

Abstract
Activation of the multicomponent enzyme NADPH oxidase requires the interaction between the tandem SH3 domain of the cytosolic subunit p47phox and the cytoplasmic tail of membrane-bound p22phox. In the resting state, p47phox exists in an autoinhibited conformation stabilized by intramolecular contacts between the SH3 domains and an adjacent polybasic region. Phosphorylation of three serine residues, Ser303, Ser304, and Ser328 within this polybasic region has been shown to be sufficient for the disruption of the intramolecular interactions thereby inducing an active state of p47phox. This active conformation is accessible to the cytoplasmic tail of p22phox and initiates the formation of the membrane-bound functional enzyme complex. Molecular dynamics simulations reveal insights in the mechanism of activation of the autoinhibited form of p47phox by in silico phosphorylation, of the three serine residues, Ser303, Ser304, and Ser328. The simulations highlight the major collective coordinates generating the opening and the closing of the two SH3 domains and the residues that cause the unmasking of the p22phox binding site.
Introduction
The phagocytic NADPH oxidase is a multisubunit enzyme that plays a vital role in innate immunity due to its ability to produce reactive oxygen species (ROS) in response to microbial infection. In the past, the microbicidal activity of ROS has been regarded as their main function and excessive ROS production has been linked to a number of disease states (1,2). However, over recent years it has become evident that ROS produced by NADPH oxidases in phagocytic and nonphagocytic cells also play important signaling roles and contribute to the regulation of a variety of cellular processes (3–6).
The phagocytic NADPH oxidase consists of a heterodimeric membrane-bound flavocytochrome b558 (containing gp91phox and p22phox) that constitutes the catalytic core and four cytosolic subunits: p40phox, p47phox, and p67phox, which form a tight heterotrimeric complex, and the small GTPase Rac. Activation requires translocation of the cytosolic components to the membrane-bound cytochrome, a process that is highly regulated, and which relies on reversible protein-protein interactions, phosphorylation, and specific recognition of phospholipids. The central player in this process is the p47phox subunit, which maintains the resting state through autoinhibitory intramolecular interactions. Stimulus-induced release of this conformation will in turn promote membrane translocation and association with the cytochrome. The p47phox subunit consists of a PX domain, a tandem SH3 domain, a polybasic/autoinhibitory region, and a proline-rich C-terminus (Fig. 1). In the inactive state, the tandem SH3 domains interact with the polybasic/autoinhibitory region in a highly unusual arrangement that has been termed the SuperSH3 domain (from now on referred to as sSH3). In this arrangement, the tandem SH3 domains cooperate to form a single ligand binding site with which the N-terminal portion of the polybasic region interacts in an intramolecular fashion. Phosphorylation of at least three serine residues (Ser303, Ser304, and Ser328) within the polybasic region has been shown to act as a switch to weaken this intramolecular interaction, thereby allowing membrane-bound p22phox to form a complex with p47phox and initiate oxidase assembly (7–12). A number of kinases have been implicated in these events, with PKC isoforms playing a particular important role. While it is not known at present if phosphorylation occurs sequentially and if conformational changes are required to allow access of the kinase, the importance of phosphorylation of Ser303, Ser304, and Ser328 has been clearly demonstrated by mutational studies. In contrast, the molecular determinants of the unmasking process have not been addressed yet, largely due to the experimental difficulties associated with producing proteins that are phosphorylated at multiple serine residues in a highly specific and stoichiometric fashion. Therefore, an in silico study of the structural and dynamic changes produced by selective phosphorylation of p47phox has been carried out, to provide a first insight into the underlying molecular mechanisms.
Figure 1.
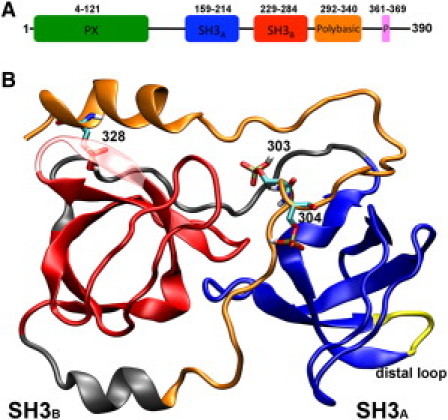
(A) Domain structure of p47phox. (B) Ribbon representation of the SH3A and SH3B domains and of the polybasic region. The phosphorylated residues are shown in licorice representation.
Previously, implicit solvent molecular dynamics (MD) simulations on the ligand recognition of sSH3 have been performed, focusing on the role of specific point mutations associated with chronic granulomatous disease (CGD), a severe immune disease caused by mutations in the NADPH oxidase, and their effect on the stability of the p47phox-p22phox complex (13). While this study targeted the mechanism related to the causes of inactivation at a submolecular level, the steps leading to the formation of the active complex starting from the autoinhibited form have not yet been addressed.
A study of phosphorylation effects on the coupling between two protein domains of Src kinases has unraveled the inactivation mechanism by a C-terminal tyrosine phosphorylation event with the help of MD simulations and has elucidated the communication between critical sites of phosphorylation by targeted MD (14). An alternative route to explore conformations accessible to a closed domain is to perform essential dynamics (ED) sampling (15) as already demonstrated for the mechanism of activation of enzymes (16,17).
We present here an in silico study which reveals the role of phosphorylation of three crucial serine residues in the polybasic region (Ser303, Ser304, and Ser328) on the dynamic coupling of the two SH3 domains in the autoinhibited form of p47phox. We focus particularly on the characterization of the release mechanism of the autoinhibited conformation caused by phosphorylation, and on the changes induced by local interactions due to this perturbation. To fully elucidate the dynamical behavior of the oxidase, we investigated the reversibility of the phosphorylation process and highlighted the principal components that drive the mechanical process restoring autoinhibition.
Methods
Modeling of the loop containing residues 198–201
p47phox crystallized as a domain-swapped dimer with the distal loop of SH3A acting as the hinge (PDB code 1NG2, see Fig. S1 in the Supporting Material). However, in solution, the crystallized fragment as well as the full-length protein are monomeric, and we have therefore used the biologically relevant monomer as the starting point for our simulations (18). We have included residues 198–201 of the distal loop (in yellow in Fig. 1), which was modeled using the modeling program MODELLER 8v2 in the biological monomer (19) with the model-loop.py script. The structure was kept rigid (except for the three residues before and after the 198–201 segment). The MODELLER library was used to generate loop conformations and the loops were scored using the dope_loopmodel script; of the obtained modeled loops, the best overall solution has been retained.
