

Abstract
The principal role of acetylcholinesterase is termination of nerve impulse transmission at cholinergic synapses, by rapid hydrolysis of the neurotransmitter acetylcholine to acetate and choline. Its active site is buried at the bottom of a deep and narrow gorge, at the rim of which is found a second anionic site, the peripheral anionic site. The fact that the active site is so deeply buried has raised cogent questions as to how rapid traffic of substrate and products occurs in such a confined environment. Various theoretical and experimental approaches have been used to solve this problem. Here, multiple conventional molecular dynamics simulations have been performed to investigate the clearance of the product, thiocholine, from the active-site gorge of acetylcholinesterase. Our results indicate that thiocholine is released from the peripheral anionic site via random pathways, while three exit routes appear to be favored for its release from the active site, namely, along the axis of the active-site gorge, and through putative back- and side-doors. The back-door pathway is that via which thiocholine exits most frequently. Our results are in good agreement with kinetic and kinetic-crystallography studies. We propose the use of multiple molecular dynamics simulations as a fast yet accurate complementary tool in structural studies of enzymatic trafficking.
Introduction
The principal role of acetylcholinesterase (AChE) is the termination of nerve impulse transmission at cholinergic synapses by rapid hydrolysis of the neurotransmitter acetylcholine (ACh) (1,2). In a first step, the enzyme is acetylated and choline (Ch) is released; a water molecule then regenerates the free enzyme with concomitant release of acetic acid (3) (see Scheme 1). In accordance with its function, AChE is an extremely rapid enzyme, especially for a serine hydrolase, operating at a rate approaching that of a diffusion-controlled reaction (3). Being an essential enzyme, AChE is the target of a variety of potent natural and synthetic toxic compounds, ranging from organophosphorus nerve agents and insecticides to the first generation of anti-Alzheimer drugs (2).
| (Scheme 1) |
The first crystal structure of an AChE, that of Torpedo californica (Tc) (4), revealed that the catalytic anionic site (CAS) nestles near the bottom of a deep and narrow gorge lined by the rings of 14 aromatic residues. At the rim of this gorge, a peripheral anionic site (PAS) contributes to catalytic efficiency by transiently binding substrates en route to the acylation site (5,6).
Due to the unusual catalytic power of AChE (1000–10,000 s−1, depending on the species), the deep confinement of its active site came as a surprise. Consequently, the trafficking of substrate and products within the active-site gorge has been the subject of intensive research, accompanied by considerable controversy, aimed at understanding how AChE's unusual structure is related to its function (7,8). Midway between the PAS and the CAS, a bottleneck is formed by the aromatic rings of the conserved aromatic residues, F330 and Y121 (residue numbers refer to TcAChE throughout the manuscript unless stated otherwise) (Fig. 1) (9,10). The cross-section of the bottleneck in the crystal structure of native TcAChE is substantially smaller than that of the Ch moiety of ACh, implying that substantial breathing motions must occur for either ACh or Ch to enter or exit the CAS (9,11). This contention was substantiated both by steered molecular dynamics (MD) simulations (12,13) and by kinetic crystallography (14). Both X-ray crystallography (5,10,15) and MD simulations (16–18) provide evidence that F330 fulfills a gating role, restricting access to the CAS when the latter is occupied.
Figure 1.
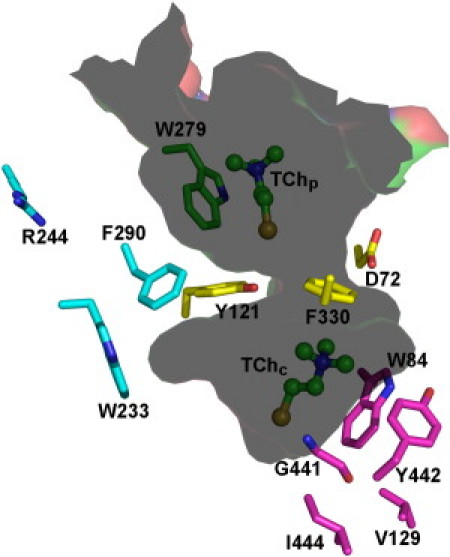
Cross-section through the active-site gorge in the crystal structure of a complex of TcAChE with TCh molecules bound at both the CAS and PAS (PDB code 2c5g). The two TCh molecules, TChC and TChP, are shown as ball-and-stick models, and the side chains of amino-acid residues situated on putative pathways of ligand trafficking are displayed as stick models. In the case of G441, the main-chain is also shown.
Aligned with the gorge is a strong electrostatic gradient (19,20), which has been suggested to be involved in attraction of the positively charged ACh toward the active site. Similarly, this gradient should restrain the exit of the product, choline (Ch) (21). This paradox led to the suggestion that an alternative back-door exit for Ch might exist, adjacent to the CAS (19,22), for which supporting evidence was obtained by MD simulations (22–24). It was proposed that the back-door would open as a result of the concerted movement of the side chains of W84, V129, and G441, generating the transient opening of a channel connecting the CAS to the bulk. Subsequent MD studies have provided further evidence for a repertoire of back- and side-doors (25,26), mainly contributed by fluctuations of residues within the Ω loop (C67–C94) (27–32). Recent kinetic crystallography studies have provided experimental evidence for the existence of both back- and side-doors (14,33).
In this study, we utilize conventional MD simulations (without applying any artificial force or potential to the simulation system) to explore the traffic of thiocholine (TCh) within the active-site gorge of TcAChE, using as a starting model the crystal structure of a complex of TcAChE in which TCh is bound at both the CAS and the PAS (Protein Data Bank (PDB) entry 2c5g). TCh is generated enzymatically by hydrolysis of the chromogenic substrate, acetylthiocholine (ATCh), which displays kinetic properties virtually identical to those of ACh, and is employed in the vast majority of kinetic studies on AChE (34). Three simulation groups were considered, in which TCh was bound at the CAS, at the PAS, or at both sites. Forty 20-ns trajectories were run for each simulation group. While the TCh at the PAS detaches in a random fashion, and is not able to penetrate to the CAS, the TCh bound at the CAS can exit the gorge via three pathways—namely, the active-site gorge, and the proposed back- and side-doors, in good agreement with the kinetic crystallography data (14,33). Interaction with the aromatic residues F330 and W84 is shown to be crucial for the selection of the exit pathways.
