

Abstract
The three-dimensional structure of a protein is formed and maintained by the noncovalent interactions among the amino-acid residues of the polypeptide chain. These interactions can be represented collectively in the form of a network. So far, such networks have been investigated by considering the connections based on distances between the amino-acid residues. Here we present a method of constructing the structure network based on interaction energies among the amino-acid residues in the protein. We have investigated the properties of such protein energy-based networks (PENs) and have shown correlations to protein structural features such as the clusters of residues involved in stability, formation of secondary and super-secondary structural units. Further we demonstrate that the analysis of PENs in terms of parameters such as hubs and shortest paths can provide a variety of biologically important information, such as the residues crucial for stabilizing the folded units and the paths of communication between distal residues in the protein. Finally, the energy regimes for different levels of stabilization in the protein structure have clearly emerged from the PEN analysis.
Introduction
The uniqueness of a protein structure, encoded in its sequence (1), is attained through noncovalent interactions among the constituent amino acids. It is becoming increasingly clear that the interactions between amino acids at the global level are the deterministic factor, and an investigation involving pairwise interactions alone is not sufficient to understand the basis of uniqueness. This consideration has led to the development of protein structure networks (PSNs). PSNs constructed at a coarse-grain level, considering Cα/Cβ atoms as nodes (PcNs), are analyzed to distinguish the features of different folds (2).
At a finer level, PSNs constructed using atomic details have been used to identify clusters of interacting residues, important for protein folding, and function (active-site) (3). A variety of properties of these networks are investigated; for instance, the global behavior of networks is characterized as random, scale-free, or small-world, etc., on the basis of properties like the degree distribution, clustering coefficient, and characteristic pathlength (2,4–6). Network properties like the phenomenon of percolation (network of connections spanning across a system), clusters, hubs, cliques, and communities are also investigated from the network at detailed atomic level, to shed light on the structural and functional determinants in protein structures (7–9). And properties like the shortest path evaluated from the network elucidate the process of allosteric communication (10).
The function of a protein is closely associated with its conformational plasticity, and the ensembles of structures generated from rigorous molecular dynamics (MD) simulations provide dynamical properties in atomistic detail. The PSNs, constructed on a single structure, have been useful in tracking changes in the dynamical properties from an ensemble of conformations (10). Coarse-grained networks like Gaussian network models, elastic network models, and anisotropic network models (11), can also provide dynamical properties like thermal fluctuations (12), and low amplitude large-scale motions relevant to function (13), from crystallographic structures. In these approaches, the Cα/Cβ atoms are generally used as nodes, and edges are made with spatial neighbors within a certain distance with a specified value of spring constant, both of which are tunable parameters. Extensive studies have been done to obtain optimal values for these parameters (14). Fluctuations from short time equilibrium simulations have also been used to obtain realistic force constants (15). Further, the relation between equilibrium fluctuations and signal propagation in proteins has been investigated from elastic network models (16).
PSNs described in literature efficiently capture the topology and associated properties at the geometric level of atom-atom contact. The chemistry, however, is not captured completely by these network representations, and a wealth of information can be extracted by incorporating the details of chemical interactions. Our study is an advance over the existing protein structure networks, in terms of edges being defined based on interaction energies among the amino acids. This interaction energy is the result of various types of interaction within a protein. Hence, we believe that using realistic interaction energies is a step toward capturing all the essential features responsible for maintaining the protein structure into a simple network.
The crucial feature of this study is to represent proteins as interaction energy weighted networks (denoted as protein energy networks (PENs)) with realistic edge-weights obtained from standard force fields, and then characterize these networks. We have derived the interaction energies from equilibrium ensembles (obtained using MD simulations) to account for the structural plasticity, crucial to elucidate the function. We have also validated the suitability of this method to study single static structures. Characterization of the nature of these networks as a function of the strength of interaction and demonstration of the utility of PENs by addressing two important problems in structural biology has been carried out.
In the first case, we have used these weighted networks to identify stabilization regions in protein structures and hierarchical organization in the folded proteins, which may provide some insights to the general mechanism of protein folding and stabilization. In the second case, we have elucidated the features of communication paths in proteins from the energy weighted networks. We have extensively discussed specific paths in the case of the PDZ domain, which is known to bring together protein binding partners that mediate various cellular processes such as signal transduction, apoptosis, and cytokinesis (17,18).
Materials and Methods
Molecular dynamics simulation
Molecular dynamics (MD) simulations were carried out on a set of six proteins (see Table S1 and Fig. S1 in the Supporting Material) for a period of 2–5 ns, using GROMACS (19). The details of the MD simulation protocol are given in Table S2. Conformational ensemble for each protein is obtained by sampling structures (every 2 ps) from 1 ns to 2 ns or from 2 ns to 5 ns, based on the root-mean-square deviation profile (detailed in Fig. S2).
Calculation of interaction energies
The nonbonded interaction energy (Eij) between any two residues (i and j) is composed of two separate energy terms: The van der Waals (vdW) interaction energy (VLJ) given by the Lennard-Jones (LJ) potential (Eq. 1) and the electrostatic interaction energy (VC) given by Coulombic potential (Eq. 2). The computed interaction energy is defined as the sum of the LJ and Coulombic interaction energies averaged over the equilibrium ensemble (Eq. 3). All favorable interaction energies are <0 kJ/mol, and hence have a negative sign.
Fluctuation of an interaction between residues i and j is defined as the summation of the fluctuations (deviations from mean interaction energies) of the vdW and electrostatic interactions. Both the interaction energies and their fluctuations were calculated using the g_energy module in GROMACS:
| (1) |
| (2) |
| (3) |
Protein energy-weighted network
In protein energy networks (PENs), the amino-acid residues are considered as nodes. A weighted edge can be made between any pair of residues i and j by considering the interaction energy (Eq. 3) as the weight. Weighted networks (PEN) are used for calculations such as the shortest path (SP), whereas unweighted networks (PENe) are created for specific maximum energy value (e) to investigate parameters such as the largest cluster, hubs, etc. PENe can be represented as an adjacency matrix (Ae; see Eq. 4).
In both PEN and PENe, we disregard edges between sequential neighbors.
| (4) |
Clusters
Clusters, identified using the Depth First Search algorithm (20), are the connected components in a network (PENe, from Eq. 4). A node belonging to one cluster is connected to other nodes of the same cluster and not to the nodes of other clusters. Largest cluster is defined as the cluster with the highest number of constituent nodes. A cluster can be classified as a secondary cluster (S) if its members (nodes/residues) are from a single secondary structure; and a cluster is termed as a super-secondary cluster (SS) if the members are from more than one secondary structure. The secondary structure assignments are obtained using the DSSP program (21).