Simulations performed and protocol
Simulations on the autoinhibited sSH3 domain and on the in silico-generated phosphorylated form (sSH3–3P) were performed with the GROMACS package (20) using the GROMOS96 force field (21). The starting structure of the sSH3 domain was generated from the biological monomer created using the coordinates of the crystal structure (PDB code: 1NG2), including the modeled distal loop of SH3A (see above). The parameters utilized for the HPO4− group substituted on the serine residue are adapted from the phosphate group of the lipid parameterization (see Table S1 in the Supporting Material). The phosphoserine coordinates have been inserted with the module Biopolymer of INSIGHT II (22).
The molecules were solvated in a box of simple-point-charge water (23) and neutralized with Na+ ions. All the simulated boxes contained ∼9500 water molecules. Simulations were carried out at a constant temperature of 300 K. The Berendsen algorithm was applied for the temperature and pressure coupling (24). After a first steepest-descent energy minimization with positional restraints on solute, the LINCS algorithm (25) was used to constrain the bonds and to carry out an initial 200 ps simulation with the positions of the solute atoms restrained by a force constant of 3000 kJ/(mol∗nm2) to let the water diffuse around the molecule and to equilibrate. The particle mesh Ewald (PME) method (26) was used for the calculation of electrostatic contribution to nonbonded interactions (grid spacing of 0.12 nm) with a cutoff of 1.4 nm and a time step of 2 fs. The trajectory length for sSH3 domain was 20 ns long. The sSH3-3P system was run for 50 ns to observe spontaneous opening and for 4 ns of ED sampling along the first eigenvector (15). From the end-point of the ED simulation, another run of 20 ns has been performed by dephosphorylating the sSH3 (sSH3-DP). In all cases, the first and the last 500 ps of simulations were used for the energy decomposition analysis.
The ED sampling has been performed according to the procedure described in Daidone et al. (27). The Dynamite server (http://s12-ap550.bioch.ox.ac.uk:8078/dynamite_html/index.html) was used to produce further principal-component analysis (PCA) of the MD trajectories. Secondary structure analysis was obtained with DSSP (28). Solvent accessibilities areas were computed with the program POPS (29,30). Images were produced with VMD 1.8.5 (31).
Results and Discussion
MD simulations of sSH3 and sSH3-3P
The purpose of our study is to investigate the effects of selected phosphorylation events on the stability and dynamics of the sSH3 structure and the possible consequences for its functional role. We therefore performed an analysis on the structural implications of the phosphorylation of three serines: Ser303, Ser304, and Ser328 of the polybasic region by an extensive MD simulations run (50 ns). We also performed a control simulation on the wild-type sSH3 domain (20 ns) as a reference, to validate the stability and dynamical behavior within the applied force field and experimental conditions.
The analysis of the root mean-square fluctuation for the simulated systems (see Fig. S2 A) reveals that in both phosphorylated and nonphosphorylated forms, higher fluctuations are observed for residues belonging to the linker connecting the two SH3 domains (residues 214–228) and within the polybasic region (residues 301–331) (see Fig. 1 for definitions). In the case of the sSH3-3P domain, higher root mean-square fluctuation values are observed for residues adjacent to the phosphorylated regions. Visual inspection of the first 20 ns of the sSH3-3P simulation reveals that in silico phosphorylations induce only partial opening of the sSH3 tandem domain. However, the underlying structural changes are noteworthy: we observe the loss of ∼70% of the intramolecular interactions between the sequence RGAPPRRSS (amino acids 296–304) of the polybasic region and the two SH3 domains (Fig. 2 A) identified in the crystal structure of the autoinhibited conformation of p47phox (18).
Figure 2.
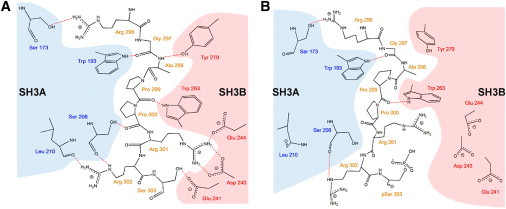
Schematic representation of the interactions observed between the motif RGAPPRRS and the tandem sSH3 domain. Residues belonging to the SH3A and SH3B domains are highlighted by shaded areas. (A) Interactions identified in the crystal structure. (B) Interaction detected for the sSH3-3P after 20 ns of MD simulation.
In particular, we lost:
-
1.
The interactions formed by the guanidinium groups of Arg301 and Arg302 with the amino acids Leu210, Asp243, and Glu244.
-
2.
The hydrogen bond between backbone carbonyl of Pro300 and side chain of Ser208.
-
3.
The hydrogen bond between the backbone of Ala298 and side chain of Tyr279.
Moreover, the presence of a phosphoserine instead of Ser303 leads to disruption of the interaction with Glu241, most probably due to charge repulsion (Fig. 2 B). To investigate this further and to ensure that the sSH3-3P domain explored other possible conformational states, a further 30 ns of simulation were performed, reaching a total of 50 ns. During these last 30 ns of simulations, the RMSD mean value of the protein (Fig. S2 B) remained stable (at ∼3.0 Å from the initial structure). No remarkable changes in the fold occurred, as confirmed by the analysis of the evolution of secondary structure elements as a function of time (Fig. S3 B), and the molecule did not open more to expose buried residues. Accordingly, no further loss of interactions with the polybasic region was observed, except for the H-bond formed between backbone carbonyl of Gly297 and the side chain of Trp193. Such a result supports reliability in the force field and simulations conditions used. Almost all intramolecular interactions identified in the crystal structure between the sequence 296RGAPPRRSS304 and the two SH3 domains are preserved.
