

Abstract
Misfolding and degradation of CFTR is the cause of disease in patients with the most prevalent CFTR mutation, an in-frame deletion of phenylalanine (F508del), located in the first nucleotide-binding domain of human CFTR (hNBD1). Studies of (F508del)CFTR cellular folding suggest that both intra- and inter-domain folding is impaired. (F508del)CFTR is a temperature-sensitive mutant, that is, lowering growth temperature, improves both export, and plasma membrane residence times. Yet, paradoxically, F508del does not alter the fold of isolated hNBD1 nor did it seem to perturb its unfolding transition in previous isothermal chemical denaturation studies. We therefore studied the in vitro thermal unfolding of matched hNBD1 constructs ±F508del to shed light on the defective folding mechanism and the basis for the thermal instability of (F508del)CFTR. Using primarily differential scanning calorimetry (DSC) and circular dichroism, we show for all hNBD1 pairs studied, that F508del lowers the unfolding transition temperature (Tm) by 6–7°C and that unfolding occurs via a kinetically-controlled, irreversible transition in isolated monomers. A thermal unfolding mechanism is derived from nonlinear least squares fitting of comprehensive DSC data sets. All data are consistent with a simple three-state thermal unfolding mechanism for hNBD1 ± F508del: N(±MgATP) ⇄ IT(±MgATP) → AT → (AT)n. The equilibrium unfolding to intermediate, IT, is followed by the rate-determining, irreversible formation of a partially folded, aggregation-prone, monomeric state, AT, for which aggregation to (AT)n and further unfolding occur with no detectable heat change. Fitted parameters indicate that F508del thermodynamically destabilizes the native state, N, and accelerates the formation of AT.
Keywords: CFTR, cystic fibrosis, NBD1, (F508del)NBD1, calorimetry, thermal denaturation, protein stability, temperature correction
Introduction
Mutations in the gene encoding the cystic fibrosis transmembrane conductance regulator (CFTR) are at the root of all CF disease and ∼1500 mutations are known to contribute to disease.1,2 The most prevalent mutation is a deletion of F508 (F508del) within the first nucleotide binding domain (hNBD1) of CFTR, which is found in about 70% of all CF chormosomes. CFTR is a member of the ABC (ATP-binding cassette) transporter superfamily. It functions as a chloride ion channel in exocrine epithelial cells of the lungs, liver, skin, digestive and reproductive tracts. Structural and functional studies on CFTR indicate that channel gating involves complex and coordinated conformational changes that require changes in domain-domain interactions, such as those between NBD1 and NBD2, transmembrane helices, the NBDs and cytosolic loops that are contiguous with the transmembrane domains, and interactions of a regulatory (R) domain only found in CFTR. Phosphorylation of the R domain by protein kinase A activates CFTR (reviewed in Ref. 3).4,5
Newly synthesized (F508del)CFTR accumulates in the endoplasmic reticulum (ER) and is eventually degraded by the ER-associated degradation pathway. At physiological temperature, none is processed normally or trafficked to the plasma membrane.3,6,7 This processing defect arises from the misfolding and/or partial folding of (F508del)CFTR, which triggers its degradation. The intracellular biosynthesis and folding pathway for CFTR is a complex process that involves both cotranslational folding of some domains, and post-translational folding and association of others.8–11
Numerous in vivo studies of CFTR intracellular biosynthesis suggest that NBD1 folds relatively early and is only modestly perturbed in (F508del) CFTR. These and other in vivo studies,2,8–10,12 as well as in vitro folding of isolated hNBD1 constructs13 point to a crucial role for F508del in the disruption of post-translational, inter-domain assembly and folding of (F508del)CFTR. Because the F508 deletion produces only local conformational changes on the surface of the crystal structure of isolated hNBD1 domains,14–17 one school of thought suggests the primary effect of F508del arises from its role in inter-domain assembly of full length CFTR.
Nevertheless, there are also considerable in vivo and in vitro data consistent with a role for alterations in the folding efficiency and/or stability of the (F508del)hNBD1 domain itself. It has long been known that biosynthesis of (F508del)CFTR is temperature-sensitive; reducing the temperature of cell growth leads to the cell surface expression of functional (F508del)CFTR,18 while transfer back to a non-permissive temperature leads to accelerated degradation of (F508del)CFTR at the cell surface.19 The mechanism behind temperature rescue of (F508del)CFTR folding and trafficking is due, in large part, to the improvement in physical properties intrinsic to the protein, whether it is folding efficiency, kinetics, or thermodynamic stability.20 Consistent with this view, small molecule chaperones, both chemical (non-specific, e.g., glycerol) and pharmacological (showing specificity for CFTR) have been shown to mimic the effects of reduced temperature by promoting trafficking to and retention at the cell surface.21,22 Glycerol and other osmolytes are well known to improve protein thermodynamic stability23–26 and there is a solid thermodynamic and structural basis for this effect (27 and references cited within). Second site mutations in (F508del)CFTR have been identified as suppressor mutations for their ability to correct the trafficking defect.28–32 It is interesting that these same mutations improve the solubility of bacterially expressed hNBD1 constructs. Furthermore, the chemical chaperone glycerol improves the yield of refolded hNBD1, apparently mimicking the effects it has in vivo on CFTR processing and maturation.31,33
Collectively, in vivo and in vitro analyses of CFTR and hNBD1 suggest that F508del perturbs either the folding efficiency (brought about by changes in the folding pathway or kinetics) and/or the native-state stability (i.e., Gibb's stabilization free energy, ΔG°) of hNBD1, independent of any subsequent inter-domain interactions of the hNBD1 domain within CFTR. However, the exact nature of this perturbation has remained unclear.13,15,16,32,34–36 The stabilization free energy has been assessed through isothermal chemical denaturation studies on numerous hNBD1 constructs of various sequences, mutations, and in the presence and absence of F508, and for the most part, these modifications do not alter the stabilization free energy, which is most commonly derived via linear extrapolation of the equilibrium constant for unfolding from moderate to zero denaturant concentration.13,15,31,37 Structures of hNBD1 domains reinforce the observations made from these stability studies. If alterations in hNBD1 stability do not account for the folding defect of (F508del)NBD1, then misfolding may arise from perturbations in the kinetics or pathway of folding/unfolding, (e.g., different intermediates may exist for (F508del)NBD1).
The apparent lack of effect of F508del on hNBD1 thermodynamic stability is the most compelling evidence against a role for the intrinsic stability of (F508del)hNBD1 on (F508del)CFTR folding and assembly, but this conclusion defies the clear effect temperature has on the folding of (F508del)hNBD1, in vitro35,36 and (F508del)CFTR, in vivo. However, virtually all measures of thermodynamic stability reported to date have been based on isothermal chemical denaturation methods. Furthermore, a quantitative evaluation of temperature's influence on a protein's thermodynamic stability cannot be extrapolated directly from isothermal denaturation experiments, even though the relationship between temperature (an intensive property of a system) and thermodynamic stability is clearly defined theoretically. Therefore, we set out to characterize the temperature-dependent unfolding of hNBD1 domains to evaluate explicitly the influence of F508del on their thermal stability and, by extrapolation, the temperature-dependent folding defect of (F508del)CFTR.
We present here a systematic differential scanning calorimetry (DSC) study of the thermal denaturation of a series of hNBD1 constructs, including experiments conducted at varying scan rate, hNBD1 protein concentration, and ATP concentration. Nonlinear least squares fitting of these extensive experimental datasets enables us to present a thermodynamic model for the thermal unfolding of (F508del) hNBD1. The accompanying paper by Wang et al.38 presents evidence for the first time that isothermal chemical unfolding occurs via a similar pathway. Together, these analyses have allowed us to propose an unfolding mechanism which reconciles previous disparate conclusions drawn from isothermal and thermal unfolding of hNBD1. While some details of thermal and isothermal may differ, the general conclusions from both studies suggest that the hNBD1 unfolding occurs from the nucleotide-free state and demonstrate that the F508del mutation substantially destabilizes the native state and accelerates the formation of an aggregation-prone, partially unfolded intermediate. It seems likely that temperature rescue of the defective in vivo processing of F508del-CFTR is due, in part, to the inhibition of this unfolding pathway of the hNBD1 domain.
Results and Discussion
Deletion of F508 uniformly reduces the thermal stability of hNBD1
The thermal stability of several sequence-matched hNBD1 pairs, differing only by the presence or absence of F508, was determined by DSC (see Table I). Heat capacity profiles shown in Figure 1 for three hNBD1 pairs are typical of those obtained for all hNBD1 pairs. Table I illustrates that all (F508del)hNBD1 constructs exhibit a fairly consistent lower unfolding temperature (Tm), averaging - 6.4 ± 0.4, compared to their sequence-matched hNBD1 partner. The hNBD1 constructs studied here were identified in a screen for mutations producing sufficient expression yields and solubility for biophysical and structural studies.15,17 Thus, an implicit goal was to avoid alteration of the core structure in the process of improving physical behavior.
