (20 mM with respect to the molecular weight of the polymer repeat unit). Films were deposited manually layer-by-layer on glass substrates according to the following general protocol optimized and characterized extensively in our past studies of this acetone-based system:26–30 1) Substrates were submerged in a solution of PEI for 20 seconds, 2) substrates were removed and immersed in an initial acetone bath for 20 seconds followed by a second acetone bath for 20 seconds, 3) substrates were submerged in a solution of PVDMA for 20 seconds, and 4) substrates were rinsed in the manner described in step 2. This cycle was repeated until the desired number of PEI/PVDMA layers was reached (typically, 50–100 bilayers; the term ‘bilayer’ as used here refers to a single PEI/PVDMA layer pair). For these experiments, the concentrations of polymer solutions were maintained by addition of acetone as needed to compensate for solvent evaporation or by the replacement of solutions of polymer with fresh solutions (i) after every dipping cycle or (ii) after every 25 dipping cycles. Films were characterized or used in subsequent experiments immediately or were dried under a stream of filtered, compressed air and stored in a vacuum desiccator until use. All films were fabricated at ambient room temperature.
Post-Fabrication Functionalization of PEI/PVDMA Films
PEI/PVDMA films were functionalized post-fabrication by immersing film-coated substrates in solutions of an amine-functionalized nucleophile (e.g., decylamine or HDFA, 1 mM in THF) at room temperature for 20 hours using methods similar to those reported in past studies.26–30 Films were rinsed with THF and acetone after functionalization and dried with filtered air. Films were stored in a vacuum desiccator after fabrication and functionalization.
Contact Angle Measurements
Contact angle measurements were made using a Dataphysics OCA 15 Plus instrument with an automatic liquid dispenser at ambient temperature. Static water contact angles were measured using a 4 μL droplet of deionized (18 MΩ) water in three different locations on a film measuring approximately 3 cm × 1 cm. Advancing and receding contact angles were measured during growth and shrinkage of 10 μL water droplets in three different locations on the film. Data are reported as the average (with standard deviation) of these individual measurements.
Results and Discussion
Fabrication and Characterization of Superhydrophobic PEI/PVDMA Films
We recently reported an approach to the fabrication and isolation of freestanding and reactive polymer multilayers30 based on methods developed, characterized, and optimized by our group for the ‘reactive’ layer-by-layer assembly of PVDMA and PEI on a broad range of surfaces and interfaces. 26–29 During the course of these past studies, we sometimes observed the appearance of these films to change from uniform and optically clear to hazy or opaque after the deposition of ~25 bilayers of PEI/PVDMA. Additional characterization of these films by SEM revealed these changes in optical appearance to result from the presence of microscale and nanoscale topographic features on the surfaces of the films.30 The work reported here sought to investigate the origin of this surface roughness and determine whether this ‘reactive’ layer-by-layer process could be exploited to fabricate films with physicochemical properties that could be used to control the wettability of surfaces coated with these materials.
During our initial investigations, we observed large variations in the physical properties (e.g., roughness) of these films depending on the batch of polymer used to fabricate the films and, more specifically, whether or not the monomer used to synthesize the polymer was freshly distilled prior to use. For example, the fabrication of 100-bilayer PEI/PVDMA films using PVDMA that was synthesized using non-distilled VDMA (referred to hereafter as PVDMA ND) resulted in films that were opaque in appearance, as observed in our past studies and shown in the digital picture in Figure 1A. In sharp contrast, 100-bilayer PEI/PVDMA films fabricated using PVDMA synthesized using freshly distilled VDMA (referred to hereafter as PVDMADIST) resulted in clear, transparent films (as shown in Figure 1B). Closer inspection of the surface morphologies of these two films using SEM revealed the films to have substantially different surface characteristics. Figures 2A and 2B show top-down, high-resolution SEM images of 100-bilayer PEI/PVDMA films fabricated using PVDMAND and PVDMADIST, respectively. These images demonstrate that the surfaces of PEI/PVDMAND films are textured with micro- and nanoscale features, but that the surfaces of PEI/PVDMADIST films are smooth and relatively featureless at this magnification.
Figure 1.

Digital photographs of 100-bilayer PEI/PVDMA films fabricated using PVDMAND (A), PVDMADIST (B), PVDMAOLIGO (C), and PVDMADIST doped with 7% (w/w) synthesized oligomer (D).
Figure 2.