Methods
The 0.195-nm resolution crystal structure of the complex of TcAChE with two TCh molecules, one bound at the CAS, and the other at the PAS (PDB entry: 2c5g) (5), provided the starting model for our simulations. Three simulation groups were considered. In group I, only the TCh molecule at the CAS, TChC, is present initially; in group II, only the TCh molecule at the PAS, TChP, is present initially; in group III, both TChC and TChP are present. The protein was inserted, together with crystal waters and the TCh molecule(s), into a box of dimensions 8.96 × 8.52 × 8.41 nm3. The box was solvated by use of a simple point charge water model (35), and the resulting solvated box was then submitted to energy minimization. Counterions were subsequently added to provide a neutral simulation system. Energy minimization was then repeated on the whole system.
After convergence had been reached, the solvent, the counterions, the protein, and the TCh molecules were sequentially coupled to a temperature bath at 300 K (36). A 20-ns simulation was then performed on the whole system. In total, forty 20-ns trajectories, in which different random initial velocities were assigned to each atom of the simulation system (36), were run for each simulation group. In group I, the simulation time was extended to 100 ns for two trajectories in order to see the full exit pathway of TChC.
To evaluate the contribution of electrostatic interactions to the trajectories of TChC and TChP, three additional 20-ns simulations were run in group III, in which the charges of the side-chain carboxyl oxygen atoms of 1), D285, 2), D72, E73, E273, E276, and D285, and 3), all acidic residues in the enzyme, were set to zero. Finally, a 100-ns simulation was run on group III, in which the quaternary nitrogen atoms of the two bound TChs were replaced by carbon atoms to yield noncharged mutated TCh molecules, thus enabling us to investigate the effect of the positive charge of TCh on its exit trajectories.
The MD simulations were carried out with the GROMACS package (37,38), using the NPT ensemble (fixed pressure, temperature, and number of atoms) and periodic boundary conditions. The GROMOS96 force field was applied to the protein (39). Topology and parameter files for TCh were generated using the PRODRG server (http://davapc1.bioch.dundee.ac.uk/cgi-bin/prodrg_beta) with the GROMOS96 force field (40). The partial charges of TCh, or of mutated TCh, were determined using the ChelpG method (41) implemented in Gaussian98 at the HF/6-31G∗∗ level (42). The pressure was maintained at 1 bar by coupling to a Berendsen barostat with τp = 1.0 ps and a compressibility of 4.5 × 10−5 bar−1 (43). The temperature was kept at 300 K by coupling to a Berendsen thermostat with a coupling time of τT = 0.1 ps (43). The LINCS method (44) was used to restrain bond lengths, allowing an integration step of 2 fs. The coordinates of the whole system were saved every 500 steps. Electrostatic interactions were calculated using the particle-mesh Ewald algorithm (45,46).
A cluster method (47) was used to classify the traffic pathways of TChC. Initially, each of the 40 trajectories in one simulation group was superimposed onto the Cα atoms of the starting structure. For each of the fitted trajectories, the coordinates of the center of mass (COM) of TChC were calculated at 1-ps intervals. Thus, 20,000 coordinates, snapshots, were generated for the COM of TChC in each trajectory. The distances of TChC from the starting position were then calculated based on these COM coordinates. If the distance of the COM of any of the generated TChCs was found to be ≤0.3 nm from its starting position, it was considered to be still trapped within the CAS, and its coordinates were excluded from the exit pathway cluster. For coordinates with a distance >0.3 nm from the starting position, those with Δx ≤ 0.01 nm, Δy ≤ 0.01, and Δz ≤ 0.01 nm were clustered and used to generate an averaged position. Connecting these average positions produced a simplified trajectory showing the exit pathway of TChC. After all the simplified trajectories had been generated, the root mean-square deviation between each pair of trajectories was calculated. If the root mean-square deviation of two such trajectories was <0.45 nm, they were assumed to be close enough to be considered as the same pathway for TChC.
Results and Discussion
In this study, three simulation groups are considered, each corresponding to a different binding scenario for the TCh/TcAChE system:
Group I refers to TcAChE with a TCh molecule bound at the CAS, i.e., TChc.
Group II refers to TcAChE with a TCh molecule bound at the PAS, i.e., TChP.
Group III refers to TcAChE complexed with two TCh molecules, i.e., both TChc at the CAS and TChp at the PAS.
In the following, the results are presented and discussed for all three simulation groups.
Behavior of TChC and TChP during the time course of the simulation
The movements of TChC and TChP in the various simulations were evaluated by estimating their distances from their starting positions as a function of time. Fig. 2 shows the time-dependent distances of TChC and TChP from their starting positions over the forty 20-ns trajectories in each simulation group. In the 40 trajectories in simulation group I, the displacement of TChC is for most of the time <0.6 nm, but peaks to ∼0.9 nm for short time intervals in a few of them (Fig. 2 A). A histogram shows that the peak of the distribution is at ∼0.2 nm (Fig. 3 A). Similar results are obtained for TChC in simulation group III, except that in two out of the 40 trajectories, brief displacements as high as ∼1.2 nm are observed (Figs. 2 B and 3 A).
Figure 2.
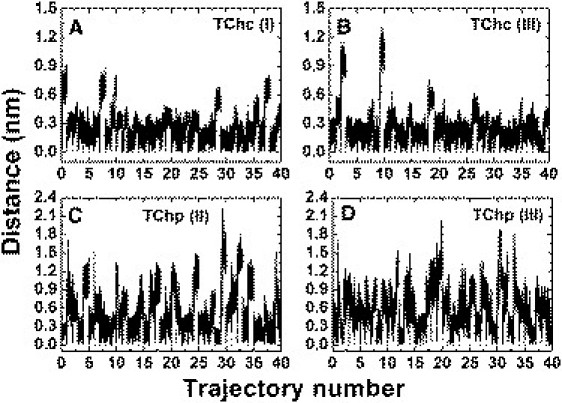
Distances of TChC and TChP from their starting points in sets of 20-ns MD simulation trajectories. Each panel shows 40 such trajectories, displayed sequentially, showing, for each trajectory, snapshots extracted at 1-ps intervals. They have the same starting coordinates, but their velocities (speed and direction) are randomly selected. (A) TChC in group I; (B) TChC in group III; (C) TChP in group II; and (D) TChP in group III. Each trajectory has a duration of 20 ns, and the distances were calculated based on the snapshots extracted at 1-ps intervals.