Structural role of phosphoserines residues
A detailed analysis of the phosphoserine contacts during the sSH3-3P trajectory, indicates that the phosphate group of pSer328 forms a hydrogen bond with the side chain of Arg267, as hypothesized by Groemping et al. (18). On the other hand, the interaction between Ser328 and Arg267, present in the initial structure of the tandem sSH3 domain, was lost after few nanoseconds of simulation. It has been previously suggested (18) that Arg316 and Arg318 in the polybasic region might be implicated in electrostatic interaction with the phosphate group of pSer303. We thus performed a detailed distance analysis to investigate the behavior of the arginines adjacent to pSer303/304, taking into account the possible competing interactions with the acidic residues in close proximity (Fig. 3 A). We calculated a scatter plot of the minimum distances between each arginine and the pSer residues on one axis and with the acidic residues on the other axis for the sSH3 and the sSH3-3P simulations (Fig. 3 B and C, respectively). We observed heterogeneous behaviors, reflecting the different environment of each residue. The highly buried Arg301 showed a restricted mobility in the two simulations, mainly due to strong interactions with both the acidic and the pSer groups.
Figure 3.
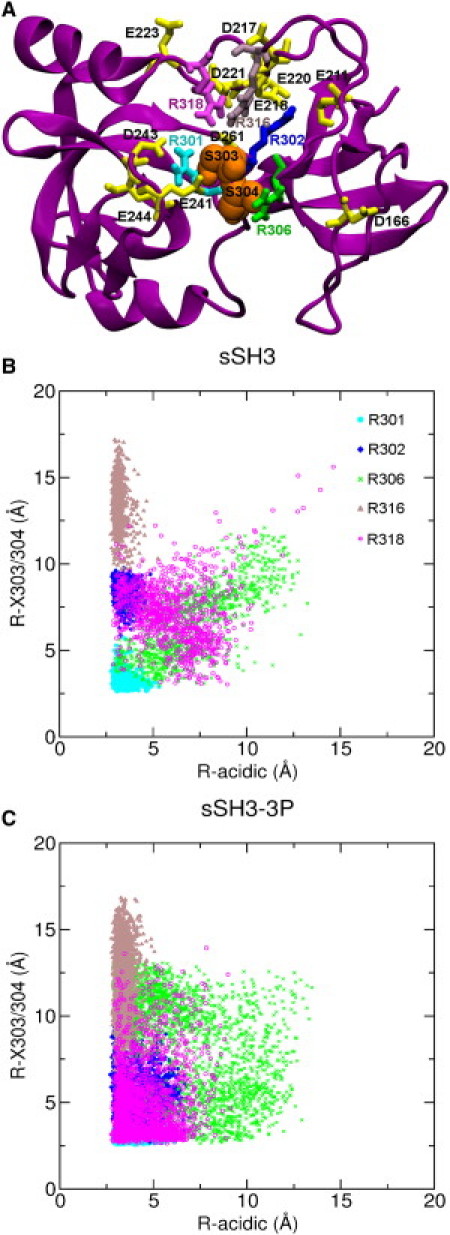
Distance analysis of selected arginine residues in the polybasic region. (A) Cartoon representation of the starting sSH3 structure. The arginine residues (R301, R302, R306, R316, and R318) are shown as licorice. The acidic residues in close proximity to the selected arginines (namely D166, E211, D217, E218, E220, D221, E223, E241, D243, E244, and D261) are represented in licorice, while the S303 and S304 residues are shown in van der Waals spheres. Scatterplots of the minimum distances between each of the selected arginines and the acidic residues shown in A (x axis) and the phosphoserine residues 303–304 (y axis) for the sSH3 (B) and sSH3-3P (C) trajectories. The minimum distances are calculated over all the possible pairs of distances between the N atoms of the guanidinium group of arginines and the side-chain nonhydrogen atoms of the other residues.
Arg302 interacted preferentially with the acidic residues in both simulations, even if it is observed to have moved closer to the phosphoserines in sSH3-3P. The highest mobility was found for the solvent-exposed Arg306, which displayed significant interactions with either the acidic or phosphoserine groups only in the sSH3-3P simulation. Arg316 was involved in strong and stable contacts with the acidic groups of the linker region in both simulations. Arg318 was very mobile in the sSH3 simulation, engaging in loose interactions with the acidic groups and the phosphoserines. Interestingly, in the sSH3-3P simulation this residue experienced a significant shift toward smaller distance values with both the acidic and phosphoserine groups (Fig. 3 C). Therefore, this is the only residue of the previously suggested ones that seems to be directly implicated in the activation of the NADPH oxidase.
Solvent organization around the sSH3 domain
To investigate the role of solvent in the structural stability and/or flexibility of the molecule, we computed the time occupancy of water molecules during the simulated time and produced a map of the hydration sites around the studied systems. In particular, the study for the native sSH3 domain allows us to highlight water molecules that may have a key role in the structural stability of the molecule and hence the preservation of the autoinhibited form. Specifically, we observe one water molecule (Fig. 4 B) with a very long occupancy time (>10 ns), caged within four residues: Gly262, Trp263, and Ser277 of the SH3B domain, and Pro299 of the polybasic region. This molecule is involved in water-mediated hydrogen bonds between the residues belonging to the SH3B domain and the backbone of Pro299, and it seems to be positioned to hold in place the polybasic region in the autoinhibited form. Interestingly, in the crystal structure of autoinhibited p47phox a water molecule was present in the same position (Fig. 4 A), supporting the notion that a water molecule in this position is necessary to fold stability.
Figure 4.
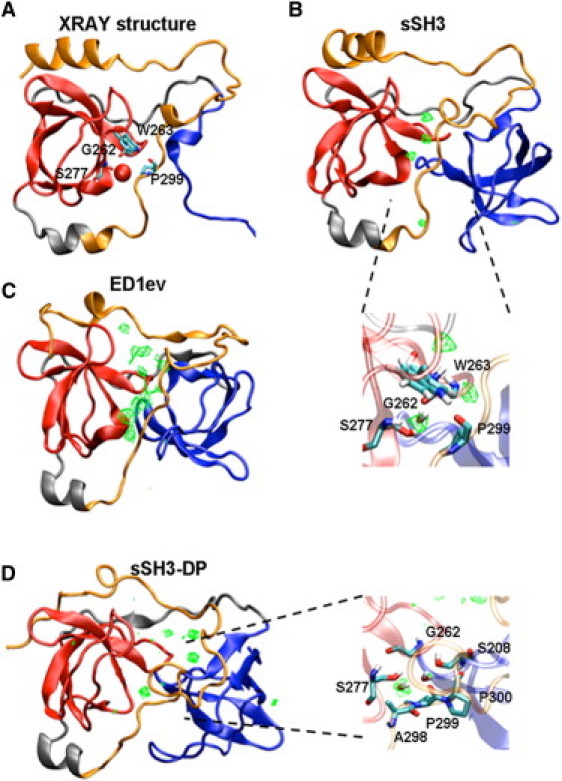
(A) Ribbon representation of the autoinhibited p47phox structure (PDB code 1NG2). (B–D) Water high residence densities represented as grids are displayed for sSH3 (calculated over the entire simulated time), for ED1ev (calculated over the last 2 ns of ED), and for sSH3-DP (calculated over the last 2 ns of MD simulation), respectively. In the close-up views (blown-up rectangles), the residues participating in interactions with the water molecule for sSH3 and sSH3-DP are shown.