Top-down SEM images of 100-bilayer PEI/PVDMA films fabricated using PVDMAND (A), PVDMADIST (B), PVDMAOLIGO (C), and PVDMADIST doped with 7% (w/w) synthesized oligomer (D). Scale bars = 2 μm.
The influence of these differences in surface morphology on the properties of these films was further manifest as large differences in the wettability of substrates coated with these two different polymer films. As shown in Figure 3, advancing water contact angles of PEI/PVDMAND films were measured to be greater than 150° (as noted above, this is the contact angle threshold generally used to classify a surface as superhydrophobic). The advancing contact angles of PEI/PVDMADIST films were significantly lower (~75°) and more closely resembled the contact angles measured for 10-bilayer PEI/PVDMA films (~65°) reported in past studies.26,27
Figure 3.

Contact angles measured for PEI/PVDMA films 100 bilayers thick fabricated using PEI and either PDVMAND, PVDMADIST, PVDMAOLIGO, or PVDMADIST doped with 7% (w/w) synthesized oligomers. Advancing contact angles are represented by the dark gray bars and receding contact angles are represented by the light gray bars.
The results described above demonstrate clearly that the history of the VDMA monomer used to synthesize PVDMA plays a significant role in determining both the morphology and the resulting physical properties of PEI/PVDMA polymer multilayers (the results in Figure 2 also demonstrate that these differences do not arise simply from the influence of solvent or other physical or mechanical aspects of our layer-by-layer approach to fabrication). Because non-distilled monomer (PVDMAND) yielded films with properties that are of potential interest in the context of developing new superhydrophobic surfaces, we conducted a series of subsequent experiments to characterize the composition of the monomer and determine whether we could establish well-defined procedures and conditions that could be used to fabricate superhydrophobic films reproducibly (that is, to design rational approaches for assembly that would not rely on the age or history of the monomer, etc.). The results of these additional investigations are described in the section below. Additional characterization of water contact angles and other surface properties are described in subsequent sections.
Influence of Azlactone-Functionalized Oligomers on PEI/PVDMA Film Morphology
Previous reports by Heilmann et al. have demonstrated that azlactone-functionalized vinyl monomers such as VDMA can undergo oligomerization reactions when stored for long periods of time in the absence of a proton scavenger, or when treated directly with catalytic amounts of acid.32 These reactions do not occur through a chain growth-type mechanism to yield short chains of PVDMA, but rather proceed by protonation of the azlactone nitrogen of VDMA followed by Michael addition to the olefin to form cyclic structures containing, most often, four, five, or six monomer units (although higher order oligomers can also be formed). A complete analysis of these oligomeric structures and a detailed discussion of the mechanism of formation of these oligomers have been reported previously (see also Figures S1 and S2 of the Supporting Information for the proposed mechanism and the structures of the oligomeric compounds that form).32 To determine whether our non-distilled supply of VDMA contained these oligomerization products, we added aliquots of a PVDMAND solution drop-wise to heptane (following a protocol reported by Heilmann et al. for the isolation of oligomers from VDMA)32 and observed the formation of a precipitate. Characterization of several different batches of monomer using this procedure revealed these samples to contain between 4% and 7% of this precipitate (w/w) depending upon the batch of monomer. Characterization of these precipitates using liquid chromatography-mass spectrometry (LC-MS) revealed a range of structures having the molecular weights shown in Table S1 of the Supporting Information. These molecular weights correspond to a mixture of VDMA-based oligomers composed of dimers through hexamers (with varying degrees of hydrolysis) that was similar to the results reported previously by Heilmann et al.32 Similar precipitation experiments using samples of freshly distilled VDMA monomer revealed these samples to be devoid of oligomers.
To investigate the potential influence of the oligomers identified above more completely, and to develop well-defined procedures for the fabrication of superhydrophobic films, we performed a series of experiments using samples of freshly distilled VDMA monomer and PVDMADIST (polymer synthesized from freshly distilled monomer) containing measured amounts of authentic, independently synthesized oligomer. We synthesized authentic samples of oligomer under controlled conditions by treating freshly distilled VDMA with TFA (see Materials and Methods for additional details).32 LC-MS characterization of samples of the resulting oligomers revealed them to be composed of a mixture of trimers, tetramers, pentamers, and hexamers (see Table S2 of the Supporting Information for additional details related to the characterization of these independently-synthesized oligomers). The composition of these mixtures was qualitatively similar to the mixtures of oligomers isolated from the non-distilled VDMA monomer (as described above) and to results reported previously by Heilmann et al. for the synthesis of these oligomers under similar controlled reaction conditions.32 These intentionally synthesized oligomers were used for all subsequent studies described below.