Figure 3.
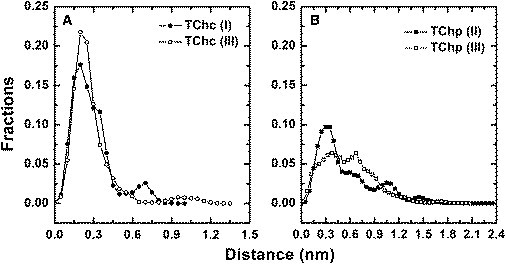
Histograms of the distances of TChC (A) and TChP (B) from their starting points. The individual distances were extracted from the MD simulations presented in Fig. 2. Each histogram thus contains 800,000 data points compiled from the 1-ps snapshots in each of the 40 MD simulations of 20-ns duration, and presents data taken from the appropriate panel in Fig. 2.
However, the displacements observed for TChP in both simulation groups are quite different (Fig. 2, C and D, and Fig. 3 B). They reach much higher values, 2 nm or more, and display much more frequent fluctuations with larger amplitudes. The histograms for TChP shown in Fig. 3 B are not as sharp as those for TChC. This suggests that a wide range of exit trajectories and/or alternative binding sites is accessible to TChP. Overall, the data indicate that TChP dissociates from the PAS frequently and randomly, while TChC remains for most of the time trapped within the active-site gorge close to the CAS. Eventually, however, TChC exits the gorge in ∼75% of the 20 ns trajectories analyzed, in both simulation groups I and III.
Exit of TChC from the CAS occurs via three routes
The CAS is the site at which TCh is generated as a result of hydrolysis of ATCh by AChE. Thus, the behavior of TChC along the course of the simulation trajectory is, from a functional perspective, more relevant than that of TChP. The CAS is buried at the bottom of the active-site gorge, below the bottleneck formed by F330 and Y121. In the crystal structure of the TCh-TcAChE complex, it can be seen that the phenyl ring of F330 rotates to cover the quaternary moiety of TChC, thus blocking access to the CAS (Fig. 1).
To identify the exit routes available to TChC, both visual inspection using the molecular graphic software VMD program (48), and a cluster method (described under Methods) were applied to the 40 trajectories in each of simulation groups I and III. Based on the distance distribution of TChC (Fig. 3 A), and as confirmed by graphic visualization, a cutoff distance of 0.3 nm was set to indicate whether TChC had remained within the CAS or had exited. Table 1 shows the statistical results for TChC within the 80 trajectories in simulation groups I and III.
Table 1.
Exit trajectories of TChC in simulation groups I and III
| Group I | Group III | |
|---|---|---|
| Exit along gorge axis | 1 (2.5%) | 2 (5.0%) |
| Exit via side-door | 3 (7.5%) | 2 (5.0%) |
| Exit through back-door | 27 (67.5%) | 26 (65.0%) |
| Remained in CAS | 9 (22.5%) | 10 (25.0%) |
Three exit pathways for TChC are seen in both group I and group III (Table 1 and Fig. 4 A). The first is along the gorge axis (Fig. 5); the second is through a channel that opens transiently within the CAS wall adjacent to W84 (Fig. 6), earlier coined the back-door (19,22,23); and the third exit pathway is via a channel that opens transiently through the Ω loop (that is, between residues V71 and D72), further up the active-site gorge, which has been referred to as the side-door (Fig. 4 A) (27–29). For TChC in simulation group I, Table 1 shows that the route along the gorge axis is followed in 1/40 trajectories; in 27/40 trajectories, exit is via the back-door; and exit by the side-door is seen in 3/40 trajectories.
Figure 4.
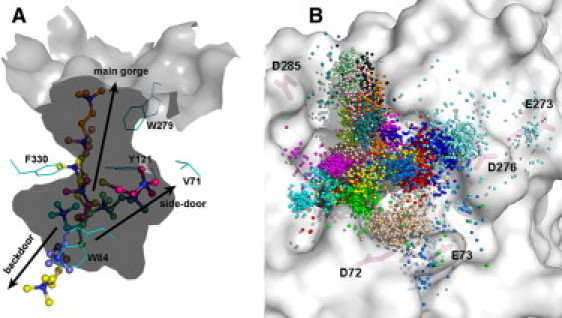
(A) Cross-section of the inner surface of the active-site gorge, displaying three putative pathways for exit of TChC from the CAS. The snapshots of TChC were extracted from MD trajectories in groups I and III. (B) Movement of TChP out of the PAS. The mouth of the active-site gorge is viewed from above. The colored balls track the positions of the quaternary nitrogens of TChP, sampled at 100-ps intervals, in each of the 40 MD trajectories in group III. Each color-coding represents one trajectory. The coordinates utilized in both panels A and B are those of PDB code 2c5g.
Figure 5.
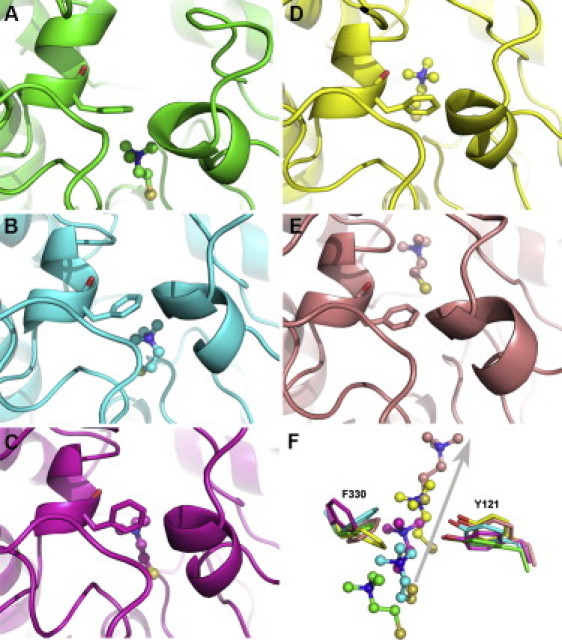
Traffic of TChC up the active-site gorge, along a MD trajectory in simulation group III. The gorge is shown as a ribbon diagram. The snapshots (A–E) display the TChC molecule as a ball-and-stick model, and the side chain of F330 as sticks. (A) The 2c5g crystal structure (green). (B) Snapshot at 2-ns simulation of the MD trajectory (cyan); (C) at 2.5 ns (magenta); (D) at 4 ns (yellow); and (E) at 12 ns (salmon). (F) Superposition of F330, Y121, and TChC extracted from the data presented in panels A–E. The arrow indicates the direction of movement of TChC. F330 and Y121 are shown as sticks, and TChC as ball-and-stick models.