Hubs
Hubs are nodes that have a higher degree or connectivity in networks. Degree (D(Vi)) of a node Vi is the total number of edges incident upon it. Hubs can be ranked based on their degree (D(Vi)) and can further be ranked based on the average interaction energy. The average interaction energy, Avg E(Vi), of a hub is given as
| (5) |
where N is the number of nodes in the PEN, Aeij is the element's value in the adjacency matrix (A) at the specific e, and Eij is the interaction energy between i and j. Hub propensity of an amino-acid type (e.g., for Ala) is defined as the fraction of that amino-acid type to occur as hub at a specific e.
Shortest path and Closeness index
Dijkstra's algorithm (20) is used to calculate the shortest path (Sij) between any two residues i and j from a PEN (algorithm is given in Fig. S3). Sij is the pathlength, which is the total number of edges traversed while moving from i to j along the calculated shortest path. Some pairs of residues may be unreachable, in which case Sij is not considered for further calculation. For computing Sij, the energy matrix is modified such that the lower (less negative) the edge weight, the costlier it is to traverse that edge. Hence, the shortest path calculated using this algorithm is the most energetically favorable path.
The Closeness index (Ci) is a property of a node, which characterizes the spread of information across the network from, to, or through that node. In a network, Ci of a node is defined as mean of the shortest paths,
| (6) |
where P is the total number of residues to which i can be connected. The residues are ranked based on Ci. The node with least Ci is ranked highest and so on.
Results and Discussion
Protein energy networks
Proteins are represented as protein energy networks (PENs) with residues as nodes and interaction energies as edge weights. Unweighted networks (PENes) can be generated from PENs using an energy cut-off e (Eq. 4). PENe is well connected when e is low (that is, less negative; e.g., –7 kJ/mol) and sparse when e is high (that is, more negative; e.g., –25 kJ/mol). In this study, we have generated PENs for all the proteins in the dataset (Table S1) and characterized the PENs using different network- and node-specific parameters. Further, we have discussed their potential applications in studying protein structure, stability, and function.
Interaction energies
Correlation between MD averages and static structures
Interaction energies between all pairs of residues in a protein are calculated as a summation of their van der Waals (Lennard-Jones potential, VLJ) and electrostatic (Coulombic potential, Vc) energies, averaged over an equilibrium ensemble obtained using MD simulations (Eq. 3). To evaluate the validity of using single structures for construction of energy-weighted networks, we compared the interaction energy values obtained from the equilibrium conformations with those from minimized crystal structures as well as with snapshots after equilibration (at 20 ps).
Reasonable correlations are seen for the proteins in the dataset, with correlation coefficients at ∼0.72–0.93 (Table S1). For proteins like signal recognition particle receptor and Barnase, the correlation coefficients, albeit low for starting structure (0.33 and 0.14, respectively), significantly improve after minimization (Table S1). Hence, energies calculated using minimized structures can provide qualitatively good information. In this work, for constructing PENs, we have used the interaction energies obtained from equilibrium ensembles.
Range of interaction energies
The interactions within proteins can be of various types such as hydrophobic interactions, hydrogen bonding (backbone, side chain, and backbone/side chain), π-π interactions, cation-π interactions, electrostatic salt bridges, etc. Samples of the possible types of interactions we observe in protein structures along with their energy values are given in Fig. S4. In general, we observe high (highly negative) interaction energies between pairs of charged residues (Fig. S4 a) and low (less negative) interaction energies between pairs of small hydrophobic residues (Fig. S4 d). The scale of energies is useful in interpreting PENs in terms of the dominant contributions, from hydrophobic, hydrogen-bonding, or electrostatic interactions, etc.
The distributions of vdW (VLJ), electrostatic (VC), and total (VLJ + VC) energies and their ranges from a typical protein (Lysozyme) are presented in Fig. 1 a. As expected, we see that the number of low energy interactions (0 to –5 kJ/mol) is high and that of high energy interactions (≤ –20 kJ/mol) is low (see Fig. 1 a; and see Fig. S5 for all six proteins in the dataset). It should be noted that the interactions between all pairs of residues could be captured by their interaction energies, irrespective of the geometric distances between the interacting pairs. Thus, the resultant interaction energy is a combination of both geometric distance and chemical nature of the interacting residues.
Figure 1.
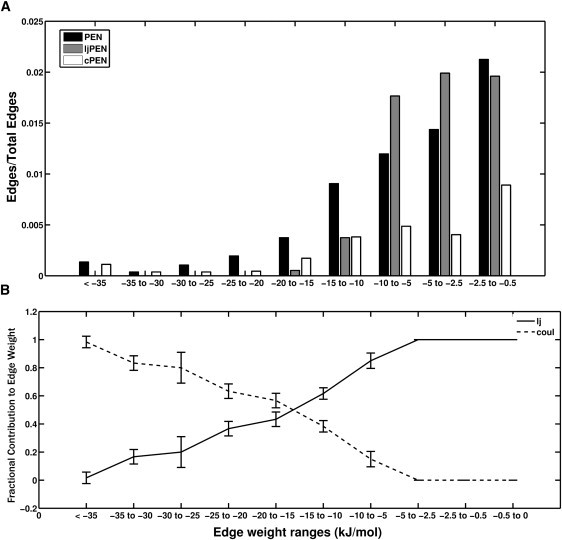
Edge-weight distribution profile in PENs (A) The bar plot shows the fraction of edges with different ranges of edge weights in Lysozyme. Fraction of edges at a specific energy range = Total number of edges within the energy range/, where N is the total number of nodes in the PEN. (B) Fractional contribution of vdW (VLJ/Eij) and electrostatic (VC/Eij) energies to the total interaction energy (Eij) at different energy ranges for all the proteins in the dataset. The error bar indicates the standard deviation of the fractional contribution values from their averages.
The distribution of the fractional contributions of vdW and electrostatic interactions to the total energy shows that the vdW dominates in the low energy region (≥–5 kJ/mol) and falls off to zero at ∼–35 kJ/mol (Fig. 1 b). This trend is reversed in the electrostatics contribution profile, where they dominate at higher interaction energy region (≤–20 kJ/mol) (Fig. 1 b). Thus, an interaction energy range of 0 to –35 kJ/mol covers a large fraction of interactions, and different interactions dominate different regions within this range. Hence, appropriate energy thresholds (e) can provide a physical basis for the type of interactions, giving insights into the structural determinants, as discussed in later sections.