We next prepared two different samples of PVDMA containing these intentionally synthesized oligomers to characterize the influence of oligomer on the properties of our PEI/PVDMA films. The first sample was prepared by the conventional free radical polymerization of a solution of freshly distilled VDMA containing 7% (w/w) of the independently synthesized oligomer mixture (this sample is referred to hereafter as PVDMAOLIGO). The second sample was prepared by the addition of 7% (w/w) of oligomer to a sample of PVDMADIST (i.e., by the addition of oligomer to a sample of polymer synthesized previously using freshly distilled monomer). These two different PVDMA samples were then used to fabricate PEI/PVDMA films using procedures identical to those used to fabricate the films shown in Figures 1A and 1B.
The images in Figures 1C and 1D show digital pictures of PEI/PVDMA films 100 bilayers thick fabricated using either PEI and PVDMA OLIGO (Figure 1C) or PEI and PVDMADIST doped with 7% oligomer (Figure 1D). Inspection of these images reveals the visual appearance of films fabricated using PVDMAOLIGO (Figure 1C) to resemble more closely the appearance of films fabricated from PVDMAND (e.g., Figure 1A). Further inspection reveals the appearance of films assembled using PVDMADIST doped with 7% oligomer after polymerization to resemble more closely the appearance of films fabricated using PVDMADIST in the absence of added oligomer (e.g., Figure 1B). Further characterization of the morphologies of these films using SEM revealed that the surfaces of PEI/PVDMAOLIGO films (Figure 2C) were rough and had micro- and nanoscale features similar to those observed for PEI/PVDMA ND films (Figure 2A). In contrast, the surfaces of films fabricated using PEI and PVDMA DIST doped with 7% oligomer (Figure 2D) were smooth and relatively featureless, similar to films fabricated using PEI and PVDMADIST (Figure 2B). Characterization of the water contact angles of these films revealed that PEI/PVDMAOLIGO films (Figures 1C and 2C) were also superhydrophobic and exhibited advancing contact angles of ~155° (Figure 3). Films assembled using PVDMADIST doped with oligomer after polymerization (Figures 1D and 2D), however, exhibited contact angles similar to those of PEI/PDVMADIST films (advancing contact angle ~72°, as shown in Figure 3).
The results of the experiments above, when combined, demonstrate that the influence of added oligomer on film roughness arises substantially from its presence during the polymerization of PVDMA, and not simply from its presence in solution during film fabrication. These results thus suggest that the polymerization of VDMA in the presence of oligomer influences the structure and properties of PVDMA in ways that influence its behavior during assembly. We note, in this context, that despite in-depth characterization of the formation of oligomers in samples of VDMA, 32 the influence of these oligomers on the polymerization of VDMA has not, to our knowledge, been broadly characterized. One possibility is that these oligomers could be incorporated into the structure of a growing polymer during polymerization in ways that alter substantially the structure or solution behavior (e.g., conformation) of the resulting polymers and, thus, the manner in which they are deposited during fabrication. (For example, the development of surface roughness in polyelectrolyte-based multilayers has been attributed in past studies to changes in the conformations of polyelectrolytes that occur upon changes in the pH and ionic strength of polymer solutions used for film fabrication.)33–35 We note in this context, that these cyclic oligomers are not functionalized with vinyl groups (e.g., see structures shown in Figure S2 of the Supporting Information). We note further, however, that Heilmann et al. have proposed enol tautomers as intermediates during the formation of these oligomers32 and that, if present, these enol tautomers could potentially be incorporated into a growing polymer chain during free radical polymerization. Another possibility is that these oligomers could behave as chain transfer or chain termination agents during polymerization. Support for this possibility is provided by the results of additional GPC characterization and the observation that PVDMA polymerized in the presence of oligomers typically yields polymers with molecular weights (e.g., Mn <20,000 g/mol) that are lower than PVDMA polymerized in the absence of oligomers (e.g., Mn >50,000 g/mol) under otherwise identical conditions. Initial characterization by nuclear magnetic resonance spectroscopy did not reveal significant differences in PVDMA synthesized in the presence or absence of added oligomer. Additional experiments will be required to establish more completely the potential influence of these oligomers on polymerization kinetics and potential changes in polymer structure and/or solution conformation.