Table I.
Thermal Stability of Paired Human NBD1 Constructs (With and Without F508) Determined by DSCa
| Pair ID No. | hNBD1 Nameb | Termini / Mutationsc | Tmd | ΔTm = TmΔ508 - Tmwt (°C) | PDB ID |
|---|---|---|---|---|---|
| 1 | hNBD1-Δ(RI,RE) 2935c46917 | 387-646[Δ405-436] | 57.7 + 0.2 | 2PZE | |
| 1 | (F508del)hNBD1Δ (RI,RE) 2935c47217 | 387-646[Δ405-436, F508del] | 51.5 + 0.3 | −6.2 + 0.3 | 2PZF |
| 2 | 387-646[Δ405-436, V510D] | 60.2 + 0.4 | |||
| 2 | 387-646[Δ405-436, V510D, F508del] | 53.0 + 0.1 | −7.2 + 0.4 | ||
| 3 | 387-646[Δ405-436, F494N, Q637R] | 59.2 | |||
| 3 | 387-646[Δ405-436, F494N, Q637R, F508del] | 52.8 | −6.4 | ||
| 4 | 387-646[Δ405-436, G550E, R553Q, R555K] | 61.7 | |||
| 4 | 387-646[Δ405-436, G550E, R553Q, R555K,F508del] | 55.7 | −6.0 | ||
| 5 | 387-678[Δ405-436] | 58.1 | |||
| 5 | 387-678[Δ405-436, F508del] | 51.7 | −6.2 | ||
| 6 | hNBDI-3152935c38217 | 389-678[F429S, F494N, Q637R] | 49.8 + 0.3 | ||
| 6 | hNBDI-3F508del152935c37117 | 389-678[F429S, F494N, Q637R, F508del] | 43.6 + 0.1 | −6.3 + 0.3 | 2BBS |
| 7 | 389-678[F429S, F494N, L636E5, Q637R] | 50.5 + 0.2 | |||
| 7 | 389-678[F429S, F494N, L636E, Q637R, F508del] | 44.9 | −6.2 + 0.2 |
DSC conducted at 1 mg/mL protein.
Nomenclature for previously published constructs is cited and given in italics. Only pair 1 constructs are given an abbreviated name in the present study for convenient reference in the body of the text.
Expression vectors for construct pairs 1–4, 6 and 7 were obtained from SGX Pharmaceuticals, Inc. Expression vectors for construct pair 5 was obtained from Dr. Julie Forman-Kay, Hospital for Sick Children University of Toronto. Mutations are changes from the wild type human sequence and represent either deletions or point mutations of surface residues; aa 405-436 and 647-678 represent the deletions associated with the RI and RE segments, respectively.
Tm is defined as the temperature at the maximum in the DSC heat capacity profile, determined after subtraction of buffer baseline and progressive baseline (see Figure 2 for example of the latter data). Average Tm values, where given, represent the average of 2–3 determinations.
Figure 1.
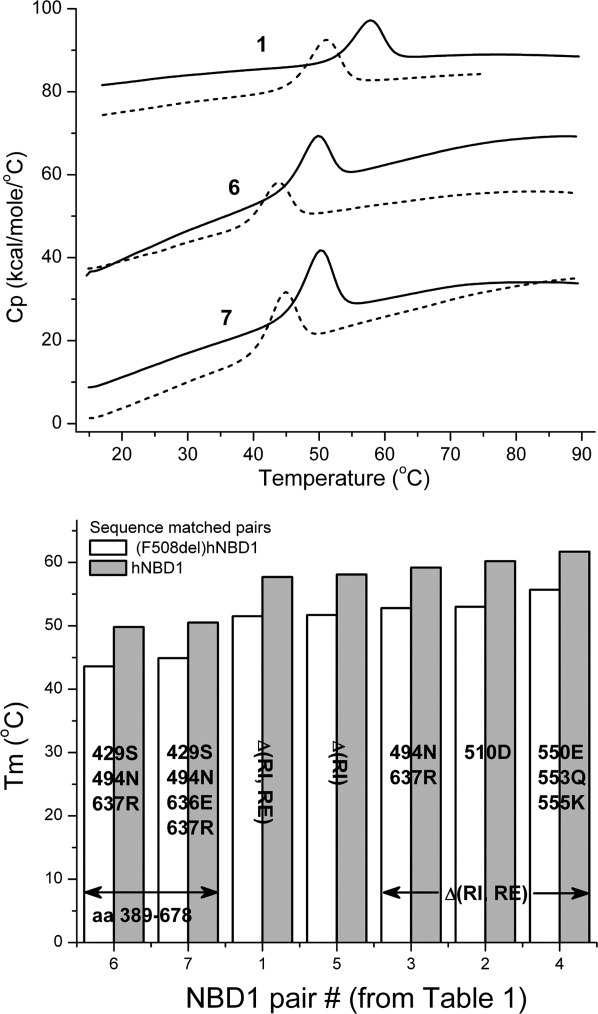
Upper Panel: DSC heat capacity profiles for hNBD1 constructs (solid lines) and the corresponding sequence-matched F508del mutant (dotted lines) in buffer containing 5 mM ATP and 10 mM MgCl2 at 2 K/min scan rate. Buffer scans are subtracted from protein scans and data are normalized to molar concentration. Protein concentrations in the calorimetric cell were 1 mg/mL. Lower Panel: The Tm values are shown for these sequence-matched pairs together with results for the other four pairs presented in Table I. Standard errors in the Tm measurements are given in Table I.
All solubilizing mutation sites are at least partially solvent exposed in natively folded hNBD1. Some of these mutations are located in disordered polypeptide segments.16,39 Biophysical studies on hNBD1 constructs containing suppressor mutations,15 known to partially correct the trafficking defect of cellular (F508del)CFTR that results from misfolding,28–30 suggest there is a systematic linkage between improved hNBD1 solubility and improved folding and/or processing of the full length CFTR.
The regulatory insertion (RI) and regulatory extension (RE) comprise 37 and 39 residues, respectively,16 and either one (hNBD1 pair 5) or both (hNBD1 pairs 1-4) are missing in some constructs studied here. These segments are so named because they are thought to undergo regulatory phosphorylation in cellular CFTR.14 Notably, the RI and RE segments are not found in any other ABC transporters and are not required for hNBD1 folding.15 Before the reported structure of mouse NBD1,14 the RE segment was thought to be part of the R domain.40 We previously showed that hNBD1 constructs with the RI and RE removed behave dramatically better during purification and biophysical characterization, including by DSC.17 In fact, the RI and RE segments are conformationally dynamic in solution, as determined from NMR39 and hydrogen/deuterium exchange mass spectrometry.15,16 Therefore, these segments do not provide critical stabilizing contacts for the native hNBD1 structure. In the present work, the core structure of hNBD1 is defined as the part that does not include RI and RE. Numerous available x-ray crystal structures (see also13), have shown that neither the surface mutations nor the presence or absence of the RI and RE segments, noticeably alter the core structure of the protein.16,17
The Tm values shown in Table I cannot be used to infer a relative thermodynamic stability for a given construct for many reasons to be discussed subsequently. Nevertheless, the Tm has been used to predict solution stability at temperatures considerably lower than Tm. It is interesting that deletion of RI has the most dramatic effect on Tm when compared to the other solubilizing mutations studied here, illustrated in the bar graph of Figure 1 (e.g., compare hNBD1 pair 5 with pairs 6 and 7) and suppressor mutations further increase the thermal stability, although to a more limited extent (compare hNBD1 pair 1 or 5 with pairs 2, 3 or 4). The Teem suppressor triplet (pair 4)29 increases Tm by 4°, the V510D mutation (pair 2)30 by 2.5°, and F494N/Q637R (pair 3) by 1.5°. These observations suggest the basis for their improved solubility may involve alteration of the unfolding kinetics and/or thermodynamics, rather than solely influencing the propensity of the folded protein to self-associate, as suggested previously.15
Consistent with the possibility of a mechanistic coupling between domain solubility and unfolding kinetics and/or thermodynamics, the F508 deletion, which we will later show reduces the thermodynamic stability of hNBD1-Δ(RI,RE), also reduces the solubility of all hNBD1 constructs.15,17 The largely invariant reduction in Tm of about 6-7°C suggests its deletion causes the same change in the folding/unfolding pathway of all the hNBD1 constructs. Likewise, the energetic consequences of the surface mutations, and the presence or absence of the regulatory segments, appear to be largely independent of the energetic consequences of the presence or absence of F508. This observation is consistent with an earlier analysis of a comprehensive set of NBD1 crystal structures suggesting no direct or allosteric coupling between the various mutations sites.16 Furthermore, the view that the NBD1 mutations reported here similarly affect the thermodynamic and/or kinetics of NBD1 unfolding in the presence and absence of F508del is directly addressed in the accompanying paper by Wang et al.38 They present the results of chemical denaturation studies for several hNBD1 pairs in Table I and demonstrate that for a given set of solubilizing mutations, similar effects on chemical denaturation are observed with and without the F508del mutation.