From the contributions of different interaction types to the total interaction energies, we observe that vdW (VLJ) energies are generally >–10 kJ/mol (Fig. 1 and Fig. S5). Therefore, we have considered two types of networks:
The total energy network (PEN), in which we take into account both vdW (packing-based) and electrostatic (charge-based) interactions.
The LJ-protein energy network (ljPEN), in which only vdW (packing-based) interactions are considered, eliminating the dominant effects of the charged interactions.
Our aim in using the second type of network (ljPEN) is to highlight packing-based hydrophobic interactions in protein structures.
Largest cluster as a function of e
In our earlier studies (4), we had shown that the size of the largest cluster (cluster with the largest number of connected nodes) as a function of contact-based interaction strength (referred to as Imin (3)) is sigmoidal with a clear transition point. Such a network provided insights into several aspects of the structure and function of proteins (22). It was also investigated from a percolation perspective in which percolation transition was rigorously evaluated (8). However, in those studies, it was not possible to examine many of the features dependent on the quantitative interaction energies, but we have overcome that here.
At low e values, the largest cluster is huge in terms of its constituent nodes (Fig. 2 a). As e increases (toward highly negative values), it starts dissolving into smaller clusters. In PENs, we observe that this decrease in the size of the largest cluster is very sharp as a function of e (Fig. 2). This small window of e (∼–10 kJ/mol to –20 kJ/mol), where the largest cluster disintegrates, is denoted as the transition region (Fig. 2 a). Interestingly, all the proteins considered for this analysis follow the same profile (Fig. 2). This point toward a universal behavior where most of the residues in proteins are connected with energies between 0 and –10 kJ/mol (where vdW values are dominant; see Fig. 1) and these connections disappears at high interaction energies (≤–20 kJ/mol, where electrostatics dominate; see Fig. 1).
Figure 2.
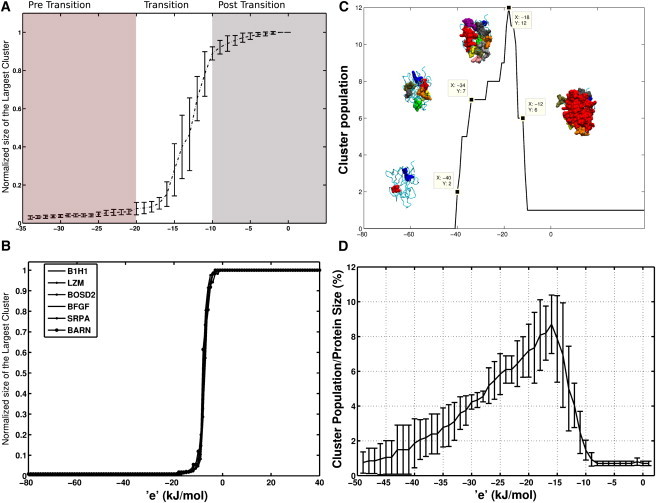
Largest cluster and cluster population as a function of e. The normalized size of the largest cluster (largest cluster size/total number of nodes in PEN) is plotted as a function of e for PENs (A) and ljPENs (B). The error bar in panel A represents standard deviation of the normalized sizes of the largest cluster from the average size (computed from all dataset proteins) at a specific e. (C) Cluster population change as a function of e is shown for B1 domain of Human Neurophilin-1 (B1H1). Clusters at a specific e (marked by ■), are highlighted as vdW spheres. (D) Cluster population changes as a function of e for all the proteins in the dataset is shown. The error bars indicate standard deviation of the cluster population, at a specific e, from the average value.
Stability and hierarchical organization
Regions of stabilization
From Fig. 2, we can see that a PEN, at the transition region, breaks up into independent clusters. Thus, it is clear that proteins are not composed of strong interactions throughout the structure. Instead, they consist of islands of highly interacting regions tethered together by weak interactions. These high-energy interacting islands of residues are vital because they are the stabilizing regions in the structure. The maximum number of such autonomous clusters, at a given e, would provide an idea about the maintenance of structural integrity in the protein.
Cluster population as a function of e (total number of clusters at a given e value) gives information about the maximum number of these isolated interaction regions. For example, Fig. 2 c shows that the B1 domain of Human Neurophilin (B1H1) has the maximum number of stabilization regions (12 clusters) at e = –18 kJ/mol. This observation correlates with our largest cluster profile (Fig. 2 a), where the cluster disintegrates completely at ∼–20 kJ/mol. Hence, maximum number of clusters is seen around the transition region and they merge to yield a single large cluster, by accruing low energy vdW interactions, as e decreases (toward less negative energy). We denote this region where the clusters have merged as the post-transition region (Fig. 2). Conversely, the number of clusters and their sizes diminish as e increases from the transition region due to smaller number of high-energy interactions (Fig. 1 a and Fig. S5), and we denote this as the pre-transition region (Fig. 2). High-energy stabilization regions in proteins are obtained in this range. This cluster population profile is similar for all proteins in the dataset (Fig. 2 d).
The network representation of proteins using interaction energies has provided such local pockets of highly stabilizing units, which is more informative than identifying pairwise stabilizing interactions. Hence, we can use PENs to study energetically important regions in protein structures. These regions can represent either possible stabilizing units in a folded structure or nucleation points during the folding process.
Hierarchical organization
From the above observations, it is clear that as e decreases, a number of strongly interacting clusters at the pre-transition region merge to form a single large cluster at the post-transition region. Also, each of the clusters in the pre-transition region represents distinct stabilizing units. By associating the stabilizing clusters with structural units (i.e., secondary structure clusters (S) comprising nodes from a single secondary structure and super-secondary structure clusters (SS) with nodes from different secondary structures), it may be possible to study the hierarchical organization of a protein structure from its structural fragments. Such a study of hierarchical assembly of a typical globular protein (Lysozyme) is given below.
The S and SS clusters in Lysozyme are analyzed as a function of e (Fig. 3). There are 10 helices (H1–H10) and three sheets (E1, E2, and E3) in Lysozyme, forming two separate domains D1 (N-terminal domain) and D2 (C-terminal domain) (Fig. 3 a; residue details in Table S3). The clusters are analyzed for six values of e ranging from –12 kJ/mol to –31 kJ/mol, covering the transition region and beyond. The number of clusters, the participating secondary structures, and the cluster location in Lysozyme are depicted as six panels in Fig. 3 b. It appears that the transition point is close to –17 kJ/mol (Fig. 3 b, panel 4), where we see the largest number (i.e., 14) of clusters, and one of the clusters (Fig. 3 b; cluster number 1 in panel 4) connects five long helices, mainly from the D2 of Lysozyme. Moreover, it also connects the residues from helix 3, which extends to the D1 (see Fig. 3 b, panel 4; clusters 3, 5, and 9).