We also considered the possibility that low molecular weight PVDMA or free oligomer could potentially diffuse freely out of a film during fabrication and form crosslinked, particulate complexes upon contact with solutions of PEI during or after each dipping cycle used to assemble the films. Such complexes could then deposit on the surface of a film during subsequent deposition cycles and contribute to the formation of surface roughness. In this context, we note that the formation and intentional incorporation of particulate interpolyelectrolyte complexes into polyelectrolyte multilayers has been used to fabricate superhydrophobic PEMs.36 To investigate this possibility, we performed additional experiments in which the PEI solutions used during fabrication were exchanged with fresh PEI solutions after the deposition of every PEI layer to reduce substantially the potential for complex formation and/or re-deposition to occur. These experimental conditions also resulted in the formation of rough, textured surfaces. These results suggest that the formation of complexes in the PEI solutions either does not contribute significantly to the surface morphologies observed, or that the formation and re-deposition of the complexes occurs rapidly (e.g., on the time scale of the 20-second polymer deposition times used to fabricate these films). Finally, we note that the results of the experiments described above demonstrating that films fabricated using PVDMADIST solutions containing free, intentionally added oligomer are smooth and featureless are also not consistent with a mechanism for the formation of roughness that is based on the diffusion of oligomer or the formation of PEI/oligomer complexes. Although additional experiments will be required to fully elucidate the underlying mechanism for the formation of these rough surfaces, we do conclude that (i) the primary influence of the oligomer arises from its presence during the polymerization of VDMA, and (ii) that the addition of controlled amounts of intentionally synthesized oligomer to samples of freshly distilled VDMA can be used to fabricate rough and superhydrophobic films reproducibly. Additional characterization of the superhydrophobic properties of these films is discussed below.
Post-Fabrication Modification of PEI/PVDMA Films with Hydrophobic Amines: Fabrication of Stable Superhydrophobic Surfaces
Additional characterization of contact angles of the rough films described above revealed that they were substantially dependent upon the structure of the last polymer deposited during fabrication. For example, whereas films terminated with a layer of PVDMA exhibited contact angles in excess of 150° (as described above), droplets of water placed on the surfaces of films terminated with a final layer of PEI spread upon contact (i.e., static contact angles were approximately 40°). These results are also consistent with the layer-by-layer nature of the assembly process used to fabricate these films. Further characterization of PVDMA-terminated films, however, also revealed the superhydrophobic properties of these films to change over time (for example, upon prolonged exposure to water; discussed in greater detail below). These results, when combined, demonstrate that surface chemistry also plays an important role in defining the superhydrophobic behavior of these rough films.
The results of past studies demonstrate that the residual azlactone functionality in PEI/PVDMA films can be exploited to chemically modify the surfaces and properties of these materials by post-fabrication treatment with a range of small molecule amines.26–30 Our next experiments sought to determine whether the treatment of rough PEI/PVDMA films with hydrophobic amines could be used to either (i) increase the superhydrophobicity of these films or (ii) create ‘stable’ films that would retain their superhydrophobic properties for prolonged periods (relative to those described above) upon exposure to water. We selected the hydrophobic amines n-decylamine and heptadecafluoroundecylamine (HDFA) for these studies based on the past use of these and similar structural motifs for the preparation of hydrophobic and superhydrophobic surfaces.3,5
We fabricated 100-bilayer PEI/PVDMADIST and PEI/PVDMAOLIGO films on glass substrates and treated these films with either decylamine or HDFA (1 mM in THF; see Materials and Methods for additional details). Figure 4 shows a plot of the dynamic contact angles measured for unmodified, decylamine-treated, and HDFA-treated PEI/PVDMADIST and PEI/PVDMAOLIGO films. Treatment of PEI/PVDMADIST films with decylamine and HDFA resulted in a moderate increase in the hydrophobicity of the surfaces (i.e., the average advancing contact angle increased from ~75° to ~95°). We note that contact angles of ~95° are similar to contact angles measured for decylamine-functionalized 10-bilayer PEI/PVDMA films fabricated on silicon substrates that we reported previously.26,27 In contrast, the advancing contact angles of already-superhydrophobic PEI/PVDMAOLIGO films treated with either decylamine or HDFA did not increase measurably (we note, however, that treatment with these hydrophobic amines did result in slightly lower contact angle hysteresis; see Figure 4).