Thermal unfolding of hNBD1 is irreversible and kinetically-controlled
We chose to focus further DSC analysis on the hNBD1-Δ(RI,RE) construct pair since crystal structures exist for this pair and they have otherwise been well-characterized.17 The differences in thermodynamic stability of these domains can be determined through a comparison of their Gibb's stabilization free energy, ΔG°, and DSC directly provides the parameters needed to calculate stabilization free energy at any temperature. However, a thermodynamic treatment of DSC data is only valid if thermal unfolding is an equilibrium process. Equilibrium is typically inferred if the unfolding transition is reversible (i.e., seen on a second heating scan). Furthermore, the data obtained must be independent of the DSC scan rate.
Unfortunately, the DSC transition is irreversible. Even under the best conditions, less than 20% reversibility is seen (data not shown). The simplest model to account for an irreversible unfolding process is the three-state model, N ⇄ U → D. This model was first proposed by Lumry and Eyring to describe the irreversible unfolding of proteins.41 According to this simple model, an equilibrium exists between the folded and unfolded states, N and U, which is followed by the irreversible formation of D, a species which is unable to refold within the time frame of the DSC experiment.
The unfolding transition temperature for both hNBD1-Δ(RI,RE) constructs is also seen to vary with scan rate (shown in Supporting Information Fig. 1). This indicates that the rate of D formation is fast compared to all scan rates tested. Therefore, under the conditions of the DSC experiment, thermal unfolding is a kinetically-controlled process and, the scan-rate dependency of this process provides information on the rate of D formation.
In fact, there is a strong theoretical foundation for extracting both kinetic and thermodynamic parameters from DSC scan-rate dependent data42–45 and these concepts have been successfully applied to numerous systems.46–50 It has been shown that thermodynamic parameters associated with the N ⇄ U equilibrium step, as well as kinetic parameters for the irreversible step U → D can be obtained (i.e., the rate constant, kirr, and activation energy Ea). However, when kirr is sufficiently faster than the rate of conversion of U back to N, then N and U are no longer in equilibrium. In this case, the unfolding reaction collapses to a two-state irreversible process between the native and denatured states, denoted by N → D. Consequently, without further analysis of the DSC data, the unfolding mechanism for the thermal unfolding of hNBD1 could be represented by either N ⇄ U → D or N → D.
The DSC unfolding transition of hNBD1 is independent of protein concentration
Aggregation is a common factor that results in irreversible unfolding. Therefore, it was necessary to determine whether or not changes in the protein concentration altered either the Tm or integrated area (ΔHcal) of the DSC unfolding transition. If aggregation occurs during the formation of D (in contrast to after the formation of D), then the rate constant for its formation, kirr, will be at least second order (e.g., in the case where a dimer is formed), if not higher. In this case, increasing protein concentrations will lead to faster rates of D formation and a lowering of the Tm.43 Even under ideal buffer conditions, isolated hNBD1 domains do not maintain solubility over prolonged periods and it is not surprising that thermal unfolding leads to aggregation that is very obvious when monitored by light scattering (see accompanying paper). Thus it was surprising to see no dependence on protein concentration of either the transition Tm or ΔH over a twenty-fold range of concentration (Table II). Even at temperatures above the DSC transition there was no heat change or detectable deviation in the instrument baseline, as is sometimes observed when post-transition aggregation leads to precipitation (see Fig. 1). A post-transition, aggregation-related, heat effect is most likely absent in the DSC heat capacity profile because of the improved cell geometry of the DSC instrument used (see Methods for further details).
Table II.
DSC Transition Tm and ΔHcal for Unfolding of hNBD1-Δ(RI,RE) and (F508del)hNBD1-Δ(RI,RE)a
|
Tm (°C) |
||
|---|---|---|
| Protein (mg/mL) | hNBD1 | (F508del)hNBD1 |
| 0.5 | 57.8 | 51.1 |
| 1 | 57.9 | 50.7 |
| 2 | 57.8 | 51.2 |
| 4–5 | 57.7 | 51.2 |
| 9–10 | 57.9 | 51.3 |
| Average Tm ± SD | 57.8 ± 0.1 | 51.1 ± 0.2 |
| Average ΔHcal (kcal/mol)b | 89.7 ± 4.3 | 76.3 ± 5.7 |
Performed at scan-rate 2°C/min at 5 mM ATP and 10 mM MgCl2.
ΔHcal is the integrated area of the DSC endothermic transition normalized to total moles of protein
Nevertheless, the significant observation from the protein concentration study is that for both hNBD1-Δ(RI,RE) constructs, aggregation occurs after formation of the rate-limiting product of thermal unfolding, which must therefore be monomeric D.44,47 A similar mechanism has been proposed for the aggregation properties of partially unfolded recombinant human interferon-γ.44,51 To better represent the concept that D is an aggregation-prone species produced from thermal unfolding, the acronym AT will be used in place of D from this point forward. Therefore, at this point, the likely thermal unfolding mechanisms for hNBD1 are represented by either N ⇄ U → AT or N → AT.
Circular dichroism (CD) spectroscopy provides evidence that AT, the product of thermal unfolding, is partially folded
Changes in the secondary and tertiary structures of the hNBD1-Δ(RI,RE) pair during thermal unfolding were investigated by monitoring far- and near-UV CD, respectively, to shed light on possible differences that may account for differences in their thermal stabilities. CD spectra of the hNBD1-Δ(RI,RE) pair are shown in Figure 2. It has been shown that the deletion of F508 produces only local conformational changes near the site of the mutation in the crystal structures of various (F508del)hNBD1 constructs; the structure is otherwise the same for all available structures of hNBD1.13–17 Therefore it is not surprising that the CD spectra are very similar, if not identical for hNBD1-Δ(RI,RE) and (F508del)hNBD1-Δ(RI,RE). Some difference in the near-UV CD spectra is not unexpected, since the protein residues that primarily contribute to the near-UV CD contain aromatic side chains, like phenylalanine, which absorbs between about 240-275 nm. Absorption of bound MgATP could also contribute in this region and thereby account for some minor variability in the near-UV CD region, which was noted depending on the ATP concentration. That is, differences in the MgATP binding constant for the two constructs could contribute to the observed differences in the spectrum near about 260 nm. It is also possible that bound ATP influences the protein spectrum, itself, for example, arising from its interaction with the tryptophan 401 side chain, which stacks against the adenine ring of ATP.15
Figure 2.

CD spectra of hNBD1-Δ(RI,RE) and (F508del)hNBD1-Δ(RI,RE) in the far-UV region (left panel) and in the near-UV region (right panel). Solid circles are the experimental data for hNBD1-Δ(RI,RE) and open circles are the experimental data for (F508del)hNBD1-Δ(RI,RE). The ATP concentration for far-UV measurements was 0.8 μM for (hNBD1-Δ(RI,RE) and 4 μm ATP for (F508del)hNBD1-Δ(RI,RE); for near-UV measurements, it was 0.125 mM ATP for both proteins. Protein concentration was 0.69 mg/mL in 0.02 cm cuvettes for far-UV CD and 1 mg/mL in 1 cm cuvettes for near-UV CD measurements.
Because thermal unfolding is influenced by the temperature scan rate, DSC and CD unfolding data were collected at the same scan rate, 2°C/min. For ease of comparison, all data are normalized to the fractional change observed (Fig. 3). Therefore, the DSC transitions have been integrated, which results in the sigmoidal progress curves seen in Figure 3. The noteworthy observations are that qualitatively similar behavior is observed in the presence or absence of the F508del mutation and the temperature-dependent loss in near-UV CD ellipticity is superimposable on the integrated DSC unfolding transition. In contrast, the major change in the far-UV CD occurs at temperatures significantly above the DSC transition, where there is no detectable change in heat capacity.
Figure 3.