Figure 3.
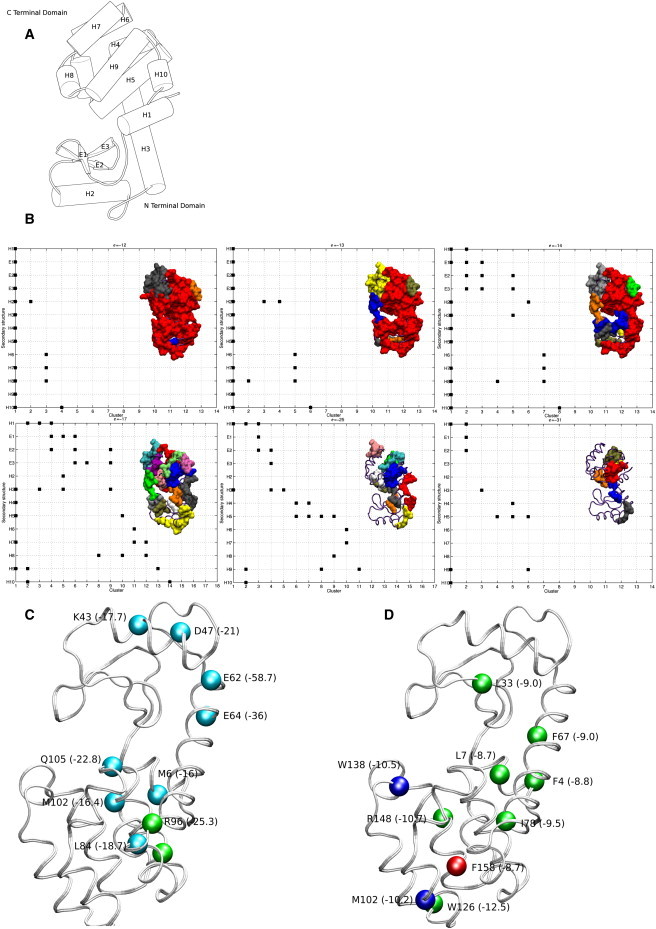
Hierarchical organization and stability regions in Lysozyme. (A) Lysozyme with secondary structures (H-helix and E-β strand) assigned according to DSSP (21) is shown. (B) The evolution of secondary (S) and super-secondary (SS) clusters as a function of e is depicted. Clusters (surf representation) and the participating secondary structures, for different e values (six panels) are shown. The clusters are arbitrarily numbered along the horizontal axis and the secondary structural units are represented along the vertical axis. Participation of secondary structures in a given cluster (Materials and Methods) is marked by ■. For example, in panel 1 (corresponding to e = –12 kJ/mol), all the secondary structures, except H6, are members of cluster 1, whereas the helices 6, 7, and 8 participate in SS cluster 3. Cluster 2 is an S cluster with participating residues from H2 only. The top 10 hubs in PEN and ljPEN of Lysozyme are shown in panels C and D. The Cα atoms of the hub residues are highlighted as vdW spheres. Residue information along with average energy per node (Eq. 5) in kJ/mol (within parentheses) is given.
Other clusters at this e value consist of residues from one to three secondary structures. Moving toward the pretransition region with e corresponding to –31 kJ/mol (Fig. 3 b; panel 6), we have six clusters with the total participating secondary structural units ranging from one to three. Interestingly, these stabilizing centers encompass most regions of the protein, with some of them connecting the secondary structural units in D1 (Fig. 3 b; cluster 2 in panel 6) and some in D2 (Fig. 3 b; clusters 5 and 6, in panel 6), and one of them (cluster 1) connecting the helices across two domains. We observe that the clusters in D2 domain are more established (in terms of their constituent nodes) than the D1 domain at this high e value. At the post-transition region (Fig. 3 b; e = –12 kJ/mol, panel 1), except for a few clusters that comprise helices toward the surface of the protein, a single large SS cluster encompassing almost all the secondary structures is seen.
Thus, PENs are an excellent means for identifying the stabilizing units and to understand the stabilizing forces that stitch together different secondary structures in proteins. Further, the possible relevance of stabilizing clusters in the folding process can be examined by comparing with experimental results. Cellitti et al. (23) have proposed an intermediate of a structured D2, with an unstructured D1 for Lysozyme, based on equilibrium native state hydrogen experiments on a permutant protein. Llinás and Marqusee (24) have observed that D2 can exist separately in a near folded state, while D1 remains largely unfolded. Our observation that D2 forms the largest cluster and that D1 has D2-dependent SS clusters (at e –14 kJ/mol (Fig. 3 b, panel 3) correlates with these experimental observations. Thus, the identification of stabilizing units can be useful in the investigation of the folding process in proteins.
Hubs
Hubs in PENs are highly connected residues, potentially important for structural stability and communication. Although the hubs represent connectivity in general, the strength of interaction can also be incorporated in weighted PENs (Eq. 5). A residue with a degree (Materials and Methods) of at least four in the post-transition region, three in the transition region, and two in the pre-transition region is considered to be a hub (see Fig. S6). The top 10 hubs for Lysozyme show that most of the highly connected hydrophobic residues (ljPEN, Fig. 3 d) are present in D2, forming a strong hydrophobic core. The PEN hubs of Lysozyme (Fig. 3 c) are positioned at the periphery of the protein, with some hub residues (E62, E64, K43, and K47, in Fig. 3 c) involved in connecting D1. These results suggest that D2 has a better hydrophobic core, in agreement with the earlier observations that D2 can autonomously fold. Further, the PEN hubs are mostly dominated by charged residues, except for Leu-84 (L84A and L84M mutant influence stability and folding (25,26)), Met-102, and Met-6. The topmost PEN hub, Arg-96, has been shown to be important for protein stability (27).
The hub propensities at different e suggest that, at the post-transition region (e ∼–4 kJ/mol), the hydrophobic and the aromatic residues have high propensity to be hubs whereas the polar/charged residues have low propensity and the trend is reversed at the pre-transition region (e at ∼–18 kJ/mol) (Fig. S7). These results indicate that the vdW-based hydrophobic interactions play a vital role in tethering different structural regions, while the charged interactions dominate the local interactions in the protein.