Figure 4.

Contact angles measured for unmodified, decylamine-treated, and HDFA-treated PEI/PVDMADIST and PEI/PVDMAOLIGO films. Advancing contact angles are represented by the dark gray bars and receding contact angles are represented by the light gray bars.
Although treatment of PEI/PVDMAOLIGO films with hydrophobic small molecules did not increase the contact angles, the long-term stability of the superhydrophobic properties of these amine-treated films was significantly improved relative to those of unmodified (i.e., azlactone-containing) films. For example, superhydrophobic PEI/PVDMAOLIGO films that were not modified by treatment with hydrophobic amines became water-absorbent after several days of submersion in water (e.g., after six days; see image in Figure 6D). This behavior is consistent with the ring-opening hydrolysis of unreacted azlactone functionality in these untreated materials and the subsequent time-dependent formation of hydrophilic carboxylate groups (discussed in more detail below). In contrast, the advancing contact angles of decylamine-treated and HDFA-treated PEI/PVDMAOLIGO films did not change significantly even after total submersion in water for at least six weeks. Figure 5 shows a plot of the dynamic contact angles of decylamine-functionalized and HDFA-functionalized films immediately after fabrication and after three or six weeks of immersion in a water bath at room temperature. The contact angle hysteresis increased slightly for decylamine-treated films over time in water and, consequently, water droplets did not roll off of these surfaces as readily as they did immediately after functionalization. The advancing and receding contact angles for PEI/PVDMAOLIGO films modified with HDFA, however, did not change substantially even after six weeks in water and water droplets rolled readily off of these films.
Figure 6.

(A–C) SEM images of 100-bilayer films fabricated from PEI and PVDMA OLIGO that were (A) unmodified, (B) treated with decylamine, or (C) treated with HDFA. The images were acquired after soaking the unmodified film in water for one week and the decylamine-treated and HDFA-treated films in water for six weeks (see text). (D–F) Images of water droplets (4 μL) on (D) an unmodified film that was immersed in water for one week, (E) a decylamine-treated film that was submerged in water for six weeks, and (F) an HDFA-treated film that was immersed in water for six weeks (F). Scale bars = 2 μm.
Figure 5.

Dynamic contact angles as a function of time for decylamine-treated and HDFA-treated PEI/PVDMAOLIGO films submerged in water. Advancing contact angles are represented by dark gray bars and receding contact angles are represented by light gray bars.
Further characterization of unmodified, decylamine-treated, and HDFA-treated films after submersion in water revealed that changes in contact angles were not a result of changes in the microstructure (i.e., roughness) of the surfaces of the films. The images in Figures 6A–C show top-down SEM images of an unmodified PEI/PVDMAOLIGO film that was submerged in water for one week (Figure 6A), and decylamine-modified (Figure 6B) and HDFA-functionalized (Figure 6C) PEI/PVDMAOLIGO films that were immersed in water for six weeks. Inspection of these images reveals that the micro- and nanoscale morphologies of the films were rough and textured, similar to that observed for PEI/PVDMAOLIGO films prior to treatment with the hydrophobic amines and exposure to water (see Figure 2C for comparison). Figures 6D–F show images of water droplets (4 μL) placed on unmodified, decylamine-treated, and HDFA-treated PEI/PVDMAOLIGO films that were soaked in water for either one week (for unmodified films) or six weeks (for treated films). As revealed by the absence of an observable water droplet in Figure 6D, unmodified PEI/PVDMA OLIGO films readily absorbed water after being exposed to water for one week.
The images shown in Figures 6E and 6F further demonstrate that decylamine-treated and HDFA-treated films continue to resist wetting by water, even after exposure to water for several weeks. We note that HDFA-treated PEI/PVDMAOLIGO films (Figure 6F) exhibited higher static contact angles than decylamine-treated films (Figure 6E) after six weeks in water and thus appear to be more durable over long periods of exposure to water. The results of these experiments demonstrate that exposure to water does not change substantially the structure or morphology of these covalently crosslinked multilayers, but that the water contact angles do change over time if the films are not treated with a hydrophobic amine. As noted above, residual azlactone functionality in PEI/PVDMA films will hydrolyze in the presence of water (or upon exposure to water vapor) to form carboxylic acid groups, and, it is therefore likely that the loss of superhydrophobicity observed for unmodified films exposed to water results from the formation of more hydrophilic functional groups on the surfaces of these films. Modification of these films with hydrophobic amines results in the formation of stable amide linkages (e.g., see Equation 1) that are less susceptible to hydrolysis and makes possible the fabrication of superhydrophobic surfaces that are stable when submerged in water for extended periods.