Thermal unfolding of hNBD1-Δ(RI,RE), left panels, and (F508del)hNBD1-Δ(RI,RE), right panels, monitored by CD and DSC at scan rate 2°C/min at 0.125 mM ATP and 0.5 mM MgCl2. CD is monitored at 297 nm (near-UV wavelength region, upper panels) and at 230 nm (far-UV wavelength region, lower panels). Protein concentration in the 1 cm cuvette was 0.125 mg/mL for far-UV and 0.5 mg/mL for near-UV measurements. The unfolding data were normalized to the fractional change observed. No baseline subtraction was performed on the spectral data shown by closed and open circles. DSC unfolding curves (represented by solid lines in the upper panels) were integrated and normalized to the unfolding calorimetric enthalpy value, ΔHT/ΔHcal; CD unfolding curves were normalized to maximal change in the CD signal, CDT/(CDinitial-CDfinal), where T = temperature of observed signal. The signal change at 230 nm for both proteins was approximately the same: starting at 20°C, θ = −6 × 103 deg cm−1 × dmole−1 and ending at 80°C, θ = −2 × 103 deg cm−1 × dmole−1. The signal change at 297 nm for hNBD1-Δ(RI,RE) was determined from the initial signal at 20°C, θ = 69 × 103 deg cm−1 × dmole−1 and ending at 60°C, θ = −58 × 103 deg cm−1 × dmole−1 ; for (F508del)hNBD1-Δ(RI,RE), initially, θ = 55 × 103 deg cm−1 × dmole−1 and ending with θ = −31 × 103 deg cm−1 × dmole−1. The insets in the upper panels are temperature unfolding of hNBD1-Δ(RI,RE), left panel, and (F508del)hNBD1-Δ(RI,RE), right panel, monitored by intrinsic Trp fluorescence with excitation wavelength at 290 nm and emission wavelength at 340 nm and DSC at scan rate 1°C/min at 0.1 mM ATP and 0.2 mM MgCl2. Trp fluorescence unfolding curves were normalized to maximal change in the Trp fluorescence signal, FLTrpT/(FLTrpinitial-FLTrpfinal), where T = temperature of observed signal. Preunfolding and postunfolding baselines of unfolding profile were subtracted using equations obtained by linear regression to obtain the data shown.
For the purpose of comparison to data collected by isothermal chemical denaturation (see accompanying paper by Wang et al.38), the change in intrinsic fluorescence was also monitored (Fig. 3, insets). Similar to the near-UV CD signal, a cooperative increase in emission at 340 nm appears coincident with the DSC transition, suggesting a change in the environment surrounding the tryptophan residues.
After heating to 80°C, the protein samples were cooled to room temperature and spectra were taken. While there is some remaining ellipticity in the sample, which is typical for thermally unfolded proteins, the spectra in both the far-UV and near-UV CD were devoid of the spectral characteristics identifiable as native-like structure (data not shown). A loss of essentially all the near-UV CD signal and a fractional loss of about 20% of the secondary structure coincide with the only thermal unfolding transition detected by DSC, indicating that AT exhibits appreciable residual structure.
Thermal unfolding of hNBD1 is strongly influenced by equilibrium binding of MgATP
Because hNBD1 constructs have improved solubility in the presence of MgATP, all studies described so far have been in the presence of at least nominally saturating amounts of MgATP. The quantitative effects of MgATP binding on thermal stability were, therefore, determined, and are shown in Figure 4. Tm and ΔHcal steeply increase at low concentrations and then rise more slowly at concentrations above complete hNBD1 saturation.
Figure 4.

DSC of hNBD1-Δ(RI,RE) and (F508del)hNBD1-Δ(RI,RE) at different concentrations of ATP at scan-rate 2 K/min. The left panel shows the temperature dependence of the molar heat capacity of (hNBD1-Δ(RI,RE) (solid lines) and (F508del)hNBD1-Δ(RI,RE) (dotted lines) at 0 mM, 0.1 mM and 5 mM ATP. The right panel represents results of DSC data analysis for hNBD1-Δ(RI,RE) (solid circles) and (F508del)hNBD1-Δ(RI,RE) (open circles) as ATP concentration dependence of Tm (top panel) and unfolding enthalpy (bottom panel) obtained from 0 mM to 20 mM ATP. Protein concentration was 1 mg/mL. The minimal detectable ATP concentration in the protein solution is 0.5 μM; see Supporting Information Methods for details.
A manifestation of Le Chatelier's Principle is the widely observed increase in protein unfolding Tm in the presence of ligand binding. Unfolding occurs at higher Tm because ligand binding to the folded protein shifts the N ⇄ U equilibrium in favor of the native state, N. Therefore, the observed influence of MgATP binding on hNBD1 thermal unfolding suggests that of the two unfolding mechanisms proposed above, the more likely is N ⇄ U → AT. In the presence of MgATP the equilibrium between bound and free hNBD1 must also be included, that is, MgATP•N ⇄ N + MgATP.
Because we know from our CD studies that AT is only partially unfolded, we propose that the N ⇄ U equilibrium in the overall unfolding process is better represented by N ⇄ IT, where IT denotes a species with a conformation intermediate between N and AT. Thus, the minimum unfolding pathway that is likely to represent the thermal unfolding of hNBD1-Δ(RI,RE) is MgATP•N ⇄ N ⇄ IT → AT. This pathway is referred to as Model ii in subsequent discussions (see Table III).
Table III.
Hypothetical Unfolding Mechanisms used to Fit DSC Dataa
| Model | Unfolding pathway |
|---|---|
| i |  |
| ii |  |
| iii(a) |  |
| iii(b) |  |
Step in brackets, [--->(AT)n], is not fit by the mathematical models; for details of the curve fitting, see Materials and Methods
DSC curve fitting is consistent with the N ⇄ IT → AT model, which includes MgATP binding equilibria to both N and IT
Two sets of DSC data were collected for each protein; the first comprising experiments at 2 mM Mg-ATP with scan-rate varying between 0.5 – 2 degree/min (Supporting Information Fig. 1) and the second comprising experiments at a constant scan rate of 2 degree/min with MgATP concentration varying between 0.088 and 10 mM for hNBD1-Δ(RI,RE) and 0.088-5 mM for hNBD1-Δ(RI,RE)-F508del (Fig. 5). For each mechanistic model used for curve-fitting (shown in Table III), the scan-rate dependent datasets were globally fit first to obtain the best fits for the kinetic parameters for formation of AT (see Table IV for fitted kinetic parameters). Subsequently, the MgATP concentration-dependent datasets were globally fit to determine the parameters for the equilibrium step(s), while constraining the kinetic parameters to a narrow range of values around the results from the first fits. The equations used to fit each model and further details on the fitting procedure can be found in the Supporting Information Materials and Methods.
Figure 5.
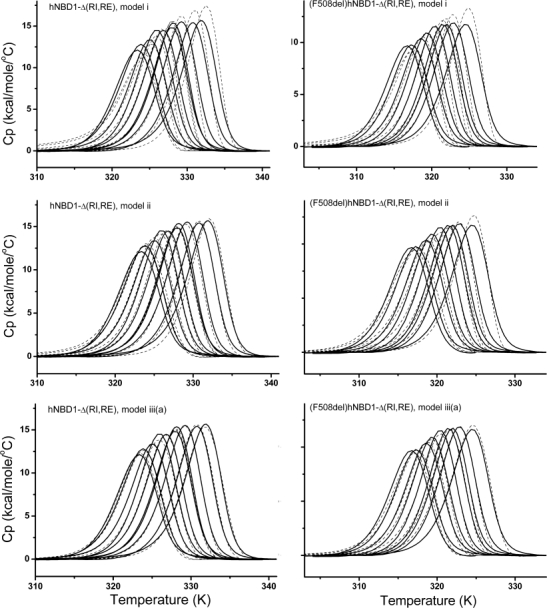
Comparison of global fits of DSC data to Models i, ii, and iii(a). Left panels: Fits for hNBD1-Δ(RI,RE). Right panels: Fit for (508del)hNBD1-Δ(RI,RE). DSC was performed at scan rate 2°C/min in buffer containing 0.088, 0.108, 0.196, 0.3, 0.46, 0.915, 0.97, 1.82, 4.7 mM ATP with 10 mM MgCl2, and 10 mM ATP with 20 mM MgCl2 for hNBD1-Δ(RI,RE) and 0.088, 0.11, 0.21, 0.27, 0.48, 0.85, 1.05, 1.7 and 4.9 mM ATP with 10 mM MgCl2 for (F508del)hNBD1-Δ(RI,RE). Solid lines are experimental data. Dashed lines are best fit curves.
Table IV.