Figure 6.
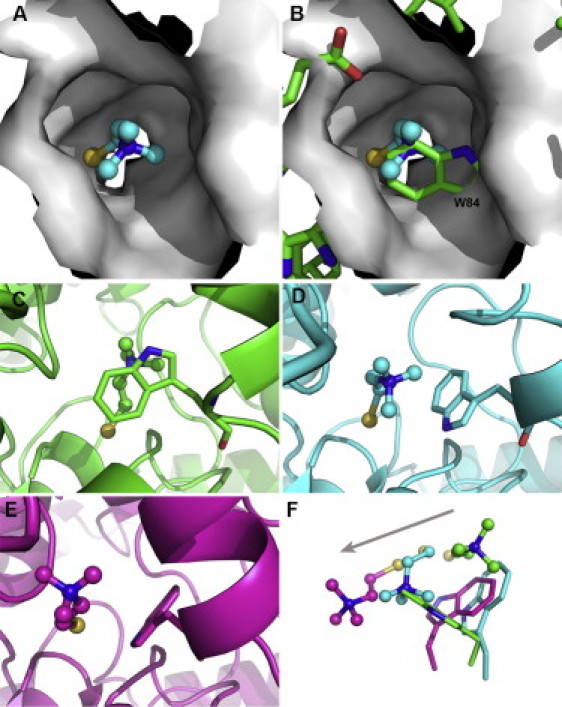
Migration of TChC through the back-door, along a MD trajectory in simulation group III. (A) Molecular surface of the back-door region in a snapshot extracted at 4.2 ns after initiation of the trajectory, looking from the protein surface into the back-door channel, and showing TChP as a ball-and-stick model. (B) Overlay of the molecular surface shown in panel A upon the crystal structure (PDB code 2c5g), some of whose residues, including W84, are displayed as sticks. It is clearly seen that, in the crystal structure, the side chain of W84 blocks the channel. (C–E) Locations of TChC and of the indole group of W84 in the crystal structure (C, green), and in snapshots extracted at 4.2 ns (D, cyan) and 17.1 ns (E, magenta) after initiation of the trajectory. The protein is displayed as a ribbon diagram, with Trp84 as a stick model, and TCh is shown as a ball-and-stick model. (F) Superposition of representations from panels C–E displaying only TChC and W84, presented and color-coded as in panels E and F. The arrow indicates the direction of movement of TChC.
In the remaining nine trajectories, the ligand does not exit the CAS during the duration of the simulation. In group III, in 2/40 cases, exit of the ligand is along the gorge axis; in 26/40 cases, exit is via the back-door; in 2/40 cases, exit is via the side-door; and in 10 trajectories, the ligand remains within the CAS. Therefore, in both simulation groups the back-door is the favored route for exit of TChC from the CAS. The behavior of TChC in the two simulation groups is quite similar (Table 1 and Figs. 2 and 3). The difference between groups I and III is that, in the latter, in addition to TChC bound at the CAS, TChP occupies the PAS. However, our simulations clearly show that the presence of TChP at the PAS does not influence the exit frequencies and pathways of TChC, and the occupation of the PAS by TChP does not appear to affect the exit of TChC along the axis of the active-site gorge.
Dissociation of TCh from the PAS is frequent and random
The PAS is located at the rim of the active-site gorge, and has a large surface exposure. Consequently, TChP can easily exit the PAS, as is seen in both simulation groups II and III (Fig. 2, C and D). Fig. 4 B shows that release of TChP occurs randomly in many directions, with the trajectories covering almost all of the area around the PAS. Interestingly, in none of the eighty 20-ns trajectories did TChP move into the bulk solvent, but always remained attached to the surface of the protein. This is also true for TChC in those trajectories in which it left the gorge. Electrostatic interactions between TCh and surface residues are most likely responsible for this, an issue further addressed below. It is also noteworthy that while TChP is observed to move toward the CAS in some simulations, it never actually crosses the bottleneck in any of them. It is possible that a simulation time of 20 ns is too short to capture such an event.
While the presence of TChP at the PAS does not appear to affect the behavior of TChC, the opposite is not true. The presence of TChC at the CAS does influence the residence time of TChP at the PAS. Fig. 3 B shows that the distance histograms for TChP in groups II and III are significantly different. TChP appears to disassociate more frequently from the PAS when TChC is present at the CAS. One reason might be that the presence of TChC substantially neutralizes the electrostatic potential along the axis of the gorge (20), thus decreasing the field restraining TChP. Such a possibility was already suggested by earlier MD simulations on complexes of AChE with tacrine and huperzine A (27,28).
Side-chain flexibility of F330 and W84
F330 and W84 are the two key players in the exit of TChC along the active-site gorge and through the back-door, respectively (Figs. 5 and 6). F330 was experimentally shown to display high conformational mobility (10,15,18). Crystallographic data provided direct evidence that binding to the CAS of either the substrate, ATCh, or the product, TCh, is accompanied by a rotation of the F330 side chain to an orientation perpendicular to the gorge axis that results in the closing of the gorge (5).