Summary and Conclusions
We have reported a facile and straightforward approach to the fabrication of superhydrophobic surface coatings that makes use of methods for the ‘reactive’ layer-by-layer assembly of azlactone-functionalized polymers. Our results demonstrate that polymer multilayers fabricated from PEI and PVDMA 100 bilayers thick exhibit micro- and nanoscale surface features that impart non-wetting, superhydrophobic behavior to film-coated substrates. These rough films exhibit advancing contact angles of ~155° without any additional surface treatment. The roughness of these films was attributed to the presence of azlactone-functionalized oligomers during the polymerization of PVDMA. Films fabricated using either (i) PVDMA synthesized in the absence of these oligomers or (ii) samples of PVDMA to which oligomers were added intentionally prior to fabrication resulted in smooth, featureless surfaces that were not superhydrophobic). Although rough PEI/PVDMA films exhibited superhydrophobic properties without any additional treatment, these properties were lost upon prolonged exposure to water. However, the reactivity of residual azlactone functionality in PEI/PVDMA films permitted facile post-fabrication modification with aliphatic and fluorinated aliphatic amines to produce surfaces that retained their superhydrophobic properties for extended periods of time (e.g., after at least six weeks of complete submersion in water).
The methods reported here do not require complicated, expensive, or harsh treatment processes, and should therefore facilitate the fabrication of superhydrophobic films on a wide range of different materials and substrates. In addition, because these methods are based on layer-by-layer assembly in organic solvents, this approach should be well suited to the assembly of superhydrophobic coatings on the surfaces of water-soluble or pH-sensitive materials that cannot be used as substrates using methods for the aqueous-based assembly of superhydrophobic polyelectrolyte multilayers.
Supplementary Material
Acknowledgments
Financial support was provided by the NSF (DMR-0520527) through a grant to the Materials Research Science and Engineering Center (MRSEC) at the University of Wisconsin, and the Alfred P. Sloan Foundation. This research made use of facilities associated with the UW Soft Materials Laboratory, which is funded in part by the NSF through the UW MRSEC and the UW Nanoscale Research Center (NSEC). We thank Dr. Steven M. Heilmann and Dr. Gerald K. Rasmussen (3M Corporation) for providing samples of 2-vinyl-4,4-dimethylazlactone. We thank Eric M. Saurer for assistance with scanning electron microscopy experiments. M.E.B. was funded in part by an NIH Chemistry Biology Interface Training Grant (NIGMS T32 GM008505).
References
- 1.Li XM, Reinhoudt D, Crego-Calama M. Chem Soc Rev. 2007;36:1350–1368. doi: 10.1039/b602486f. [DOI] [PubMed] [Google Scholar]
- 2.Roach P, Shirtcliffe NJ, Newton MI. Soft Matter. 2008;4:224–240. doi: 10.1039/b712575p. [DOI] [PubMed] [Google Scholar]
- 3.Zhang X, Shi F, Niu J, Jiang YG, Wang ZQ. J Mater Chem. 2008;18:621–633. [Google Scholar]
- 4.Nosonovsky M, Bhushan B. Curr Opin Colloid Interface Sci. 2009;14:270–280. [Google Scholar]
- 5.Crick CR, Parkin IP. Chem Eur J. 2010;16:3568–3588. doi: 10.1002/chem.200903335. [DOI] [PubMed] [Google Scholar]
- 6.Decher G. Science. 1997;277:1232–1237. [Google Scholar]