Best Fit Parameters for Unfolding Mechanism Model iii(a) from Global Fit of DSC Dataa
| Parameters |
hNBD1-Δ(RI,RE) |
(F508del)hNBD1-Δ(RI,RE) |
|
|---|---|---|---|
| Process | χ2 | 2.9E + 04 | 5.9E + 04 |
| N ⇄ IT | Ku at 45°C | 1.0 ± 0.04 | 2.8 ± 0.6 |
| Hv,r at respective Tm (kcal/mol) | 70.6 ± 2.5 | 57.2 ± 3.9 | |
| MgATP • N ⇄ N + MgATP | Kd1 at 25 °C (μM) | 0.51 ± 0.16 | 1.0 ± 0.5 |
| Hd1 at 25 °C (kcal/mol) | 20.6 ± 2.8 | 25.6 ± 5.0 | |
| MgATP • IT ⇄ IT + MgATP | Kd2 at 52.8 °C (μM) | (6.1 ± 0.2) × 103 | (2.5 ± 0.5) × 103 |
| Hd2 at 52.8 °C (kcal/mol) | −6.9 ± 2.0 | −10 ± 4.2 | |
| kirr at 58 °C (min−1) | 3.1 ± 0.5 | 11.5 ± 6.9 | |
| IT → AT | Ea (kcal/mol) | 33.5 ± 1.0 | 23.9 ± 2.3 |
| Hd,irr at 58 °C (kcal/mol) | −15.8 ± 1.1 | −18.4 ± 2.5 |
Estimated standard errors in each parameter value are based on 95% confidence level.
We evaluated the mechanistic models based on three criteria, summarized in Supporting Information Table I. First, we compared the χ2 values for the fitted curves; the smaller the value, the better the fit. The χ2 for the fit to a given model is seen to decrease as the complexity and associated number of fitting parameters increase, that is, from Model i to Model iii(b), χ2 becomes progressively smaller. However, the better fit may only be the result of the greater number of fitting parameters, and would not necessarily mean the model with the lowest χ2 is the correct model. Therefore two other methods were used to discriminate among the models, the statistical F-test and the Akaike's Information Criterion (AIC), based on information theory. These evaluation methods take into account the number of fitting parameters in computing the relative probability that a given model is the best fit. The detailed methods for quantitative comparison of F-ratios and the AIC can be found in the legend to Supporting Information Table I and Materials and Methods. From this analysis, Model ii was clearly better than Model i. In Model ii, the IT state does not bind MgATP; however there is no structural or mechanistic reason to rule out ATP binding by state IT, so a model incorporating ATP binding to IT, Model iii(a), was tested and the fit was improved further and also deemed to be more likely correct based on both the F-ratio and the AIC. The fitted curves for Models i, ii and iii(a) are shown with the DSC data in Figure 5. A variant of Model iii(a), denoted iii(b), switches N ⇄ IT from an on-pathway (with respect to AT formation) to an off-pathway step. This variant was suggested from the early work of Thomas and coworkers31 who suggested that the formation of an aggregation-prone intermediate may not be on the unfolding pathway. The χ2 value for Model iii(b) is similar to that for Model iii(a). However, F-test and AIC analysis suggests Model iii(b) is, by far, less likely to be correct than Model iii(a). Incorporation of additional states (and the requisite additional parameters) into the unfolding model did not result in improved fits based on any of our evaluation criteria and were mechanistically unnecessary to account for the unfolding data.
We conclude that of the mechanistic models tested, Model iii(a) is the most likely to represent the thermal unfolding mechanism for hNBD1. Table IV gives the values obtained for the parameters fit to this model and Supporting Information Figure 2 shows plots of the fraction of each species present as a function of temperature at two different ATP concentrations.
Of critical importance for any proposed model, the values obtained for all parameters must be physically reasonable and this is confirmed for Model iii(a) as shown in Table IV. The fitted value for hNBD1-Δ(RI,RE) binding to MgATP, Kd1 = 0.5 μM at 25°C and 0.04 μM at 4°C, are within the range of the value measured by surface plasmon resonance at 4°C, 0.8 μM.17 The corresponding fitted value for (F508del)hNBD1-Δ(RI,RE) is similar. There is no published value for this construct but for a different sequence pair, the binding constants for NBD1 and for (F508del)NBD1 are similar.31 The remaining parameters will be discussed in more detail below.
Conclusions and Interpretations
Taken on face value, these results represent a new perspective on hNBD1 stability and provide significant insights into the unfolding mechanism. The new information provided by these studies are summarized as follows: (1) the native state of (F508del)hNBD1 is thermodynamically less stable than hNBD1; (2) an intermediate, IT, exists in the unfolding pathway and retains weak affinity for MgATP; (3) a kinetically-trapped, partially unfolded state, AT, is the aggregation-prone conformational state in hNBD1 thermal unfolding; (4) lower temperature will reduce the extent of hNBD1 unfolding and the aggregation resulting from this pathway; (5) since temperature affects CFTR folding, it is reasonable to suggest that in the context of the CFTR protein, reduced-temperature rescue of folded (F508del)hNBD1 results from less AT formation and consequently less non-native and counter-productive interactions with other CFTR domains; and (6) all the unfolding models tested here support the proposal that lower temperature will reduce the aggregation resulting from hNBD1 unfolding in a quantitatively predictable manner. The latter inference is supported by the comparison of the kinetic parameters for the formation of AT derived by each model shown in Table III. Regardless of the model used, the derived rate, kirr, for formation of AT is significantly faster for (F508del)hNBD1-Δ(RI,RE) (see Supporting Information Table I). As illustrated in Figure 6, as temperature is lowered from Tm to below 37°C, the rate of AT formation slows dramatically.
Figure 6.
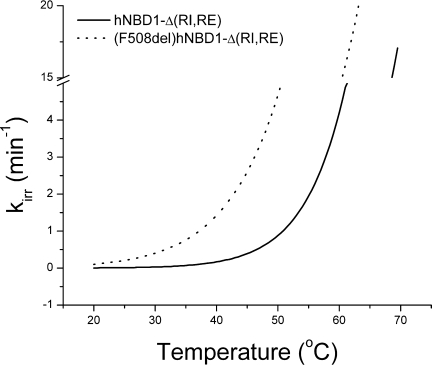
Temperature-dependence of the rate constant, kirr, for the IT → AT unfolding step for hNBD1-Δ(RI,RE) and (F508del)hNBD1-Δ(RI,RE). Fitted parameters in Table IV were used to construct the plot.
Before the studies presented here, and in the accompanying paper,38 all published chemical denaturation studies have been performed at a fixed temperature, near ambient. In those studies, the observed unfolding reaction has been modeled as a simple, two-state transition between a native and unfolded state and a stabilization free energy, ΔG°, was determined by linear extrapolation of ΔG from moderate denaturant concentration to 0 M denaturant. The difference in ΔG° (ΔΔG°) between hNBD1 and (F508del)hNBD1 determined from those previous studies was negligible (≪1 kcal/mol), giving rise to the interpretation that F508del does not alter the stability of hNBD1. From those studies, the ΔG° values for hNBD1 have ranged, depending on the construct, from about 3.7 kcal/mol13,37 to 11 kcal/mol.15 On the other hand, we show that thermal unfolding is under kinetic control and is clearly not two-state. As we will show in the accompanying paper,38 a re-examination of hNBD1 chemical denaturation suggests it is also not two-state.
For the present studies, the only thermodynamically valid unfolding ΔG° one may calculate is for the N ⇄ IT step; ΔG° = 3.2 kcal/mol and 2.1 kcal/mol at 25°C, for hNBD1-Δ(RI,RE) and (F508del)hNBD1-Δ (RI,RE), respectively. Because IT is not completely unfolded nor is it the final unfolded species produced by thermal denaturation, it is not surprising that these values are rather small. However, the difference between them, which is ΔΔG° = 1.1 kcal/mol, is larger than the negligible difference found in previous chemical denaturation studies (by as much as fourfold). Furthermore, the fractional difference is even bigger, that is, (F508del)hNBD1-Δ(RI,RE) is about one third less stable than hNBD1-Δ(RI,RE).