It is thus conjectured that the exit of TChC up the active-site gorge requires that the F330 side chain reverts to its open conformation, with an orientation parallel to the gorge axis. In order to investigate whether and how the exit of TChC along the axis of the active-site gorge is correlated with the side-chain conformations of F330 and Y121, namely, the two bottleneck residues, their χ1 and χ2 angles were plotted as function of simulation time for one of the two trajectories in which migration of TChC from the CAS to the PAS had been observed (simulation group III; Fig. 7 A). The χ1 and χ2 angles of Y121, and the χ1 angle of F330, are stable along the simulation trajectory, whereas the χ2 angle of F330 exhibits large fluctuations. Snapshots along this MD trajectory are shown in Fig. 5. They provide evidence that exit of TChC along the axis of the active-site gorge is directly associated with changes in the side-chain conformation of F330.
Figure 7.
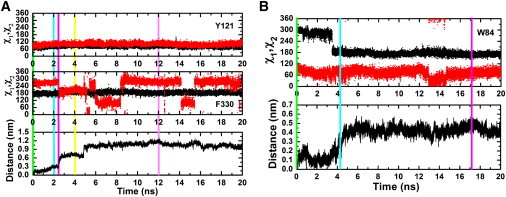
Changes in the χ1 and χ2 angles of Y121, F330, and W84, and in the distance of TChC from its starting position, in two trajectories taken from simulation group III. (A) Changes in the χ1 and χ2 angles of Y121 and F330 during exit of ThCc along the gorge axis. (B) Change in the χ1 and χ2 angles of W84 during exit of ThCc through the back-door. Black traces show χ1 angles and red traces show χ2 angles. The colored bars in panels A and B mark the simulation times at which the snapshots shown in Figs. 5 and 6, respectively, were taken.
At the start of the simulation, the aromatic ring of F330 caps the quaternary moiety of TChC, with which it interacts through a cation-π interaction. After 2 ns, TChC has moved toward the bottleneck, with concomitant initiation of side-chain rotation of F330. As TChC moves further up the gorge, the side chain of F330 achieves its open conformation, resulting in a widening of the bottleneck, thus allowing the passage of TChC. This correlates with the observation of the distance of TChC jumping from ∼0.3 to ∼0.6 nm just a few hundred picoseconds after the reorientation of the F330 side chain (i.e., the second and the third bars in Fig. 7 A). Thus, the exit of TChC, when it occurs along the axis of the active-site gorge, critically depends on the conformation of the side chain of F330. Fluctuations in the χ2 angle of F330 are observed even after TChC has passed the bottleneck, in agreement with earlier studies that revealed the intrinsically high conformational freedom associated with this angle (5,10,12,15–18).
Our simulations also confirm that TChC can exit the CAS via the proposed back-door, adjacent to W84. It is noteworthy that this is the principal exit pathway for TChC in both simulation groups I and III. Fig. 6, A and B, illustrates the sealing of the back-door by the side chain of W84 in the native TcAChE structure. We investigated the putative correlation between the side-chain conformation of W84 and the movement of TChC through the back-door by examining changes in the χ1 and χ2 angles of the side chain of W84 and the distance of TChC as a function of simulation time (Fig. 7 B). Both χ1 and χ2 angles changed during the 20-ns simulations in which TChC exited via the back-door, with the changes in χ1 being larger than those in χ2. Notably, a sharp change of ∼120° in χ1 preceded the increase in the distance of TChC. From inspection of the snapshots in Fig. 6, it is appears that the rotation of W84 χ1 is important for exit of TChC to occur through the back-door.
Electrostatic interactions between TCh and TcAChE
As mentioned in the Introduction, extensive studies have been carried out to calculate the electrostatic potential along the active-site gorge, and to explore the effect of the potential gradient on the traffic of charged ligands (19–21,49,50). Here, we analyze how individual acidic residues affect the movement of TCh bound at the CAS or at the PAS, of TChC and TChP. Electrostatic interactions that might affect clearance of TChC and TChP from the CAS and the PAS, respectively, were examined by calculating the minimal distance between their quaternary nitrogen atoms and the carboxylate oxygen atoms of acidic residues lining the gorge and on the surface adjacent to the mouth of the gorge. Table 2 lists the frequency of such interactions in the three simulation groups, using a cutoff distance of 0.6 nm.
Table 2.
Frequency of carboxyl oxygens of acidic residues making electrostatic interactions with the quaternary groups of TChC or TChP in the three simulation groups
| TChC (I) | TChC (III) | TChP (II) | TChP(III) | |
|---|---|---|---|---|
| D72 | 204,012 | 140,762 | 436,350 | 270,416 |
| E73 | 13,286 | 1920 | 101,465 | 161,444 |
| E82 | 4268 | |||
| E199 | 643,868 | 645,377 | ||
| E247 | 14,207 | 562 | ||
| E273 | 1395 | 4206 | ||
| D276 | 118,740 | 81,120 | ||
| E278 | 36,208 | 3937 | ||
| D285 | 21,820 | 593,531 | 586,572 | |
| E327 | 122 | 106 | ||
| E443 | 9060 | 19,638 | ||
| E445 | 60,269 | 78,047 |
Cutoff for the maximal distance between the quaternary nitrogen and the carboxyl oxygen is 0.6 nm.
The interactions were measured based on snapshots extracted every picosecond along the trajectories, yielding a total of 800,000 snapshots for each simulation group. The calculations show that the carboxyl groups of D72, E73, E199, D276, D285, E327, E443, and E445 interact electrostatically with TChC, while those of D72, E73, E82, E247, E273, D276, E278, and D285 interact with TChP. Among these residues, E199 and D285 have the highest interaction time with TChC and TChP, respectively. Interestingly, D72, a residue located midway up the gorge, is that with the second highest capacity to interact electrostatically with both TChC and TChP. Fig. S1 in the Supporting Material displays locations of the acidic residues interacting with the two TCh molecules: E73, E247, E273, D276, and D285 are on the surface of the protein around the rim of the gorge; D72 and E278 are above F330, and are thus assigned to the PAS; E199, E327, and E443 are at the bottom of the gorge, close to the CAS; E445 is a solvent-exposed residue, located on the outside of the back-door.