- 7.Bertrand P, Jonas A, Laschewsky A, Legras R. Macromol Rapid Comm. 2000;21:319–348. [Google Scholar]
- 8.Hammond PT. Adv Mater. 2004;16:1271–1293. [Google Scholar]
- 9.Peyratout CS, Dahne L. Angew Chem Int Ed. 2004;43:3762–3783. doi: 10.1002/anie.200300568. [DOI] [PubMed] [Google Scholar]
- 10.Tang ZY, Wang Y, Podsiadlo P, Kotov NA. Adv Mater. 2006;18:3203–3224. [Google Scholar]
- 11.Boudou T, Crouzier T, Ren K, Blin G, Picart C. Adv Mater. 2010;22:441–467. doi: 10.1002/adma.200901327. [DOI] [PubMed] [Google Scholar]
- 12.Soeno T, Inokuchi K, Shiratori S. Appl Surf Sci. 2004;237:543–547. [Google Scholar]
- 13.Zhai L, Cebeci FC, Cohen RE, Rubner MF. Nano Lett. 2004;4:1349–1353. [Google Scholar]
- 14.Zhang X, Shi F, Yu X, Liu H, Fu Y, Wang ZQ, Jiang L, Li XY. J Am Chem Soc. 2004;126:3064–3065. doi: 10.1021/ja0398722. [DOI] [PubMed] [Google Scholar]
- 15.Jisr RM, Rmaile HH, Schlenoff JB. Angew Chem Int Ed. 2005;44:782–785. doi: 10.1002/anie.200461645. [DOI] [PubMed] [Google Scholar]
- 16.Shi F, Wang ZQ, Zhang X. Adv Mater. 2005;17:1005–1009. [Google Scholar]
- 17.Zhao N, Shi F, Wang ZQ, Zhang X. Langmuir. 2005;21:4713–4716. doi: 10.1021/la0469194. [DOI] [PubMed] [Google Scholar]
- 18.Ji J, Fu JH, Shen JC. Adv Mater. 2006;18:1441–1444. [Google Scholar]
- 19.Bravo J, Zhai L, Wu ZZ, Cohen RE, Rubner MF. Langmuir. 2007;23:7293–7298. doi: 10.1021/la070159q. [DOI] [PubMed] [Google Scholar]
- 20.Ogawa T, Ding B, Sone Y, Shiratori S. Nanotechnology. 2007;18 [Google Scholar]
- 21.Zhang LB, Chen H, Sun JQ, Shen JC. Chem Mater. 2007;19:948–953. [Google Scholar]
- 22.Liao KS, Wan A, Batteas JD, Bergbreiter DE. Langmuir. 2008;24:4245–4253. doi: 10.1021/la703730b. [DOI] [PubMed] [Google Scholar]
- 23.Zhang LB, Li Y, Sun JQ, Shen JC. J Colloid Interface Sci. 2008;319:302–308. doi: 10.1016/j.jcis.2007.11.020. [DOI] [PubMed] [Google Scholar]
- 24.Zhao Y, Li M, Lu QH, Shi ZY. Langmuir. 2008;24:12651–12657. doi: 10.1021/la8024364. [DOI] [PubMed] [Google Scholar]
- 25.For leading references and a review on covalent layer-by-layer assembly see Bergbreiter DE, Liao KS. Soft Matter. 2009;5:23–28.
- 26.Buck ME, Zhang J, Lynn DM. Adv Mater. 2007;19:3951–3955. [Google Scholar]
- 27.Buck ME, Breitbach AS, Belgrade SK, Blackwell HE, Lynn DM. Biomacromolecules. 2009;10:1564–1574. doi: 10.1021/bm9001552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Buck ME, Lynn DM. Adv Mater. 2010;22:994–998. doi: 10.1002/adma.200903054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Buck ME, Lynn DM. ACS Appl Mater Interfaces. 2010;2:1421–1429. doi: 10.1021/am1000882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Buck ME, Lynn DM. Langmuir. 2010;26:16134–16140. doi: 10.1021/la103009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heilmann SM, Rasmussen JK, Krepski LR. J Polym Sci Part A. 2001;39:3655–3677. [Google Scholar]
- 32.Heilmann SM, Moren DM, Krepski LR, Rasmussen JK, Gaddam BN, Roscoe SB, Lewandowski KM, McIntosh LH, Roberts RR, Fansler DD, Szekely GG, Weil DA, Thakur KA, Pathre SV, Battiste JL, Hanggi DA. J Macromol Sci, Part A: Pure Appl Chem. 2003;A40:755–790. [Google Scholar]
- 33.Ruths J, Essler F, Decher G, Riegler H. Langmuir. 2000;16:8871–8878. [Google Scholar]
- 34.McAloney RA, Sinyor M, Dudnik V, Goh MC. Langmuir. 2001;17:6655–6663. [Google Scholar]
- 35.DeLongchamp DM, Kastantin M, Hammond PT. Chem Mater. 2003;15:1575–1586. [Google Scholar]
- 36.Liu XK, Dai BY, Zhou L, Sun JQ. J Mater Chem. 2009;19:497–504. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.