Communication paths
Allosteric communications form a crucial component of the functioning of a large number of proteins. The concepts of ligand-induced conformational changes (MWC and KNF models (28,29)) and their equilibrium populations (30) have stimulated a number of experimental and theoretical studies (10,12,16,18,31–36) to elucidate the mechanisms of allosteric communication. The ideas such as anisotropic flow of energy between distal sites and presence of specific transport channels along which local structural perturbations propagate through proteins have also been discussed (18). Fundamental issues such as the efficiency of energy flow in terms of the depth and the curvature of potential wells have been probed (37).
Experimental studies such as ultrafast spectroscopy (38), NMR measurements (39), and mutational effects (17), as well as theoretical correlations obtained from MD simulations (32,34), normal mode analysis (40), and statistical coupling analysis (33) have been performed to study paths of communication. Multiple preexisting pathways have been suggested from protein structure analyses (41).
Unraveling the energy transport channels and flow pathways in individual proteins and connecting them to their functions are presently considered the main challenges (18). Our group has been involved in elucidating the communication paths in proteins by integrating the concept of structure network constructed on the basis of geometric contact of noncovalent interactions with ligand-induced conformational changes obtained through MD simulations (10). In this study, we consider the contacts and communication paths on the basis of energies of interactions, which provide a more rigorous estimate of the strength of interactions and their changes induced upon ligand binding.
First, we present statistical features of parameters such as the shortest path and Closeness index. Secondly, we present our studies on the details of specific paths in the PDZ2 domain, which has been extensively investigated by a variety of techniques (17,32–34,41,42).
Shortest/optimal paths
Several factors such as the physical distance between end members, interaction strength, and/or the magnitude of fluctuations of connecting bonds (37) are involved in communication paths. It is reasonable to assume that the bonds should be strong enough to maintain the path and at the same time be flexible for transmission of information. Thus, it is likely that the communication paths are optimal in terms of both strength and stiffness. However, a clear understanding of the basis of the optimal path requires extensive theoretical studies.
As a first step toward this goal, here we have analyzed the relation between bond (noncovalent) strength and stiffness in protein structures. We have evaluated the bond stiffness (fluctuations from MD simulations) in the proteins from our dataset and compared them with their bond strengths (interaction energies). Interestingly, we find a high correlation (correlation coefficient of 0.83 (Fig. S8)) between interaction energies of the bonds and their fluctuations. This observation shows that the higher (highly negative) the interaction energy, the lower the bond stiffness. The amplitude of fluctuations is higher for electrostatic interactions than for vdW interactions. This unexpected result may be due to the fact that many of the electrostatic interactions are on the periphery of the proteins. It is to be noted, however, that this observed correlation is for all possible individual bonds in the proteins and feasible paths may involve bonds with a range of interaction energies. The communication paths are likely to be short (in terms of pathlength), with the strength and stiffness of the connecting bonds optimal.
Also, physically meaningful conditions such as the inclusion of conserved residues, or dynamically correlated residues can be enforced while identifying the shortest path (10) as optimal paths. However, the definition of connection between nodes remains the important parameter. Physical connection (contact-based) between spatially proximal residues, considered as edges in PSN, is often employed for the calculation of the shortest path. In PENs, the edges are weighed on the basis of interaction energies, which provide a more realistic estimate of the communication paths in proteins by identifying energetically favorable paths.
The shortest paths (SPs) between all pairs of residues in PENs of the proteins in Table S1 are calculated using Djikstra's Algorithm (see Materials and Methods) and are compared with those derived from Cα-Cα distance-based networks (PcNs) (Fig. S9). There is no good correlation between the two networks in terms of the pathlengths and their propensities. Thus, the SPs evaluated from PENs are substantially different from those evaluated using PcNs. Although the gross topological features are characterized by the SPs of PcNs, accurate energy-based SPs from PENs are more suitable for investigating finer details such as communication paths between distal residues.
Closeness index
The shortest path is dependent on two nodes (end members). However, the potential of each node to take part in communication (shortest paths) between a large set of nodes in the network can be evaluated by a node-specific parameter called the Closeness index (Ci) (see Materials and Methods). Ci differs from the degree of a node, because it quantifies the influence of all the nodes in the network. The top 10 residues with least Ci obtained for the PEN of proteins in the dataset are given in Table S4. The Ci of the residues in Lysozyme from PEN, ljPEN, and PcN are depicted in Fig. S10. By and large, the expected features such as the smaller Ci of the central residues and a larger Ci of the peripheral residues are reproduced by all PENs, but they differ in their details and also depend upon the topology of the protein (Fig. S11). The residues with lower Ci values (highly ranked) generally hold the interaction network together at various points. Thus, the Ci may be associated with diverse structural and functional implications. The importance of residues with the least Ci in Lysozyme and Barnase is provided in Table S5 (43–48).
Communication paths in PDZ2 domain
PDZ domains are modular proteins, involved in mediating interactions between protein partners (49). These proteins are shown to exhibit allosteric activity, and have been extensively studied through various approaches like chemical shifts upon ligand binding using NMR (50), changes in energetic coupling between interacting residues (32), pump-probe MD simulations (34), anisotropic thermal diffusion (51), normal mode analysis (40), and statistical coupling analysis (33). Different allosteric pathways have been proposed by the above-mentioned studies (41).
The communication paths between allosterically important residues may be influenced by factors such as the binding of ligands and an analysis of such paths can offer insights into the function of proteins. In this section, we have investigated SPs and Ci in the PENs of PDZ2 domain from human phosphatase, for which NMR structures of both the apo- and ligand-bound forms (Fig. S12) are available (17,32,42). PENs for both these forms were generated from 2 ns MD simulations and energetically favorable paths of communications between key residues in allosteric communication were obtained in both forms.
Ligand-induced changes in shortest path and closeness index
The ligand binding has induced a definite rewiring in the PEN of PDZ2 domain. Interactions were gained or lost upon ligand binding and overall changes in the interaction energies were observed (Fig. S13). This alteration in the interaction energies in PDZ2 by the ligand has introduced rerouting of communication between residues (Fig. S14). Communications (shortest paths) that were absent in the apo form were established between residues by ligand binding (Table S6 and Fig. S14). Although most of the shortest paths (in terms of pathlengths given by the total number of edges in the path) remain unchanged, a fraction of shortest paths have either lengthened or shortened upon ligand binding (Table S6 and Fig. S14).