Qu and Thomas37 were the first to report a modest 3°C difference in the Tm between protein constructs containing an incomplete hNBD1 domain in the absence of presence of the F508del mutation, but they discounted the significance of this difference, given the inconsequential ΔΔG° they obtained by chemical denaturation. There is a two-part explanation for the unexpectedly large difference in thermal stabilities that we see in the present studies between complete hNBD1 domain constructs in the absence of presence of the F508del. As shown in Figure 7, for (F508del)hNBD1-Δ(RI,RE), both ΔH and ΔS are smaller for the N ⇄ IT unfolding step, which translate a modest ΔΔG° at 25°C into a demonstrably significant thermal stability difference. That is, there is a 4 degree difference in Tm (read from the temperature at which ΔG = 0 for each construct). The faster rate of the irreversible step IT → AT also independently lowers Tm for (F508del)hNBD1-Δ(RI,RE). Together these two unfolding steps produce the observed ΔTm of ∼6 – 7°C. As a result, significantly more IT and AT are formed for (F508del) hNBD1-Δ(RI,RE) at every temperature. For instance, at 37°C, 3.3 times more IT is formed, which is irreversibly converted to AT with a rate that is 10 times faster than the rate for hNBD1-Δ(RI,RE) (see Fig. 6). As the temperature increases, the formation of IT and the rate of AT formation also rise, resulting in more and more AT formation at higher temperatures.
Figure 7.

Plots of ΔG, ΔH, and TΔS versus temperature for the N ⇄ IT transition. The fitted ΔCp values of 1.86 kcal/mol/K, and 2.13 kcal/mol/K, for hNBD1-Δ(RI,RE) and (F508del) hNBD1-Δ(RI,RE), respectively, with other fitted parameters found in Table IV were used to construct the plots.
Given conclusions (5) and (6) above, it is tempting to speculate that the AT conformation identified in these studies is equivalent to the aggregation-prone, partially folded state previously proposed by Thomas and coworkers31 and now characterized in detail in the isothermal chemical denaturation studies presented in the accompanying paper by Wang et al.38 It is also possible that the loss of secondary structure observed above the DSC transition temperature represents the major transition observed in chemical denaturation. In fact, the results we present in Wang et al.38 support this hypothesis. We show that under conditions of low MgATP concentration, chemical denaturation is most likely represented by a two-step unfolding pathway, a simplified representation of which is given by MgATP•N ⇄ N → [AC]n→ U. We propose that the structural transition of the first step in isothermal chemical denaturation is similar if not identical to the structural transition observed by DSC. This step leads to only a minor loss in average secondary structure and the formation of a partially folded, aggregation-prone intermediate, AC. The second step of chemical denaturation is likely to be similar to the thermal transition following that detected by DSC, which does not involve a significant enthalpy change, but as with chemical denaturation, produces a species with minimal secondary structure as monitored by CD. It is tempting to speculate38 that AC and AT share some properties that are predicted for a molten globule, for example, presence of secondary structure but no apparent tertiary structure (as probed by CD) and a low structural cooperativity resulting in the absence of a calorimetric transition.52–54 The full details and experimental support for the isothermal chemical denaturation pathway, and the associated structural transitions proposed for hNBD1, are presented in our accompanying paper.38
In summary, the studies reported here represent the first thermodynamic description of hNBD1 thermal unfolding. One plausible explanation exists for the increased solubility of all “solubilizing” mutations, regardless of origin. While improvement in solubility could arise from the properties of the folded domain, in all cases studied here, the likely alternative explanation is that aggregation is initiated from the partially unfolded AT state, not the folded state, and solubilizing mutations act by inhibiting the formation of AT. Furthermore, the inclusion of high MgATP concentration or osmolytes like glycerol and other so-called “stabilizing” additives lead to improved solubility by inhibiting the formation of AT.
These studies may also provide a highly relevant model for the thermal stability of CFTR during its biosynthesis, folding, and domain assembly. It is likely that temperature- and chaperone-rescue of the (F508del)CFTR trafficking defect results from the increased thermodynamic stability of the (F508del) hNBD1 domain and the reduced rate of formation of an aggregation-prone, partially folded (F508del) hNBD1 domain. The correlation between improved hNBD1 domain solubility and improved folding and/or processing of full length CFTR that has been observed in the presence of suppressor mutations may be likewise explained, and has been recently extended to a study of (F508del)CFTR containing the RI deletion.55
Material and Methods
Protein purification
Protein purification was conducted as previously described,15 yielding protein in 150 mM NaCl, 20 mM HEPES pH 7.5, 10% glycerol, 10% ethylene glycol, 1 mM Tris(2-carboxymethyl)phosphine (TCEP), 2 mM ATP, 3 mM magnesium chloride. Proteins were >98% pure as judged by Coomassie Blue staining of SDS-PAGE gels, showed no evidence of aggregation and ran as monomers during gel filtration. Protein concentration in the absence of ATP was measured spectrophotometrically using an extinction coefficient for hNBD1-Δ(RI,RE) equal to 0.9 OD280 nm/mg/mL, and in the presence of ATP concentration, was measured with the Bio-Rad Protein assay in microplate format, calibrated using B. subtilis NAD synthetase.
Differential scanning calorimetry (DSC)
Calorimetry was carried out on the Nano-DSC II (CNC, U.S.A.) in 0.299 mL cells or on the VP-Capillary DSC System (MicroCal Inc., GE HealthCare, U.S.A.) in 0.130 mL cells at heating rates from 0.5 K/min to 2 K/min. An external pressure of 2.0 atm was maintained during all DSC runs to prevent possible degassing of the solutions on heating. Unless otherwise indicated, the buffer for DSC and CD experiments was 20 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM TCEP, 10% glycerol and 10% ethylene glycol, containing ATP at up to 20 mM (as noted) and MgCl2 up to 40 mM (as noted). The reversibility of unfolding was checked by sample reheating after cooling in the calorimetric cell. The heat capacity curves were corrected for the instrumental baseline. DSC data were analyzed with the MicroCal Origin software, from which the unfolding temperature (Tt), and the calorimetric (ΔHcal) and van't Hoff (ΔHvH) unfolding enthalpies were obtained. Mathematical equations describing possible models were written in a format recognizable by Origin, based on the equations described under DSC analysis, and used for non-linear least-squared curve-fitting.
The DSC cells used for these studies have a capillary design, which is known to help minimize the heat effects accompanying sample precipitation. This seems to be the most likely explanation for the lack of post-transition baseline effects (see Fig. 1). Heat capacity profiles of hNBD1-Δ(RI,RE) acquired with a DSC instrument equipped with cylindrical cells, exhibit a post-transition baseline anomaly, consistent with the precipitation event (data not shown).
Circular dichroism (CD)
CD spectra were recorded on an Olis RSM 1000 CD spectrophotometer (OLIS) or a CD Spectrophotometer (JASCO, Japan) equipped with the thermostatic platform and programmable circulating water bath. The cells had a path length of 0.02 and 1.0 cm, respectively, for far-UV spectra at 0.5–1 mg/mL and for near-UV spectra at 0.8 – 1.6 mg/mL. Spectra were recorded from 260 to 200 nm for far-UV and from 320 to 260 nm for near-UV in 0.5-nm steps. The molar ellipticity values were calculated according to the following expression: [θ] = (θ) (100 × MRW/lc), where θ is the measured ellipticity in degrees, MRW is the mean residues molecular weight in grams per mole (111.3 and 111.2 for hNBD1-Δ(RI,RE) and (F508del)hNBD1-Δ(RI,RE), respectively), l is the path length in centimeters, and c is the concentration of the protein in grams per liter. The value [θ] has the units of degrees•cm2/decimole-residue. For protein unfolding monitored by CD, a cell with a path length of 1.0 cm was used. The temperature was increased at a scan rate of 1 or 2 °C/min, the sample solution was stirred at 250 rpm, and the CD intensity was recorded continuously at ∼230 nm or ∼290 nm.
Intrinsic fluorescence
A PolarStar Optima (BMG LABTECH) fluorescence plate reader was equipped with 290 ± 5 nm excitation and 340 ± 5 nm emission filters and an integrated heating block fabricated to hold a 384 microtiter plate. Thermal protein unfolding was conducted at a 1°C/min heating rate and monitored by following the change in intrinsic fluorescence intensity at 340 nm. Due to technical limitations, CD, DSC, and fluorescence could not all be monitored at the same scan rate. Experiments were conducted at 0.4 mg/mL protein concentrations in 20 mM HEPES pH 7.5, 10% glycerol, 10% ethylene glycol, 150 mM NaCl, 0.1 mM ATP, 0.2 mM MgCl2, 1 mM TCEP; 15 μL oil (chill-out liquid clear, Bio-Rad Lab, Inc.) was placed over the 30 μL sample to prevent evaporation during heating. Data were collected and analyzed using in-house software written for this purpose.
DSC data analysis by nonlinear curve fitting
Data were fit using the non-linear regression algorithm of Levenberg-Marquardt with Origin 7.0 (OriginLab Corporation, Northhamption, MA 01060). Origin C, a programming language included in Origin, was used to write mathematical equations describing the various unfolding models. For details, see Supporting Information Material and Methods.