For the sSH3-3P molecule, the insertion of a water molecule between residues Ser208 of SH3A and Asp261 of SH3B is detected, and persists in the same position for most of the simulated time. This water molecule is hydrogen-bonded to the two residues and locks in place the hinge region between the SH3A and SH3B domains, impeding thereby a further opening of the cage (data not shown). Under these conditions, it appears that the sSH3-3P domain simulation has reached a local minimum, most probably a plateau in the associated energy landscape. To study the mechanism of opening of the two domains to expose residues necessary to the binding, we therefore resorted to perform enhanced sampling by means of ED sampling simulations (see also ED Sampling: Opening of the sSH3 Domain).
The critical role of the solvent in the structural stability of the sSH3 domain is also highlighted by the analysis of the energy components for the moieties composing the tandem domain. The presence of the phosphorylated residues affects mostly the electrostatic components (Coulomb term) relative to the interactions between the polybasic moiety and the solvent, when compared to the sSH3 20 ns simulation (see Table S2). The sSH3-3P simulation shows favored intra-SH3 domains interactions, possibly as a result of the enhanced correlation in the two domains' motion, as will be described in the next paragraph. During these fluctuations, the SH3A interacts more favorably with the polybasic region, losing interactions with the solvent. For the sSH3-3P (ED1) simulation aimed at encouraging the opening, the ED sampling manages to overcome the intramolecular attraction of the two domains previously observed by exposing them more to the solvent.
Generally, the SH3B domain shows a more favorable interaction with the solvent than SH3A in all the simulations. As already discussed, the site harboring the highly time-resident water molecule creates a constraint between the polybasic region and the SH3B moiety; therefore, simulations that favor the opening of the sSH3 domain cause a rearrangement of the solvent around this region to compensate the loss of favorable local interactions.
Principal component analysis of sSH3 and sSH3-3P
Principal component analysis (PCA) is a powerful tool for finding global, correlated motions in atomic simulations of macromolecules (32,33). PCA is often used to reduce the complexity of the data obtained by MD simulations trajectories. The motion is decomposed in the principal components associated to an eigenvector and an eigenvalue. For PCA (34) and quasiharmonic analyses (35–38) it has been shown that >90% of the total atomic motion is described by <5% of all degrees of freedom (33,39–41). In particular, the essential subspace explored by the PCA modes, i.e., the one contributing most to the atomic displacement, can be exploited in directing MD simulations toward a desired state. This can be achieved by the use of different approaches for sampling enhancement like ED sampling (15), metadynamics (42), conformational flooding (43), umbrella sampling (44), adaptive biasing force (45), and ED replica exchange (46) methods. Here we used ED sampling simulations (described in the next section) aimed at opening the sSH3 domain and exposing new residues for competitive binding.
PCA analyses on our system showed that, in both the phosphorylated and nonphosphorylated forms, the motion of Cα atoms is almost fully described by the first 10 eigenvectors. In particular, the first eigenvector accounts for ∼40% and 45% of the total amount of the motion for the sSH3 and sSH3-3P, respectively (see Fig. S4).
Projection of the trajectories along the first two eigenvectors for the sSH3-3P simulation (Fig. 5) shows that the protein explores three conformational states and remains trapped in the last explored state until the end of the simulation. During the first five nanoseconds, the conformational transition occurs along both eigenvectors 1 and 2. However, during the following 15 ns, conformational changes occurs only along the first eigenvector; once transition to the last state had occurred, the system did not show any significant changes along the two analyzed eigenvectors for the last 30 ns.
Figure 5.
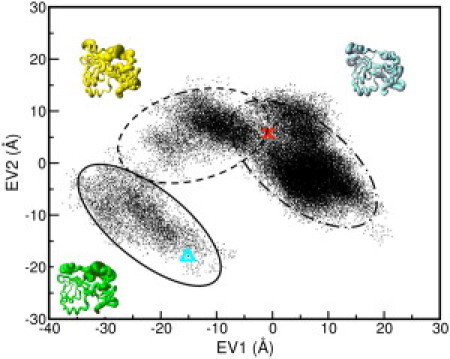
Projections of the trajectory of the sSH3-3P simulation onto the first (EV1) and second (EV2) eigenvectors. (Triangle and the cross) Projection of the starting and final structure onto the first and the second eigenvectors, respectively. The conformational space explored during the first 5 ns (solid lines), between 5 and 15 ns (dashed lines), and the last 30 ns (dash-dotted lines) is defined by the circles. The range of motion observed along the first eigenvector for each conformational subspace sampling is displayed by the sausage plots representation, in which the thickness of the sausage is proportional to the fluctuation from the average structure of the underlying ensemble.
Fig. 5 reports also sausage plot representations embedding the mobility observed along the first eigenvector calculated for the three conformational states. The plots highlight that a large mobility is initially associated with the linker loop region and the areas adjacent to the phosphorylated residues, resulting in thicker sausage diameter. They also clearly show that the fluctuations of the linker loop are sensibly reduced after 20 ns (thinner sausage diameter), whereas the polybasic region and the SH3 domains show an evident flexibility throughout the performed simulations. A representation of correlated motions in the sSH3 and sSH3-3P molecules is shown in Fig. 6. As expected, in the sSH3, a mesh of lines interconnects the Cα atoms comprised within secondary structural elements of each domain (Fig. 6 A), consistent with locally correlated motions. Interestingly, in the sSH3-3P, the two domains appear to be strongly correlated in their motion, as highlighted by the relative correlation plot (Fig. 6 B) with mesh lines enclosing the two domains. By using porcupine plots that graphically display the direction of the motion of the Cα atoms along the first eigenvector, in the sSH3-3P simulation we observe a twisting movement of the two SH3 domains, which rotate in opposite directions around an ideal axis connecting them (Fig. 7, A and B).