The changes in the shortest paths result in alteration of the Ci of residues. For most of the residues, Ci increases upon ligand binding (Fig. 4 a). However, in a few cases, the Ci decreased (Table S7 and Fig. 4 a), showing that these residues might become more favorably involved in allosteric communication upon binding to the ligand. Most of the residues that have decreased Ci upon ligand binding have been reported, in other studies (Table S7 and Table S8), to be important for communication.
Figure 4.
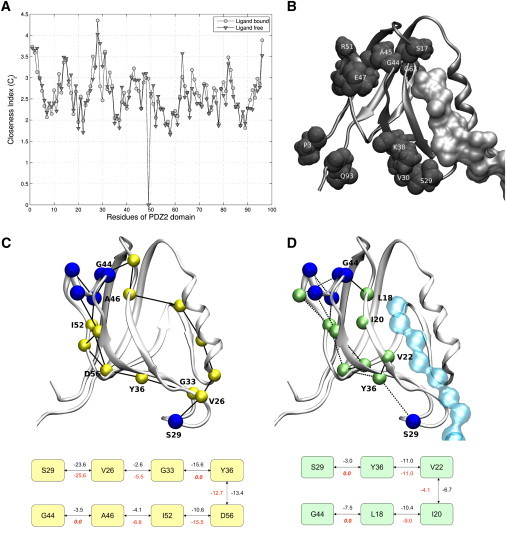
Effect of changes in the shortest paths upon binding of ligand to PDZ2 domain. The figure highlights the effect of the changes in the SPs in PEN of PDZ2 domain upon ligand binding. (A) The plot shows the Ci of residues in the apo (▾) and the ligand-bound forms (○) of PDZ2. (B) Residues whose Ci have decreased (>0.2) upon ligand binding, are highlighted. (C) SPs between S29, and residues G44, A45, A46, E47, and S48 (SPS29,α1) in the apo form are shown. (D) SPS29,α1 for the ligand-bound form. The residue identity for those that lie in SPS26,G44 are marked in both apo (C) and ligand-bound (D) forms. Interaction energies between the residues are provided in the graph/network below the cartoon figures. The interaction energies which are given below the edges are obtained from the PEN of ligand-bound (C) and the apo (D) PDZ2 domain. They indicate the interactions gained or lost upon ligand binding.
The residues whose Ci decreased upon ligand binding are from the α1, β1-β2 loop (which is a conserved glycine-rich motif (32)) regions (Fig. 4 b). They form part of pathway I (from the binding site to residues in the α1 helix) proposed on the basis of NMR chemical shifts (42). The Ci of residues from the β2-β3 loop and C- and N-terminal residues has also decreased upon ligand binding. These residues lie in the pathway II (starts from the binding site and goes perpendicularly across strands β2, β3, β4, β6, and β1), as proposed by Kong and Karplus (32).
The above observations on changes in SPs and Ci suggest that the information is channeled through fewer paths and does not diffuse throughout the PDZ2 domain, complementing the earlier observation by energetic coupling (32) and energy flow studies, which consider proteins as a network of sites in a percolation cluster (18).
Communication between binding site and other distal sites—allosteric pathways
The regions such as α1 helix (G44–S48) and β1-β2 loop (K13–S17) have been shown to change their conformations upon ligand binding (17,42). They were found to be energetically coupled to spatially distal residues such as those from β2-β3 loop (V26 and S29) and C-terminal residue A69 of α2 helix, respectively (32). Hence, we have evaluated the shortest paths from S29 to the α1 helix (SPS29,α1) and from A69 to β2-β3 loop (SPA69,β1-β2) in both the apo and the ligand-bound forms (Fig. 4, c and d, Fig. S15 and Table S9). Strikingly, the paths like SPS29,G44 take entirely different routes, and the SPS29,G44 pathlength (both in terms of the number of edges and interaction energies) decreases upon ligand binding. In the apo state, the path consists of peripheral residues such as V26, D56, and I52; however, in the ligand-bound state, it penetrates through the core of the protein via residues L18, I20, and V22 of central β-sheet (see Fig. 4, c and d; see also Fig. S16, which shows the SPs obtained from single snapshot PENs). The shortening is due to the gain of new energetically favorable connections like L18-G44 and S29-Y36, which was indirectly connected in the apo form (see Fig. 4, c and d). Further, one of the paths identified between S29 and the residues of α1 helix in the apo form passing through the residues of α2 helix (which belongs to another energetically coupled cluster (32)) completely disappears in the ligand-bound form. SPA69,β1-β2 also undergo similar changes upon ligand binding (Fig. S15).
Some of the residues, which appear in SPS29,α1, have been identified as important for allosteric communication from earlier studies. For example, residues L18 (backbone interacts with the carbonyl end of the last residue of the ligand) and I20 (also SPV26,α1, Table S9) were shown to have a change in side-chain dynamics upon ligand binding, from NMR studies (17). V22 was found to have altered dynamics upon I35V mutation that changes the binding affinity of PDZ2 to the ligand (17). The residues I52 and A46, which were identified to be important for communication, also emerge as residues involved in allostery by chemical shift mapping (17,41,42). The SPA69,β1-β2 passes through H71, and V75, which were known to be important for allosteric communication in the PDZ3 domain of PSD-95 (33). L78 shows changes in side-chain dynamics upon ligand binding (17,42). L18, which was involved in SPS29,α1, also plays a vital role in SPA69,β1-β2.
The ligand-induced changes in the PEN of PDZ2 domain resulted in changes in the communication paths between various residues. These changes are not confined to residues just around the binding site, but encompass distal sites like the α1 helix. This clearly indicates that perturbation at the binding site have energetic repercussions at distal sites in PDZ2 domain, indicating allosteric behavior. The global energetic changes in PEN upon ligand binding are reflected in the changes in Ci of residues. The decrease of Ci in only a fraction of residues upon ligand binding indicates channeling of information. Furthermore, PEN is able to capture details of the paths of possible communication, their associated energies, and alternate paths taken upon ligand binding. Such detailed elucidation of the pathways compliments the vast body of knowledge from other studies and is likely to inspire further investigations.
Conclusions
In this study, we have constructed weighted protein structure networks (PEN) based on noncovalent interaction energies. This is an advance over the existing protein structure networks in considering not only the geometry, but also the chemistry of interacting amino acids, and has the potential to provide more detailed insights into protein structure, stability, and function. The PENs are constructed to take into account all components of the energy term and ljPENs account only for the van der Waals interactions. The unweighted PENes, at desired energy values (e), are used to investigate the network behavior at different energy levels. The PENs are analyzed using different network parameters like the largest cluster, cluster population, hubs, shortest paths, and closeness indices.