Quantitative discrimination among models used to fit the DSC data
A comparison of models was made using statistical methods, that is, goodness-of-fit as quantified by the residual χ2, the F test, and a method based on information theory, Akaike's Information Criterion, (AIC). For a description of all of these methods see reference.56 The F test can only be applied to nested models, that is, in cases where the complex model containing more parameters can be transformed into the simpler model by setting one or more of the parameters to either 0 or 1. The AIC can be used to compare both nested and non-nested models. Details on how these methods were applied can be found in the Supporting Information Materials and Methods.
Acknowledgments
The authors thank Dr. Julie Forman-Kay, Hospital for Sick Children and the University of Toronto, Toronto, Ontario, Canada, for the generous gift of two hNBD1-Δ(RI) expression vectors. They also thank Dr. JianLi An for assistance in the expression and purification of proteins used in this study.
References
- 1.Zielenski J, Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet. 1995;29:777–807. doi: 10.1146/annurev.ge.29.120195.004021. [DOI] [PubMed] [Google Scholar]
- 2.Riordan JR. CFTR function and prospects for therapy. Annu Rev Biochem. 2008;77:701–726. doi: 10.1146/annurev.biochem.75.103004.142532. [DOI] [PubMed] [Google Scholar]
- 3.Riordan JR. Assembly of functional CFTR chloride channels. Annu Rev Physiol. 2005;67:701–718. doi: 10.1146/annurev.physiol.67.032003.154107. [DOI] [PubMed] [Google Scholar]
- 4.Mornon JP, Lehn P, Callebaut I. Atomic model of human cystic fibrosis transmembrane conductance regulator: membrane-spanning domains and coupling interfaces. Cell Mol Life Sci. 2008;65:2594–2612. doi: 10.1007/s00018-008-8249-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Serohijos AW, Hegedus T, Aleksandrov AA, He L, Cui L, Dokholyan NV, Riordan JR. Phenylalanine-508 mediates a cytoplasmic-membrane domain contact in the CFTR 3D structure crucial to assembly and channel function. Proc Natl Acad Sci USA. 2008;105:3256–3261. doi: 10.1073/pnas.0800254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O'Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- 7.Kopito RR. Biosynthesis and degradation of CFTR. Physiol Rev. 1999;79:S167–S173. doi: 10.1152/physrev.1999.79.1.S167. [DOI] [PubMed] [Google Scholar]
- 8.Cheung JC, Deber CM. Misfolding of the cystic fibrosis transmembrane conductance regulator and disease. Biochemistry. 2008;47:1465–1473. doi: 10.1021/bi702209s. [DOI] [PubMed] [Google Scholar]
- 9.Chen EY, Bartlett MC, Loo TW, Clarke DM. The DeltaF508 mutation disrupts packing of the transmembrane segments of the cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2004;279:39620–39627. doi: 10.1074/jbc.M407887200. [DOI] [PubMed] [Google Scholar]
- 10.Du K, Sharma M, Lukacs GL. The DeltaF508 cystic fibrosis mutation impairs domain-domain interactions and arrests post-translational folding of CFTR. Nat Struct Mol Biol. 2005;12:17–25. doi: 10.1038/nsmb882. [DOI] [PubMed] [Google Scholar]
- 11.Du K, Lukacs GL. Cooperative assembly and misfolding of CFTR domains in vivo. Mol Biol Cell. 2009;20:1903–1915. doi: 10.1091/mbc.E08-09-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui L, Aleksandrov L, Chang XB, Hou YX, He L, Hegedus T, Gentzsch M, Aleksandrov A, Balch WE, Riordan JR. Domain interdependence in the biosynthetic assembly of CFTR. J Mol Biol. 2007;365:981–994. doi: 10.1016/j.jmb.2006.10.086. [DOI] [PubMed] [Google Scholar]
- 13.Thibodeau PH, Brautigam CA, Machius M, Thomas PJ. Side chain and backbone contributions of Phe508 to CFTR folding. Nat Struct Mol Biol. 2005;12:10–16. doi: 10.1038/nsmb881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lewis HA, Buchanan SG, Burley SK, Conners K, Dickey M, Dorwart M, Fowler R, Gao X, Guggino WB, Hendrickson WA, Hunt JF, Kearins MC, Lorimer D, Maloney PC, Post KW, Rajashankar KR, Rutter ME, Sauder JM, Shriver S, Thibodeau PH, Thomas PJ, Zhang M, Zhao X, Emtage S. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator. EMBO J. 2004;23:282–293. doi: 10.1038/sj.emboj.7600040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewis HA, Zhao X, Wang C, Sauder JM, Rooney I, Noland BW, Lorimer D, Kearins MC, Conners K, Condon B, Maloney PC, Guggino WB, Hunt JF, Emtage S. Impact of the deltaF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure. J Biol Chem. 2005;280:1346–1353. doi: 10.1074/jbc.M410968200. [DOI] [PubMed] [Google Scholar]
- 16.Lewis HA, Wang C, Zhao X, Hamuro Y, Conners K, Kearins MC, Lu F, Sauder JM, Molnar KS, Coales SJ, Maloney PC, Guggino WB, Wetmore DR, Weber PC, Hunt JF. Structure and dynamics of NBD1 from CFTR characterized using crystallography and hydrogen/deuterium exchange mass spectrometry. J Mol Biol. 2010;396:406–430. doi: 10.1016/j.jmb.2009.11.051. [DOI] [PubMed] [Google Scholar]
- 17.Atwell S, Brouillette CG, Conners K, Emtage S, Gheyi T, Guggino WB, Hendle J, Hunt JF, Lewis HA, Lu F, Protasevich II, Rodgers LA, Romero R, Wasserman SR, Weber PC, Wetmore D, Zhang FF, Zhao X. Structures of a minimal human CFTR first nucleotide-binding domain as a monomer, head-to-tail homodimer, and pathogenic mutant. Protein Eng Des Sel. 2010;23:375–384. doi: 10.1093/protein/gzq004. [DOI] [PubMed] [Google Scholar]
- 18.Denning GM, Anderson MP, Amara JF, Marshall J, Smith AE, Welsh MJ. Processing of mutant cystic fibrosis transmembrane conductance regulator is temperature-sensitive. Nature. 1992;358:761–764. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]
- 19.Sharma M, Benharouga M, Hu W, Lukacs GL. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J Biol Chem. 2001;276:8942–8950. doi: 10.1074/jbc.M009172200. [DOI] [PubMed] [Google Scholar]
- 20.Wang X, Koulov AV, Kellner WA, Riordan JR, Balch WE. Chemical and biological folding contribute to temperature-sensitive DeltaF508 CFTR trafficking. Traffic. 2008;9:1878–1893. doi: 10.1111/j.1600-0854.2008.00806.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Varga K, Goldstein RF, Jurkuvenaite A, Chen L, Matalon S, Sorscher EJ, Bebok Z, Collawn JF. Enhanced cell-surface stability of rescued DeltaF508 cystic fibrosis transmembrane conductance regulator (CFTR) by pharmacological chaperones. Biochem J. 2008;410:555–564. doi: 10.1042/BJ20071420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loo TW, Bartlett MC, Clarke DM. Correctors promote folding of the CFTR in the endoplasmic reticulum. Biochem J. 2008;413:29–36. doi: 10.1042/BJ20071690. [DOI] [PubMed] [Google Scholar]
- 23.Back JF, Oakenfull D, Smith MB. Increased thermal stability of proteins in the presence of sugars and polyols. Biochemistry. 1979;18:5191–5196. doi: 10.1021/bi00590a025. [DOI] [PubMed] [Google Scholar]
- 24.Gekko K, Timasheff SN. Thermodynamic and kinetic examination of protein stabilization by glycerol. Biochemistry. 1981;20:4677–4686. doi: 10.1021/bi00519a024. [DOI] [PubMed] [Google Scholar]
- 25.Davis-Searles PR, Saunders AJ, Erie DA, Winzor DJ, Pielak GJ. Interpreting the effects of small uncharged solutes on protein-folding equilibria. Annu Rev Biophys Biomol Struct. 2001;30:271–306. doi: 10.1146/annurev.biophys.30.1.271. [DOI] [PubMed] [Google Scholar]
- 26.Yadav JK, Prakash V. Thermal stability of alpha-amylase in aqueous cosolvent systems. J Biosci. 2009;34:377–387. doi: 10.1007/s12038-009-0044-0. [DOI] [PubMed] [Google Scholar]