Combination of the data on the traffic of TChC and of that on its electrostatic interactions with acidic residues point to E199 being the major contributor to the electrostatic attraction of TChC, a fact that may explain why TChC remains at the CAS during most of the simulation time. The observation that the shortest distance between both E199Oɛ1 and E199Oɛ2 and the quaternary nitrogen atom of TChC is only ∼0.3 nm, may explain the strong tendency of TChC to remain within the CAS for a prolonged period. The exit of TChC from the CAS involves interactions with both E199 and E445 if it exits via the back-door, and with E199 and D72 if it exits via the gorge axis. It is plausible that when rotation of the side chain of F330 opens the bottleneck, the negative charge of D72 will attract TChC toward the rim of the gorge. Analogously, after the required conformational change in the side chain of W84 has taken place, electrostatic attraction by E445 may assist exit of TChC via the back-door.
The data presented in Fig. 4 B suggest that TChP does not move into the bulk solvent after its exit from the PAS, but instead remains on the protein surface. The calculation of minimal distances between the quaternary nitrogen of TChP and the carboxyl oxygen atoms of aspartate and glutamate residues indicates that TChP makes strong electrostatic interactions with acidic residues on the protein surface. In the native protein, most of these interactions are with D285 (Table 2 and Fig. 8). To verify whether the electrostatic interaction with D285 is the main force that maintains TChP tethered to the protein surface, the charges of its carboxyl oxygen atoms were set to zero at the beginning of an independent 20-ns simulation.
Figure 8.
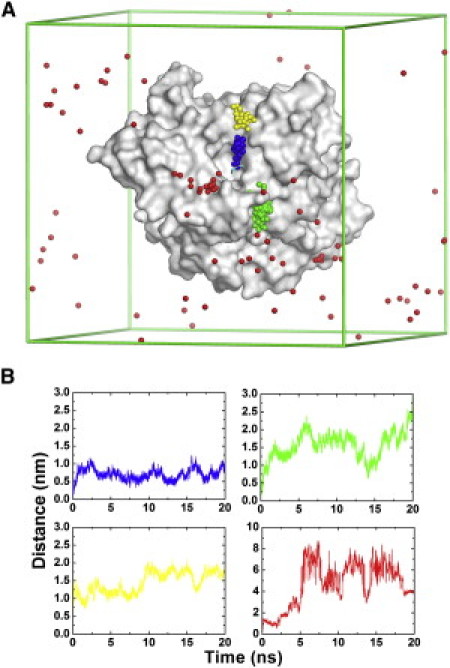
MD trajectories for movement of TChP in native and mutated TcAChE in which acidic residues had been neutralized. (A) The movement of TChP as extracted, at 200-ps intervals, from four MD trajectories, is shown, with the individual balls representing the nitrogen of TChP. The blue trajectory is that of one of the forty trajectories in simulation group III. The simulation conditions in the other three trajectories were similar to those for the blue trajectory, except that the charges of side-chain carboxyl oxygen atoms were set to zero for D285 (green trajectory), for D72, E73, E273, E276, and D285 (yellow trajectory), and for all aspartate and glutamate residues (red trajectory). The molecular surface of the protein (PDB code 2c5g) is displayed in light gray, and the lines of the simulation box are depicted in green. (B) The distance of TChP from its binding site at the PAS in the four trajectories displayed in Fig. 8A, with the same color coding (blue, green, yellow, and red).
In this simulation, instead of remaining on the protein surface near D285 (as in Fig. 8 A and in the blue trajectory, upper left, in Fig. 8 B), TChP moved to another surface area, behind the back- and side-doors, making a strong electrostatic interaction with E82 (Fig. 8 A, and in the green trajectory, upper right, in Fig. 8 B). In an additional simulation, the charges of the five acidic residues that interact strongly with TChP, i.e., D72, E73, E273, E276, and D285 (see Table 2), were all neutralized. Consequently, TChP interacted with E247, a residue also located at the mouth of the active site gorge, and remained in its vicinity throughout most of the duration of the simulation (Fig. 8 A, and in the yellow trajectory, lower left, in Fig. 8 B). We went on to design two extreme scenarios, in which either all the acidic residues of the protein were neutralized, or the charge of TChP was abolished.
In the former case, TChP moved into the bulk solution within a few nanoseconds (Fig. 8 A, and in the red trajectory, lower right, in Fig. 8 B) and did not reassociate with the protein. In the latter, the mutated TCh detached from the protein surface within ∼37 ns (Fig. S2). This set of independent simulations thus confirms that electrostatic attraction to acidic residues at the gorge entrance, notably E285, is the main force that tethers TChP in its vicinity and prevents its movement into the bulk solvent. Our findings are not in good agreement with experimental mutagenesis data that revealed only a modest contribution of surface electrostatic residues to the catalytic efficiency of AChE (51); this discrepancy may, however, be due to fact that the experimental data were acquired at higher ionic strength and on much longer timescales than our simulation data.
Conclusions
Multiple conventional MD simulations were utilized to study the traffic of the enzymatic hydrolysis product, TCh, within the active-site gorge of TcAChE. Overall, our results are in good agreement with experimental data obtained in kinetic and kinetic-crystallography studies. To our knowledge, this is the first computational study to show that the hydrolysis product can exit through back- and side-doors, and not only through the active-site gorge. Thus, MD simulations provide a fast and accurate theoretical approach for studying the traffic of substrates, products, and other ligands within enzymes and other proteins.
Acknowledgments
We thank Douglas Tobias for critical reading of the manuscript.
This study was supported by the National Natural Science Foundation of China (grant No. 20720102040), the State Key Program of Basic Research of China (grant No. 2009CB918501), the National S&T Major Project (grant Nos. 2009ZX09501-001 and 2009ZX09301-001), by grants to M.W. from the Délégation Générale pour l'Armement (No. DGA-REI 2009-34-0023) and the Agence Nationale de la Recherche (No. ANR-09-BLAN-0192-04), by the European Commission VIth Framework Research and Technological Development Program, “SPINE2-COMPLEXES” Project (under contract No. 031220), and “Teach-SG” Project (under contract No. ISSG-CT-2007-037198), the Kimmelman Center for Biomolecular Structure and Assembly, the Benoziyo Center for Neurosciences, the Divadol Foundation, the Nalvyco Foundation, the Bruce Rosen Foundation, the Jean and Julia Goldwurm Memorial Foundation, the Neuman Foundation, the Kalman and Ida Wolens Foundation, and a research grant from Erwin Pearl. J.L.S. is the Morton and Gladys Pickman Professor of Structural Biology. Computational resources were supported by the Computer Network Information Center of the Chinese Academy of Sciences, and by the Shanghai Supercomputing Center, and were sponsored by a grant from the Information Construction Project of the Chinese Academy of Sciences during the period of the 11th Five-Year Plan.