Figure 6.
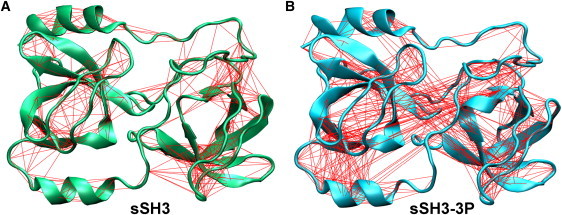
Correlated motions of Cα residues belonging to SH3A and SH3B domains in (A) sSH3 simulation and (B) sSH3-3P simulation. Motions with >50% correlation are indicated by lines connecting the involved residues.
Figure 7.
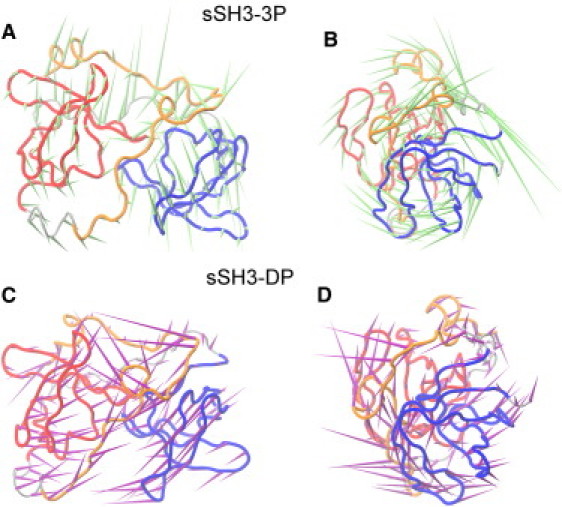
Porcupine images of the motions corresponding to the first eigenvector for simulations of (A and B) sSH3-3P and (C and D) sSH3-DP. Each Cα atom has a cone attached pointing in the direction of motion described by the eigenvector corresponding to that atom.
ED sampling: opening of the sSH3 domain
Starting from the sSH3-3P conformation, 4 ns of ED sampling were performed along the first eigenvector of the PCA. The ED sampling technique was employed to encourage the protein to move along this coordinate direction and to explore a large region of space that is not explored in the free MD simulations (15).
By comparing the structure of the sSH3-3P before and at the end of the ED sampling, one can observe that the two SH3 domains seem to have moved away from each other during the simulation (Fig. S5). The buried cage connecting the SH3 domains appears to have significantly opened and the polybasic region has dislocated from the initial position, leaving free accessibility to the binding site (Fig. S5 B). The time evolution of the secondary structures shows that the content in β-sheet and α-helical secondary structure elements is maintained along the trajectory (Fig. S3 C), confirming that the opening does not significantly alter the main structure of the protein. Widening the cage implies free accessibility of the region connecting the domains and several water molecules are now free to insert between them as shown in Fig. 4 C, where a map of the highest occupancy hydration sites is reported as a grid density mesh. It is worth noting that the opening of the cage also leads to the diffusion of the water molecule described above that prevented the complete opening of the cage through hydrogen bonds between residues Ser208 and Asp261 of the two domains.
To obtain a quantitative measure of the sSH3 domain opening with consequent dislocation of the polybasic region from the initial position, the solvent accessible surface area (SASA) of the residues in contact with the polybasic region (within 5 Å) has been calculated (29,30). These residues (displayed in Fig. S5) also include the residues of the SH3 domains responsible for the interaction with p22phox (residues 167, 193, 204, 204, 209, 243, 244, 263, and 276 highlighted in green) (18). The value of total accessible area calculated for the selected residues after the ED simulation is 1039 Å2, while for sSH3 and sSH3-3P it is 867 and 870 Å2, respectively. These values confirm the earlier observation that sSH3-3P simulation alone was not able to significantly open the structure and simulate the activation mechanism. For the ED simulation, on the other hand, the previously named residues become definitely exposed to the solvent. These observations confirm that the previous equilibrium sSH3-3P simulation was necessary:
-
1.
to define the principal components for the ED sampling; and
-
2.
to initiate some key rearrangements that are better explored by the ED.
Nevertheless, some of the p22phox interacting residues that became slightly exposed during the sSH3-3P simulation continue to expose themselves during the ED simulation. The amount of solvent accessibility is quantified in Table 1 where the SASA of the residues responsible for the interaction with p22phox is reported. Of particular note are the more exposed residues Trp193, Trp204, Pro206, and Phe209, belonging to SH3A, and Asp243 belonging to SH3B.
Table 1.
SASA (Å2) of the residues responsible for the interaction with p22phox
| SH3A |
SH3B |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Y167 | W193 | W204 | P206 | F209 | D243 | E244 | W263 | P276 | |
| sSH3-3P | 47 | 21 | 38 | 15 | 19 | 56 | 30 | 54 | 15 |
| sSH3-3P (ED1) | 46 | 70 | 59 | 24 | 37 | 83 | 25 | 35 | 10 |
| sSH3-DP | 24 | 41 | 86 | 17 | 17 | 82 | 20 | 27 | 13 |
Solvent-accessible surface areas (SASA) of the residues in contact with p22phox were computed with the program POPS (29,30). Boldface represents the most important changes observed.
The analysis of the contacts formed by the phosphoserine residues indicates that, during the ED trajectory, two out of three interactions previously created during the sSH3-3P simulation (see MD Simulations of sSH3 and sSH3-3P) are broken, encouraging this way the dislocation of the polybasic region. These interactions are the ones formed by pSer303 with Arg318 and by pSer328 with Arg267, while the electrostatic interaction between pSer304 and Arg301 (residue belonging to polybasic region) is conserved throughout the trajectory. This implies that the interactions internal to the polybasic region are conserved at the expense of polybasic-SH3 domain interactions.