- 27.Street TO, Bolen DW, Rose GD. A molecular mechanism for osmolyte-induced protein stability. Proc Natl Acad Sci USA. 2006;103:13997–14002. doi: 10.1073/pnas.0606236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Teem JL, Berger HA, Ostedgaard LS, Rich DP, Tsui LC, Welsh MJ. Identification of revertants for the cystic fibrosis delta F508 mutation using STE6-CFTR chimeras in yeast. Cell. 1993;73:335–346. doi: 10.1016/0092-8674(93)90233-g. [DOI] [PubMed] [Google Scholar]
- 29.DeCarvalho AC, Gansheroff LJ, Teem JL. Mutations in the nucleotide binding domain 1 signature motif region rescue processing and functional defects of cystic fibrosis transmembrane conductance regulator delta f508. J Biol Chem. 2002;277:35896–35905. doi: 10.1074/jbc.M205644200. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Loo TW, Bartlett MC, Clarke DM. Correctors promote maturation of cystic fibrosis transmembrane conductance regulator (CFTR)-processing mutants by binding to the protein. J Biol Chem. 2007;282:33247–33251. doi: 10.1074/jbc.C700175200. [DOI] [PubMed] [Google Scholar]
- 31.Qu BH, Strickland EH, Thomas PJ. Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. J Biol Chem. 1997;272:15739–15744. doi: 10.1074/jbc.272.25.15739. [DOI] [PubMed] [Google Scholar]
- 32.Pissarra LS, Farinha CM, Xu Z, Schmidt A, Thibodeau PH, Cai Z, Thomas PJ, Sheppard DN, Amaral MD. Solubilizing mutations used to crystallize one CFTR domain attenuate the trafficking and channel defects caused by the major cystic fibrosis mutation. Chem Biol. 2008;15:62–69. doi: 10.1016/j.chembiol.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 33.Zhang XM, Wang XT, Yue H, Leung SW, Thibodeau PH, Thomas PJ, Guggino SE. Organic solutes rescue the functional defect in delta F508 cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2003;278:51232–51242. doi: 10.1074/jbc.M309076200. [DOI] [PubMed] [Google Scholar]
- 34.Richardson JM, Thibodeau PH, Watson J, Thomas PJ. Pediatric Pulmonol. 2007;42:203. [Google Scholar]
- 35.Brouillette C, Protasevich I, Yang Z, Atwell S, Zhao X, Emtage S. Differential scanning calorimetry detects a difference in the stability of the ΔF508 mutant of the nucleotide binding domain 1 of CFTR. Pediatric Pulmonol. 2008;43:227. [Google Scholar]
- 36.Protasevich I, Yang Z, Wang C, Atwell S, Zhao X, Emtage S, Wetmore D, Hunt J, Brouillette C. Analysis of the thermal unfolding of CFTR nucleotide binding domain 1 by differential scanning calorimetry. Pediatric Pulmonol. 2009;44:227. [Google Scholar]
- 37.Qu BH, Thomas PJ. Alteration of the cystic fibrosis transmembrane conductance regulator folding pathway. J Biol Chem. 1996;271:7261–7264. doi: 10.1074/jbc.271.13.7261. [DOI] [PubMed] [Google Scholar]
- 38.Wang C, Protasevich I, Yang Z, Seehausen D, Skalak T, Zhao X, Atwell S, Emtage JS, Wetmore DR, Brouillette CG, Hunt JF. Integrated biophysical studies implicate partial unfolding of NBD1 of CFTR in the molecular pathogenesis of F508del cystic fibrosis. Protein Sci. 2010;19:1932–1947. doi: 10.1002/pro.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baker JM, Hudson RP, Kanelis V, Choy WY, Thibodeau PH, Thomas PJ, Forman-Kay JD. Nat Struct Mol Biol. 2007;14:738–745. doi: 10.1038/nsmb1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen JM CC, Jacques C, Boeuf G, Denamur E, Lecointre G, Mercier B, Cramb G, Férec C. A combined analysis of the cystic fibrosis transmembrane conductance regulator: implications for structure and disease models. Mol Biol Evol. 2001;18:1771–1788. doi: 10.1093/oxfordjournals.molbev.a003965. [DOI] [PubMed] [Google Scholar]
- 41.Lumry R, Eyring H. Conformation changes of protein. J Phys Chem. 1954;58:110–120. [Google Scholar]
- 42.Sanchez-Ruiz JM. Theoretical analysis of Lumry-Eyring models in differential scanning calorimetry. Biophys J. 1992;61:921–935. doi: 10.1016/S0006-3495(92)81899-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lepock JR, Ritchie KP, Kolios MC, Rodahl AM, Heinz KA, Kruuv J. Influence of transition rates and scan rate on kinetic simulations of differential scanning calorimetry profiles of reversible and irreversible protein denaturation. Biochemistry. 1992;31:12706–12712. doi: 10.1021/bi00165a023. [DOI] [PubMed] [Google Scholar]
- 44.Kendrick BS, Cleland JL, Lam X, Nguyen T, Randolph TW, Manning MC, Carpenter JF. Aggregation of recombinant human interferon gamma: kinetics and structural transitions. J Pharm Sci. 1998;87:1069–1076. doi: 10.1021/js9801384. [DOI] [PubMed] [Google Scholar]
- 45.Remmele RL, Jr., Zhang-van Enk J, Dharmavaram V, Balaban D, Durst M, Shoshitaishvili A, Rand H. Scan-rate-dependent melting transitions of interleukin-1 receptor (type II): elucidation of meaningful thermodynamic and kinetic parameters of aggregation acquired from DSC simulations. J Am Chem Soc. 2005;127:8328–8339. doi: 10.1021/ja043466g. [DOI] [PubMed] [Google Scholar]
- 46.Singh N, Liu Z, Fisher HF. The existence of a hexameric intermediate with molten-globule-like properties in the thermal denaturation of bovine-liver glutamate dehydrogenase. Biophys Chem. 1996;63:27–36. doi: 10.1016/s0301-4622(96)02192-8. [DOI] [PubMed] [Google Scholar]
- 47.Roberts CJ. Kinetics of irreversible protein aggregation: Analysis of extended Lumry - Eyring models and implications for predicting protein shelf life. J Phys Chem B. 2003;107:1194–1207. [Google Scholar]
- 48.Arroyo-Reyna A, Tello-Solis SR, Rojo-Dominguez A. Stability parameters for one-step mechanism of irreversible protein denaturation: a method based on nonlinear regression of calorimetric peaks with nonzero deltaCp. Anal Biochem. 2004;328:123–130. doi: 10.1016/j.ab.2004.02.021. [DOI] [PubMed] [Google Scholar]
- 49.Stirpe A, Guzzi R, Wijma H, Verbeet MP, Canters GW, Sportelli L. Calorimetric and spectroscopic investigations of the thermal denaturation of wild type nitrite reductase. Biochim Biophys Acta. 2005;1752:47–55. doi: 10.1016/j.bbapap.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 50.Grinberg VY, Burova TV, Grinberg NV, Shcherbakova TA, Guranda DT, Chilov GG, Svedas VK. Thermodynamic and kinetic stability of penicillin acylase from Escherichia coli. Biochim Biophys Acta. 2008;1784:736–746. doi: 10.1016/j.bbapap.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 51.Kendrick BS C, J S, Cleland JL, Randolph TW. A transient expansion of the native state precedes aggregation of recombinant human interferon-γ. Proc Natl Acad Sci USA. 1998;95:14142–14146. doi: 10.1073/pnas.95.24.14142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Xie D, Freire E. Molecular basis of cooperativity inprotein folding. V. Thermodynamic and structural conditions forthe stabilization of compact denatured states. Proteins. 1994;19:291–301. doi: 10.1002/prot.340190404. [DOI] [PubMed] [Google Scholar]
- 53.Griko YV, Privalov PL. Thermodynamic puzzle of apomyoglobin unfolding. J Mol Biol. 1994;235:1318–1325. doi: 10.1006/jmbi.1994.1085. [DOI] [PubMed] [Google Scholar]
- 54.Pfeil W BV, Ptitsyn OB. Physical nature of the phase transition in globular proteins. Calorimetric study of human alpha-lactalbumin. FEBS Lett. 1986;198:287–291. doi: 10.1016/0014-5793(86)80422-7. [DOI] [PubMed] [Google Scholar]
- 55.Aleksandrov A, Kota P, Aleksandrov L, He L, Jensen T, Cui L, Gentzsch M, Dokholyan N, Riordan J. Regulatory insertion removal restores maturation, stability and function of ΔF508 CFTR. J Mol Biol. 2010;401:194–210. doi: 10.1016/j.jmb.2010.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Motulsky HJ, Christopoulos A. 2003. Fitting models to biological data using linear and nonlinear regressiong. A protical guide to curve fitting. GraphPad Software, Inc. Available at http://www.graphpad.com.