Contributor Information
Yechun Xu, Email: ycxu@mail.shcnc.ac.cn.
Joel L. Sussman, Email: joel.sussman@weizmann.ac.il.
Supporting Material
References
- 1.Rosenberry T.L. Acetylcholinesterase. Adv. Enzymol. Relat. Areas Mol. Biol. 1975;43:103–218. doi: 10.1002/9780470122884.ch3. [DOI] [PubMed] [Google Scholar]
- 2.Silman I., Sussman J.L. Acetylcholinesterase: ‘classical’ and ‘non-classical’ functions and pharmacology. Curr. Opin. Pharmacol. 2005;5:293–302. doi: 10.1016/j.coph.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 3.Quinn D.M. Acetylcholinesterase: enzyme structure, reaction dynamics, and virtual transition states. Chem. Rev. 1987;87:955–979. [Google Scholar]
- 4.Sussman J.L., Harel M., Silman I. Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science. 1991;253:872–879. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- 5.Colletier J.P., Fournier D., Weik M. Structural insights into substrate traffic and inhibition in acetylcholinesterase. EMBO J. 2006;25:2746–2756. doi: 10.1038/sj.emboj.7601175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenberry T.L., Sonoda L.K., Johnson J.L. Analysis of the reaction of carbachol with acetylcholinesterase using thioflavin T as a coupled fluorescence reporter. Biochemistry. 2008;47:13056–13063. doi: 10.1021/bi8015197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Szegletes T., Mallender W.D., Rosenberry T.L. Nonequilibrium analysis alters the mechanistic interpretation of inhibition of acetylcholinesterase by peripheral site ligands. Biochemistry. 1998;37:4206–4216. doi: 10.1021/bi972158a. [DOI] [PubMed] [Google Scholar]
- 8.Stojan J., Brochier L., Fournier D. Inhibition of Drosophila melanogaster acetylcholinesterase by high concentrations of substrate. Eur. J. Biochem. 2004;271:1364–1371. doi: 10.1111/j.1432-1033.2004.04048.x. [DOI] [PubMed] [Google Scholar]
- 9.Zhou H.X., McCammon J.A. The gates of ion channels and enzymes. Trends Biochem. Sci. 2010;35:179–185. doi: 10.1016/j.tibs.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kryger G., Silman I., Sussman J.L. Structure of acetylcholinesterase complexed with E2020 (Aricept): implications for the design of new anti-Alzheimer drugs. Structure. 1999;7:297–307. doi: 10.1016/s0969-2126(99)80040-9. [DOI] [PubMed] [Google Scholar]
- 11.Zhou H.X., Wlodek S.T., McCammon J.A. Conformation gating as a mechanism for enzyme specificity. Proc. Natl. Acad. Sci. USA. 1998;95:9280–9283. doi: 10.1073/pnas.95.16.9280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu Y., Shen J., Jiang H. How does huperzine A enter and leave the binding gorge of acetylcholinesterase? Steered molecular dynamics simulations. J. Am. Chem. Soc. 2003;125:11340–11349. doi: 10.1021/ja029775t. [DOI] [PubMed] [Google Scholar]
- 13.Niu C., Xu Y., Jiang H. Dynamic mechanism of E2020 binding to acetylcholinesterase: a steered molecular dynamics simulation. J. Phys. Chem. B. 2005;109:23730–23738. doi: 10.1021/jp0552877. [DOI] [PubMed] [Google Scholar]
- 14.Colletier J.P., Royant A., Weik M. Use of a ‘caged’ analogue to study the traffic of choline within acetylcholinesterase by kinetic crystallography. Acta Crystallogr. D Biol. Crystallogr. 2007;63:1115–1128. doi: 10.1107/S0907444907044472. [DOI] [PubMed] [Google Scholar]
- 15.Harel M., Schalk I., Sussman J.L. Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA. 1993;90:9031–9035. doi: 10.1073/pnas.90.19.9031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tai K., Shen T., McCammon J.A. Analysis of a 10-ns molecular dynamics simulation of mouse acetylcholinesterase. Biophys. J. 2001;81:715–724. doi: 10.1016/S0006-3495(01)75736-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shen T., Tai K., McCammon J.A. Molecular dynamics of acetylcholinesterase. Acc. Chem. Res. 2002;35:332–340. doi: 10.1021/ar010025i. [DOI] [PubMed] [Google Scholar]
- 18.Xu Y., Colletier J.P., Sussman J.L. Flexibility of aromatic residues in the active-site gorge of acetylcholinesterase: x-ray versus molecular dynamics. Biophys. J. 2008;95:2500–2511. doi: 10.1529/biophysj.108.129601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ripoll D.R., Faerman C.H., Sussman J.L. An electrostatic mechanism for substrate guidance down the aromatic gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. USA. 1993;90:5128–5132. doi: 10.1073/pnas.90.11.5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Felder C.E., Botti S.A., Sussman J.L. External and internal electrostatic potentials of cholinesterase models. J. Mol. Graph. Model. 1997;15:318–327. doi: 10.1016/s1093-3263(98)00005-9. 335–337. [DOI] [PubMed] [Google Scholar]
- 21.Botti S.A., Felder C.E., Silman I. A modular treatment of molecular traffic through the active site of cholinesterase. Biophys. J. 1999;77:2430–2450. doi: 10.1016/S0006-3495(99)77080-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Axelsen P.H., Harel M., Sussman J.L. Structure and dynamics of the active site gorge of acetylcholinesterase: synergistic use of molecular dynamics simulation and x-ray crystallography. Protein Sci. 1994;3:188–197. doi: 10.1002/pro.5560030204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gilson M.K., Straatsma T.P., Sussman J.L. Open “back door” in a molecular dynamics simulation of acetylcholinesterase. Science. 1994;263:1276–1278. doi: 10.1126/science.8122110. [DOI] [PubMed] [Google Scholar]
- 24.Kovach I.M., Qian N., Bencsura A. Efficient product clearance through exit channels in substrate hydrolysis by acetylcholinesterase. FEBS Lett. 1994;349:60–64. doi: 10.1016/0014-5793(94)00580-x. [DOI] [PubMed] [Google Scholar]