After ED sampling: closure of the sSH3 domain
To assess whether we could observe reversibility in the sSH3 domain opening-closing process, we dephosphorylated the final structure obtained from ED1ev and performed 20 ns of MD simulations on this system (sSH3-DP). The structural response of the protein upon dephosphorylation can be observed by the time evolution of the radius of gyration of the sSH3-DP (Fig. 8 A). The Rg of the protein rapidly decreases during the first 5 ns of simulation from 16.8 to 16.1 Å and then fluctuates around this value until end of trajectory. The value at which this measure finally converges is almost the same of that of the native sSH3 domain, suggesting that, once the phosphates are removed, the protein rearranges itself toward a structure close to the native one.
Figure 8.
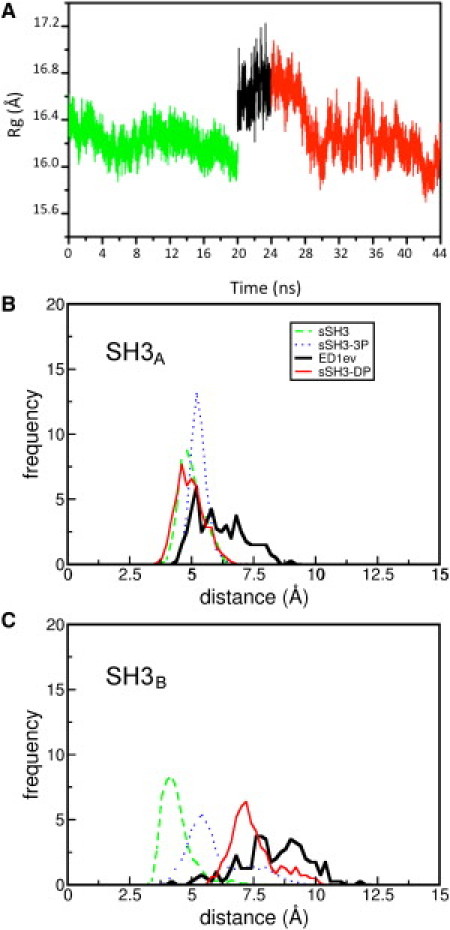
(A) Radius of gyration of Cα atoms during the simulated time for sSH3 (0-20 ns), ED1ev (20-24 ns), and sSH3-DP (24-44 ns). Distributions of the SH3A-polybasic region (B) and of the SH3B-polybasic region (C) minimum distances for the sSH3 (dashed line), sSH3-3P (dotted line), ED1ev (thick solid line), and sSH3-DP (thinner solid line) trajectories. Minimum distances are calculated over Cα atoms of either the SH3A or the SH3B domain and the R301-S304 segment of the polybasic region.
To compare the effects of dephosphorylation on the principal motions of the protein after the ED sampling, we investigated the most significant collective modes of motion occurring during the sSH3-DP simulation. We decomposed the global motion of the proteins into the few principal motions and identified the ones contributing most to the closure of the two domains. We found that the first eigenvector accounts for >50% of the total amount of the motion for sSH3-DP simulation. The directions of motion described in the first eigenvector for sSH3-DP is shown in the porcupine plots in Fig. 7, C and D. It can be seen that the motion described in this case is different from the one observed in sSH3-3P before the ED (Fig. 7, A and B). The principal motion of sSH3-DP can be depicted as a motion that brings the two domains closer.
To quantify the amount of closure of the sSH3 domain in the dephosphorylated form, we have calculated the SASA of the residues in contact with the polybasic region (within 5 Å) and compared them to the same values in the native sSH3, the sSH3-3P and the ED1ev structure. The calculated total area for the selected residues after the sSH3-DP simulation is 818 Å2. This value is close to the values calculated for the sSH3 and sSH3-3P (867 and 870 Å2, respectively), indicating that the movement of the domains leads to the restoring of a nativelike solvent accessibility of the residues in contact with the polybasic region. The SASA of the residues responsible for the interaction with p22phox for sSH3-DP are shown in Table 1, along with the values calculated for sSH3-3P and ED1ev for comparison. The analysis of the data reveals that some of the most exposed residues during the ED1ev become less exposed to the solvent after the sSH3-DP trajectory. These residues, all belonging to SH3A, are Tyr167, Trp193, Pro206, and Phe209. On the other hand, two residues, Trp204 and Asp243, exposed to the solvent during the ED1ev, remain exposed to the solvent also during the sSH3-DP simulation. In addition, the changes in the interactions between the SH3 domains and the polybasic region observed in the ED1ev structure turned out to be only partially reversible upon dephosphorylation (Fig. S5 C).
To better characterize the extent of native contacts restored upon dephosphorylation, we analyzed the distribution of the distances between the 301–304 segment of the polybasic region with either the SH3A or the SH3B domain. For the SH3A domain (Fig. 8 B), it is possible to see that the ED1ev (thick solid line) distance profile is shifted toward larger values than the sSH3 one (dashed line), indicating a loss of interactions between the polybasic segment and the SH3 domain. However, lost contacts seem to be restored after dephosphorylation, because the SH3-DP profile (thinner solid line) is well superimposed onto the sSH3 one. The same observations hold for the SH3B domain (Fig. 8 C), although in this case the distance of the polybasic region with SH3B domain is only partially recovered by the SH3-DP simulation (thinner solid line). It should be pointed out that the increment of the distance between the polybasic region and both the SH3 domains can already be observed in the unbiased sSH3-3P simulation (dotted line), indicating that, to some extent, the destabilization of the interactions occurs as an intrinsic consequence of the phosphorylation and it is not an artifact of the ED sampling procedure.
To analyze the overall contact rearrangements occurring in the different simulations, we compared the conservation of both short- and long-range native contacts found in the final conformations with respect to the x-ray structure (Fig. S6). Contacts are generally well preserved within each of the SH3 domains in all cases (shown in blue scale). For the unbiased sSH3-3P simulation (Fig. S6, short range), the most important short-range contact losses are mainly found in the interdomain linker (shown in red scale) and in the region close to the pSer303 and pSer304 residues. The rearrangements observed at the end of the ED1ev trajectory involve more extensively the polybasic region and the flexible C-terminal helix (Fig. S6). After dephosphorylation, the long-range contacts are partly recovered (Fig. S6, long range), suggesting that the system is relaxing back toward the starting sSH3 conformation. The rearrangement of the two domains to restore the native structure has, as a consequence, the entrapment of a water molecule by residues Gly262, Ser277, and Pro299 the same residues which were surrounding the water molecule in the x-ray structure highlighted in Fig. 4 A.