The PENs exhibit three distinct behaviors as a function of e. The pre-transition region (<–20 kJ/mol) comprises smaller clusters with mainly charged and polar residues as hubs. Crucial topological changes take place in the transition region (–10 kJ/mol to –20 kJ/mol), where the smaller clusters aggregate, through low energy vdW interactions, to form a single large cluster in the post-transition region (>–10 kJ/mol). These behaviors reinforce the concept that hydrophobic interactions hold together local clusters of highly interacting residues, keeping the protein topology intact.
Clusters represent possible stabilizing units in a folded structure, or nucleation points during the folding process. By associating these clusters with their structural units, we have studied the hierarchical assembly of a model protein, relating the observations on the secondary and super-secondary clusters to the stability of the domains in Lysozyme.
Communication paths in protein structures have been evaluated as shortest paths (SPs) between functionally important residues. PENs provide a distinct advantage over contact-based networks, by identifying energetically favorable paths. The effect of global changes in shortest paths on a residue is given as a node-specific parameter termed the Closeness index (Ci). The structural and functional implications of residues with low Ci values are evident from correlations with experimental observations in Lysozyme and Barnase.
In the final section, we examined allosteric communications in PDZ domains using PENs. Changes in PEN upon ligand binding, resulting in alterations in SPs and Ci of a small fraction of residues, indicate that allosteric communication is anisotropic in PDZ. Our observations establish that the SPs between functionally important sites traverse through key residues in PDZ2 domain. Such a detailed elucidation of pathways at the energy level has been attempted for the first time, to our knowledge, in this study.
In summary, the study of structure networks based on interaction energies can effectively bring out the factors responsible for structural organization and stability. In addition, they can highlight subtle changes leading to allosteric communications responsible for the functioning of several proteins.
Acknowledgments
We thank Moitrayee Bhattacharrya for her help in manuscript preparation.
We acknowledge the support from the Department of Science and Technology (DST Mathematical Biology grant, No. DST0773) and the Department of BioTechnology (India).
Supporting Material
References
- 1.Anfinsen C.B. Principles that govern the folding of protein chains. Science (NY) 1973;181:223–230. doi: 10.1126/science.181.4096.223. [DOI] [PubMed] [Google Scholar]
- 2.Bagler G., Sinha S. Network properties of protein structures. Physica A. 2004;346:27–33. [Google Scholar]
- 3.Kannan N., Vishveshwara S. Identification of side-chain clusters in protein structures by a graph spectral method. J. Mol. Biol. 1999;292:441–464. doi: 10.1006/jmbi.1999.3058. [DOI] [PubMed] [Google Scholar]
- 4.Brinda K.V., Vishveshwara S. A network representation of protein structures: implications for protein stability. Biophys. J. 2005;89:4159–4170. doi: 10.1529/biophysj.105.064485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.del Sol A., Fujihashi H., Nussinov R. Residue centrality, functionally important residues, and active site shape: analysis of enzyme and non-enzyme families. Protein Sci. 2006;15:2120–2128. doi: 10.1110/ps.062249106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greene L.H., Higman V.A. Uncovering network systems within protein structures. J. Mol. Biol. 2003;334:781–791. doi: 10.1016/j.jmb.2003.08.061. [DOI] [PubMed] [Google Scholar]
- 7.Brinda K.V., Vishveshwara S., Vishveshwara S. Random network behavior of protein structures. Mol. Biosys. 2010;6:391–398. doi: 10.1039/b903019k. [DOI] [PubMed] [Google Scholar]
- 8.Deb D., Vishveshwara S., Vishveshwara S. Understanding protein structure from a percolation perspective. Biophys. J. 2009;97:1787–1794. doi: 10.1016/j.bpj.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ghosh A., Vishveshwara S. Variations in clique and community patterns in protein structures during allosteric communication: investigation of dynamically equilibrated structures of methionyl tRNA synthetase complexes. Biochemistry. 2008;47:11398–11407. doi: 10.1021/bi8007559. [DOI] [PubMed] [Google Scholar]
- 10.Ghosh A., Vishveshwara S. A study of communication pathways in methionyl- tRNA synthetase by molecular dynamics simulations and structure network analysis. Proc. Natl. Acad. Sci. USA. 2007;104:15711–15716. doi: 10.1073/pnas.0704459104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bahar I., Lezon T.R., Eyal E. Global dynamics of proteins: bridging between structure and function. Annu. Rev. Biophys. 2010;39:23–42. doi: 10.1146/annurev.biophys.093008.131258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atilgan A.R., Durell S.R., Bahar I. Anisotropy of fluctuation dynamics of proteins with an elastic network model. Biophys. J. 2001;80:505–515. doi: 10.1016/S0006-3495(01)76033-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bahar I., Atilgan A.R., Erman B. Vibrational dynamics of proteins: significance of slow and fast modes in relation to function and stability. Phys. Rev. Lett. 1998;80:2733–2736. [Google Scholar]
- 14.Yang L., Song G., Jernigan R.L. Protein elastic network models and the ranges of cooperativity. Proc. Natl. Acad. Sci. USA. 2009;106:12347–12352. doi: 10.1073/pnas.0902159106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moritsugu K., Smith J.C. Coarse-grained biomolecular simulation with REACH: realistic extension algorithm via covariance Hessian. Biophys. J. 2007;93:3460–3469. doi: 10.1529/biophysj.107.111898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chennubhotla C., Bahar I. Signal propagation in proteins and relation to equilibrium fluctuations. PLOS Comput. Biol. 2007;3:1716–1726. doi: 10.1371/journal.pcbi.0030172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fuentes E.J., Gilmore S.A., Lee A.L. Evaluation of energetic and dynamic coupling networks in a PDZ domain protein. J. Mol. Biol. 2006;364:337–351. doi: 10.1016/j.jmb.2006.08.076. [DOI] [PubMed] [Google Scholar]
- 18.Leitner D.M. Energy flow in proteins. Annu. Rev. Phys. Chem. 2008;59:233–259. doi: 10.1146/annurev.physchem.59.032607.093606. [DOI] [PubMed] [Google Scholar]