- 25.Enyedy I.J., Kovach I.M., Brooks B.R. Alternate pathways for acetic acid and acetate ion release from acetylcholinesterase: a molecular dynamics study. J. Am. Chem. Soc. 1998;120:8043–8050. [Google Scholar]
- 26.Van Belle D., De Maria L., Wodak S.J. Pathways of ligand clearance in acetylcholinesterase by multiple copy sampling. J. Mol. Biol. 2000;298:705–726. doi: 10.1006/jmbi.2000.3698. [DOI] [PubMed] [Google Scholar]
- 27.Wlodek S.T., Clark T.W., McCammon J.A. Molecular dynamics of acetylcholinesterase dimer complexed with tacrine. J. Am. Chem. Soc. 1997;119:9513–9522. [Google Scholar]
- 28.Tara S., Helms V., McCammon J.A. Molecular dynamics of mouse acetylcholinesterase complexed with huperzine A. Biopolymers. 1999;50:347–359. doi: 10.1002/(SICI)1097-0282(19991005)50:4<347::AID-BIP1>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 29.Tara S., Straatsma T.P., McCammon J.A. Mouse acetylcholinesterase unliganded and in complex with huperzine A: a comparison of molecular dynamics simulations. Biopolymers. 1999;50:35–43. doi: 10.1002/(SICI)1097-0282(199907)50:1<35::AID-BIP4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 30.Bui J.M., McCammon J.A. Protein complex formation by acetylcholinesterase and the neurotoxin fasciculin-2 appears to involve an induced-fit mechanism. Proc. Natl. Acad. Sci. USA. 2006;103:15451–15456. doi: 10.1073/pnas.0605355103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tai K., Shen T., McCammon J.A. Mechanism of acetylcholinesterase inhibition by fasciculin: a 5-ns molecular dynamics simulation. J. Am. Chem. Soc. 2002;124:6153–6161. doi: 10.1021/ja017310h. [DOI] [PubMed] [Google Scholar]
- 32.Bui J.M., Tai K., McCammon J.A. Acetylcholinesterase: enhanced fluctuations and alternative routes to the active site in the complex with fasciculin-2. J. Am. Chem. Soc. 2004;126:7198–7205. doi: 10.1021/ja0485715. [DOI] [PubMed] [Google Scholar]
- 33.Colletier J.P., Bourgeois D., Weik M. Shoot-and-Trap: use of specific x-ray damage to study structural protein dynamics by temperature-controlled cryo-crystallography. Proc. Natl. Acad. Sci. USA. 2008;105:11742–11747. doi: 10.1073/pnas.0804828105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellman G.L., Courtney K.D., Feather-Stone R.M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 1961;7:88–95. doi: 10.1016/0006-2952(61)90145-9. [DOI] [PubMed] [Google Scholar]
- 35.Berendsen H.J.C., Postma J.P.M., Hermans J. Intermolecular Forces. In: Pullman B., editor. Reidel, Dordrecht; The Netherlands: 1981. pp. 331–342. [Google Scholar]
- 36.van der Spoel D., van Buuren A.R., Berendsen H.J.C. Department of Physics, Groningen University, Groningen; The Netherlands: 2001. GROMACS User Manual. [Google Scholar]
- 37.Berendsen H.J.C., van der Spoel D., van Drunen R. GROMACS: a message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995;91:43–56. [Google Scholar]
- 38.Lindahl E., Hess B., van der Spoel D. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Model. 2001;7:306–317. [Google Scholar]
- 39.van Gunsteren W.F., Billeter S.R., Tironi I.G. Vdf Hochschulverlag AG an der ETH Zurich; Zurich, Switzerland: 1996. Biomolecular Simulations: The GROMOS96 Manual and User Guide. [Google Scholar]
- 40.van Aalten D.M., Bywater R., Vriend G. PRODRG, a program for generating molecular topologies and unique molecular descriptors from coordinates of small molecules. J. Comput. Aided Mol. Des. 1996;10:255–262. doi: 10.1007/BF00355047. [DOI] [PubMed] [Google Scholar]
- 41.Breneman C.M., Wiberg K.B. Determining atom-centered monopoles from molecular electrostatic potentials. The need for high sampling density in formamide conformational analysis. J. Comput. Chem. 1990;11:361–373. [Google Scholar]
- 42.Frisch M.J., Trucks G.W., Pople J.A. Gaussian Inc.; Pittsburgh, PA: 1998. Gaussian 98, Rev. A.6. [Google Scholar]
- 43.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 44.Hess B., Bekker B., Fraaije J.G.E.M. LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 1997;18:1463–1472. [Google Scholar]
- 45.Darden T., York D., Pedersen L. Particle mesh Ewald: an Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 46.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald potential. J. Chem. Phys. 1995;103:8577–8592. [Google Scholar]
- 47.Shao J., Tanner S.W., Cheatham T.E. Clustering molecular dynamics trajectories. 1. Characterizing the performance of different clustering algorithms. J. Chem. Theory Comput. 2007;3:2312–2334. doi: 10.1021/ct700119m. [DOI] [PubMed] [Google Scholar]
- 48.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 49.Antosiewicz J., Wlodek S.T., McCammon J.A. Acetylcholinesterase: role of the enzyme's charge distribution in steering charged ligands toward the active site. Biopolymers. 1996;39:85–94. doi: 10.1002/(SICI)1097-0282(199607)39:1%3C85::AID-BIP9%3E3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 50.Wlodek S.T., Shen T., McCammon J.A. Electrostatic steering of substrate to acetylcholinesterase: analysis of field fluctuations. Biopolymers. 2000;53:265–271. doi: 10.1002/(SICI)1097-0282(200003)53:3<265::AID-BIP6>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 51.Shafferman A., Ordentlich A., Velan B. Electrostatic attraction by surface charge does not contribute to the catalytic efficiency of acetylcholinesterase. EMBO J. 1994;13:3448–3455. doi: 10.1002/j.1460-2075.1994.tb06650.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.