Conclusions
In this work we have addressed the effect of site-specific phosphorylation on the dynamic coupling of the tandem SH3 domains in the autoinhibited form of p47phox, using MD simulations and ED sampling. This approach proved very useful in driving our simulations toward the opening of the sSH3 structure and in obtaining a conformation that mimics the activated state. Most importantly, the simulations highlight the mechanical driving force toward the activated form caused by phosphorylation, and the key residues exposed during the process. The interesting result is the observed reversibility of the process: dephosphorylation of the opened sSH3 structure induces a partial closure of the domain and a reorganization of the secondary structure elements toward the native ones. Particularly remarkable is the behavior of a crucial water molecule observed to maintain the two SH3 domains in the autoinhibited form that reversibly positions itself in the same site after dephosphorylation and closure of the activated state. Furthermore, in characterizing the molecular determinants of the activation process, we identified specific hydration sites crucial to the stability of p47phox that may play a pivotal role in the SH3A-SH3B domains opening and closing mechanism. The computational results highlight a predominant role of SH3A in the function of p47phox as the opening process leads to an increased exposure of residues in this domain. This observation is in strong agreement with previous experimental evidence, which showed that SH3A plays a key role in the interaction with p22phox (18).
Taken together, the data presented here show that MD simulations are a powerful approach to the study of biological processes that are not easily accessible by experimental approaches, and can provide useful insights into the molecular determinants that drive phosphorylation-induced conformational changes.
Acknowledgments
B.P. was funded by a short-term fellowship from the Federation of the Societies of Biochemistry and Molecular Biology. A.F. and F.F. were funded by the Leverhulme Trust (grant No. F/07 040/AL), and K.R. and F.F. were funded by the Medical Research Council.
Footnotes
This is an Open Access article distributed under the terms of the Creative Commons-Attribution Noncommercial License (http://creativecommons.org/licenses/by-nc/2.0/), which permits unrestricted noncommercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Contributor Information
Katrin Rittinger, Email: katrin.rittinger@nimr.mrc.ac.uk.
Franca Fraternali, Email: franca.fraternali@kcl.ac.uk.
Supporting Material
References
- 1.Babior B.M. Phagocytes and oxidative stress. Am. J. Med. 2000;109:33–44. doi: 10.1016/s0002-9343(00)00481-2. [DOI] [PubMed] [Google Scholar]
- 2.Benard V., Bokoch G.M., Diebold B.A. Potential drug targets: small GTPases that regulate leukocyte function. Trends Pharmacol. Sci. 1999;20:365–370. doi: 10.1016/s0165-6147(99)01367-x. [DOI] [PubMed] [Google Scholar]
- 3.Finkel T. Oxidant signals and oxidative stress. Curr. Opin. Cell Biol. 2003;15:247–254. doi: 10.1016/s0955-0674(03)00002-4. [DOI] [PubMed] [Google Scholar]
- 4.Lee S.R., Kwon K.S., Rhee S.G. Reversible inactivation of protein-tyrosine phosphatase 1B in A431 cells stimulated with epidermal growth factor. J. Biol. Chem. 1998;273:15366–15372. doi: 10.1074/jbc.273.25.15366. [DOI] [PubMed] [Google Scholar]
- 5.Meng T.C., Fukada T., Tonks N.K. Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol. Cell. 2002;9:387–399. doi: 10.1016/s1097-2765(02)00445-8. [DOI] [PubMed] [Google Scholar]
- 6.Brown D.I., Griendling K.K. Nox proteins in signal transduction. Free Radic. Biol. Med. 2009;47:1239–1253. doi: 10.1016/j.freeradbiomed.2009.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sumimoto H., Kage Y., Takeshige K. Role of Src homology 3 domains in assembly and activation of the phagocyte NADPH oxidase. Proc. Natl. Acad. Sci. USA. 1994;91:5345–5349. doi: 10.1073/pnas.91.12.5345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.el Benna J., Faust L.P., Babior B.M. The phosphorylation of the respiratory burst oxidase component p47phox during neutrophil activation. Phosphorylation of sites recognized by protein kinase C and by proline-directed kinases. J. Biol. Chem. 1994;269:23431–23436. [PubMed] [Google Scholar]
- 9.El Benna J., Faust R.P., Babior B.M. Phosphorylation of the respiratory burst oxidase subunit p47phox as determined by two-dimensional phosphopeptide mapping. Phosphorylation by protein kinase C, protein kinase A, and a mitogen-activated protein kinase. J. Biol. Chem. 1996;271:6374–6378. doi: 10.1074/jbc.271.11.6374. [DOI] [PubMed] [Google Scholar]
- 10.Fontayne A., Dang P.M., El-Benna J. Phosphorylation of p47phox sites by PKCα, βII, δ, and ζ: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry. 2002;41:7743–7750. doi: 10.1021/bi011953s. [DOI] [PubMed] [Google Scholar]
- 11.Inanami O., Johnson J.L., Babior B.M. Activation of the leukocyte NADPH oxidase by phorbol ester requires the phosphorylation of p47phox on serine 303 or 304. J. Biol. Chem. 1998;273:9539–9543. doi: 10.1074/jbc.273.16.9539. [DOI] [PubMed] [Google Scholar]
- 12.Park J.W., Hoyal C.R., Babior B.M. Kinase-dependent activation of the leukocyte NADPH oxidase in a cell-free system. Phosphorylation of membranes and p47phox during oxidase activation. J. Biol. Chem. 1997;272:11035–11043. doi: 10.1074/jbc.272.17.11035. [DOI] [PubMed] [Google Scholar]
- 13.Watanabe Y., Tsuboi H., Miyamoto A. Molecular dynamics study on the ligand recognition by tandem SH3 domains of p47phox, regulating NADPH oxidase activity. Comput. Biol. Chem. 2006;30:303–312. doi: 10.1016/j.compbiolchem.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 14.Young M.A., Gonfloni S., Kuriyan J. Dynamic coupling between the SH2 and SH3 domains of c-Src and Hck underlies their inactivation by C-terminal tyrosine phosphorylation. Cell. 2001;105:115–126. doi: 10.1016/s0092-8674(01)00301-4. [DOI] [PubMed] [Google Scholar]
- 15.Amadei A., Linssen A.B.M., Berendsen H.J. An efficient method for sampling the essential subspace of proteins. J. Biomol. Struct. Dyn. 1996;13:615–625. doi: 10.1080/07391102.1996.10508874. [DOI] [PubMed] [Google Scholar]
- 16.Daidone I., Roccatano D., Hayward S. Investigating the accessibility of the closed domain conformation of citrate synthase using essential dynamics sampling. J. Mol. Biol. 2004;339:515–525. doi: 10.1016/j.jmb.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 17.Friedman R., Caflisch A. Pepsinogen-like activation intermediate of plasmepsin II revealed by molecular dynamics analysis. Proteins. 2008;73:814–827. doi: 10.1002/prot.22105. [DOI] [PubMed] [Google Scholar]
- 18.Groemping Y., Lapouge K., Rittinger K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell. 2003;113:343–355. doi: 10.1016/s0092-8674(03)00314-3. [DOI] [PubMed] [Google Scholar]
- 19.Sali A., Blundell T.L. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 20.Van Der Spoel D., Lindahl E., Berendsen H.J. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 21.Daura X., Jahn B., Mark A.E. Parametrization of aliphatic CHn united atoms of GROMOS96 force field. J. Comput. Chem. 1998;19:535–547. [Google Scholar]
- 22.INSIGHT II. Molecular Modeling Software, Accelrys, San Diego, CA.