- 19.van der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 20.Cormen T.H. MIT Press; Cambridge, MA: 2001. Introduction to Algorithms. [Google Scholar]
- 21.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 22.Vishveshwara S., Ghosh A., Hansia P. Intra and inter-molecular communications through protein structure network. Curr. Protein Pept. Sci. 2009;10:146–160. doi: 10.2174/138920309787847590. [DOI] [PubMed] [Google Scholar]
- 23.Cellitti J., Bernstein R., Marqusee S. Exploring subdomain cooperativity in T4 lysozyme II: uncovering the C-terminal subdomain as a hidden intermediate in the kinetic folding pathway. Protein Sci. 2007;16:852–862. doi: 10.1110/ps.062632807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Llinás M., Marqusee S. Subdomain interactions as a determinant in the folding and stability of T4 lysozyme. Protein Sci. 1998;7:96–104. doi: 10.1002/pro.5560070110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gassner N.C., Baase W.A., Matthews B.W. Methionine and alanine substitutions show that the formation of wild-type-like structure in the carboxy-terminal domain of T4 lysozyme is a rate-limiting step in folding. Biochemistry. 1999;38:14451–14460. doi: 10.1021/bi9915519. [DOI] [PubMed] [Google Scholar]
- 26.Gassner N.C., Baase W.A., Matthews B.W. Multiple methionine substitutions are tolerated in T4 lysozyme and have coupled effects on folding and stability. Biophys. Chem. 2003;100:325–340. doi: 10.1016/s0301-4622(02)00290-9. [DOI] [PubMed] [Google Scholar]
- 27.Mooers B.H., Tronrud D.E., Matthews B.W. Evaluation at atomic resolution of the role of strain in destabilizing the temperature-sensitive T4 lysozyme mutant Arg 96→His. Protein Sci. 2009;18:863–870. doi: 10.1002/pro.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monod J., Wyman J., Changeux J.P. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 29.Koshland D.E., Jr., Némethy G., Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 30.Weber G. Ligand binding and internal equilibria in proteins. Biochemistry. 1972;11:864–878. doi: 10.1021/bi00755a028. [DOI] [PubMed] [Google Scholar]
- 31.del Sol A., Fujihashi H., Nussinov R. Residues crucial for maintaining short paths in network communication mediate signaling in proteins. Mol. Sys. Biol. 2006;2:0019. doi: 10.1038/msb4100063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kong Y., Karplus M. Signaling pathways of PDZ2 domain: a molecular dynamics interaction correlation analysis. Proteins. 2009;74:145–154. doi: 10.1002/prot.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lockless S.W., Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science (NY) 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- 34.Sharp K., Skinner J.J. Pump-probe molecular dynamics as a tool for studying protein motion and long range coupling. Proteins. 2006;65:347–361. doi: 10.1002/prot.21146. [DOI] [PubMed] [Google Scholar]
- 35.Tsai C.J., del Sol A., Nussinov R. Allostery: absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol. 2008;378:1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cui Q., Karplus M. Allostery and cooperativity revisited. Protein Sci. 2008;17:1295–1307. doi: 10.1110/ps.03259908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Piazza F., Sanejouand Y.H. Long-range energy transfer in proteins. Phys. Biol. 2009;6:046014. doi: 10.1088/1478-3975/6/4/046014. [DOI] [PubMed] [Google Scholar]
- 38.Kukura P., McCamant D.W., Mathies R.A. Structural observation of the primary isomerization in vision with femtosecond-stimulated Raman. Science (NY) 2005;310:1006–1009. doi: 10.1126/science.1118379. [DOI] [PubMed] [Google Scholar]
- 39.Brüschweiler S., Schanda P., Tollinger M. Direct observation of the dynamic process underlying allosteric signal transmission. J. Am. Chem. Soc. 2009;131:3063–3068. doi: 10.1021/ja809947w. [DOI] [PubMed] [Google Scholar]
- 40.De Los Rios P., Cecconi F., Juanico B. Functional dynamics of PDZ binding domains: a normal-mode analysis. Biophys. J. 2005;89:14–21. doi: 10.1529/biophysj.104.055004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.del Sol A., Tsai C.J., Nussinov R. The origin of allosteric functional modulation: multiple pre-existing pathways. Structure. 2009;17:1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fuentes E.J., Der C.J., Lee A.L. Ligand-dependent dynamics and intramolecular signaling in a PDZ domain. J. Mol. Biol. 2004;335:1105–1115. doi: 10.1016/j.jmb.2003.11.010. [DOI] [PubMed] [Google Scholar]
- 43.Poteete A.R., Hardy L.W. Genetic analysis of bacteriophage T4 lysozyme structure and function. J. Bacteriol. 1994;176:6783–6788. doi: 10.1128/jb.176.22.6783-6788.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dong F., Zhou H.X. Electrostatic contributions to T4 lysozyme stability: solvent-exposed charges versus semi-buried salt bridges. Biophys. J. 2002;83:1341–1347. doi: 10.1016/S0006-3495(02)73904-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Poteete A.R., Rennell D., Hardy L.W. Alteration of T4 lysozyme structure by second-site reversion of deleterious mutations. Protein Sci. 1997;6:2418–2425. doi: 10.1002/pro.5560061115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xu J., Baase W.A., Matthews B.W. The response of T4 lysozyme to large-to-small substitutions within the core and its relation to the hydrophobic effect. Protein Sci. 1998;7:158–177. doi: 10.1002/pro.5560070117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nolde S.B., Arseniev A.S., Billeter M. Essential domain motions in barnase revealed by MD simulations. Proteins. 2002;46:250–258. doi: 10.1002/prot.10030. [DOI] [PubMed] [Google Scholar]
- 48.Zegers I., Deswarte J., Wyns L. Trimeric domain-swapped barnase. Proc. Natl. Acad. Sci. USA. 1999;96:818–822. doi: 10.1073/pnas.96.3.818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fanning A.S., Anderson J.M. PDZ domains: fundamental building blocks in the organization of protein complexes at the plasma membrane. J. Clin. Invest. 1999;103:767–772. doi: 10.1172/JCI6509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Walma T., Spronk C.A., Vuister G.W. Structure, dynamics and binding characteristics of the second PDZ domain of PTP-BL. J. Mol. Biol. 2002;316:1101–1110. doi: 10.1006/jmbi.2002.5402. [DOI] [PubMed] [Google Scholar]
- 51.Ota N., Agard D.A. Intramolecular signaling pathways revealed by modeling anisotropic thermal diffusion. J. Mol. Biol. 2005;351:345–354. doi: 10.1016/j.jmb.2005.05.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.