- 23.Berendsen H.J.C., Postma J.P.M., Hermans J. In: Interaction Models for Water in Relation to Protein Hydration. Pullman B., editor. 1981. pp. 331–342. [Google Scholar]
- 24.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 25.Hess B., Bekker H., Fraaije J. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. doi: 10.1021/ct700200b. [DOI] [PubMed] [Google Scholar]
- 26.Darden T., Perera L., Pedersen L. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Structure. 1999;7:55–60. doi: 10.1016/s0969-2126(99)80033-1. [DOI] [PubMed] [Google Scholar]
- 27.Daidone I., Amadei A., Nola A.D. Molecular dynamics simulation of protein folding by essential dynamics sampling: folding landscape of horse heart cytochrome c. Biophys. J. 2003;85:2865–2871. doi: 10.1016/S0006-3495(03)74709-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kabsch W., Sander C. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 1983;22:2577–2637. doi: 10.1002/bip.360221211. [DOI] [PubMed] [Google Scholar]
- 29.Fraternali F., Cavallo L. POPS: a fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 2002;30:2950–2960. doi: 10.1093/nar/gkg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cavallo L., Kleinjung J., Fraternali F. POPS: a fast algorithm for solvent accessible surface areas at atomic and residue level. Nucleic Acids Res. 2003;31:3364–3366. doi: 10.1093/nar/gkg601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 32.Fujiwara S., Amisaki T. Molecular dynamics study of conformational changes in human serum albumin by binding of fatty acids. Proteins. 2006;64:730–739. doi: 10.1002/prot.21053. [DOI] [PubMed] [Google Scholar]
- 33.Amadei A., Linssen A.B., Berendsen H.J. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 34.Lange O.F., Grubmüller H. Can principal components yield a dimension reduced description of protein dynamics on long time scales? J. Phys. Chem. B. 2006;110:22842–22852. doi: 10.1021/jp062548j. [DOI] [PubMed] [Google Scholar]
- 35.Karplus M., Kushick J. Method for estimating the configurational entropy of macromolecules. Macromolecules. 1981;14:325–332. [Google Scholar]
- 36.Levy R., Karplus M., Perahia D. Evaluation of the configurational entropy for proteins: application to molecular dynamics simulations of an α-helix. Macromolecules. 1984;17:1370–1374. [Google Scholar]
- 37.Levy R.M., Srinivasan A.R., McCammon J.A. Quasi-harmonic method for studying very low frequency modes in proteins. Biopolymers. 1984;23:1099–1112. doi: 10.1002/bip.360230610. [DOI] [PubMed] [Google Scholar]
- 38.Teeter M., Case D. Harmonic and quasiharmonic descriptions of crambin. J. Phys. Chem. 1990;94:8091–8097. [Google Scholar]
- 39.Berendsen H.J., Hayward S. Collective protein dynamics in relation to function. Curr. Opin. Struct. Biol. 2000;10:165–169. doi: 10.1016/s0959-440x(00)00061-0. [DOI] [PubMed] [Google Scholar]
- 40.Hayward S., Kitao A., Gō N. Effect of solvent on collective motions in globular protein. J. Mol. Biol. 1993:1207–1217. doi: 10.1006/jmbi.1993.1671. [DOI] [PubMed] [Google Scholar]
- 41.Kitao A., Gō N. Investigating protein dynamics in collective coordinate space. Curr. Opin. Struct. Biol. 1999;9:164–169. doi: 10.1016/S0959-440X(99)80023-2. [DOI] [PubMed] [Google Scholar]
- 42.Spiwok V., Lipovová P., Králová B. Metadynamics in essential coordinates: free energy simulation of conformational changes. J. Phys. Chem. B. 2007;111:3073–3076. doi: 10.1021/jp068587c. [DOI] [PubMed] [Google Scholar]
- 43.Lange O.F., Schäfer L.V., Grubmüller H. Flooding in GROMACS: accelerated barrier crossings in molecular dynamics. J. Comput. Chem. 2006;27:1693–1702. doi: 10.1002/jcc.20473. [DOI] [PubMed] [Google Scholar]
- 44.Joshi H.V., Jedrzejas M.J., de Groot B.L. Domain motions of hyaluronan lyase underlying processive hyaluronan translocation. Proteins. 2009;76:30–46. doi: 10.1002/prot.22316. [DOI] [PubMed] [Google Scholar]
- 45.Henin J., Fiorin G., Klein M.L. Exploring multidimensional free energy landscapes using time-dependent biases on collective variables. J. Chem. Theory Comput. 2010;6:35–47. doi: 10.1021/ct9004432. [DOI] [PubMed] [Google Scholar]
- 46.Kubitzki M.B., de Groot B.L. Molecular dynamics simulations using temperature-enhanced essential dynamics replica exchange. Biophys. J. 2007;92:4262–4270. doi: 10.1529/biophysj.106.103101. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.