

Abstract
Adipose tissue-derived adipokines play important roles in controlling systemic insulin sensitivity and energy balance. Our recent efforts to identify novel metabolic mediators produced by adipose tissue have led to the discovery of a highly conserved family of secreted proteins, designated as C1q/TNF-related proteins 1–10 (CTRP1 to -10). However, physiological functions regulated by CTRPs are largely unknown. Here we provide the first in vivo functional characterization of CTRP3. We show that circulating levels of CTRP3 are inversely correlated with leptin levels; CTRP3 increases with fasting, decreases in diet-induced obese mice with high leptin levels, and increases in leptin-deficient ob/ob mice. A modest 3-fold elevation of plasma CTRP3 levels by recombinant protein administration is sufficient to lower glucose levels in normal and insulin-resistant ob/ob mice, without altering insulin or adiponectin levels. The glucose-lowering effect in mice is linked to activation of the Akt signaling pathway in liver and a marked suppression of hepatic gluconeogenic gene expression. Consistent with its effects in mice, CTRP3 acts directly and independently of insulin to regulate gluconeogenesis in cultured hepatocytes. In humans, alternative splicing generates two circulating CTRP3 isoforms differing in size and glycosylation pattern. The two human proteins form hetero-oligomers, an association that does not require interdisulfide bond formation and appears to protect the longer isoform from proteolytic cleavage. Recombinant human CTRP3 also reduces glucose output in hepatocytes by suppressing gluconeogenic enzyme expression. This study provides the first functional evidence linking CTRP3 to hepatic glucose metabolism and establishes CTRP3 as a novel adipokine.
Keywords: Adipose Tissue, Adipose Tissue Metabolism, Diabetes, Gluconeogenesis, Insulin Resistance, Metabolism, Metabolic Regulation, Obesity, Adipokine
Introduction
As the largest endocrine organ, adipose tissue secretes many bioactive molecules that circulate in blood, collectively termed adipokines (1). These proteins include leptin, adiponectin, resistin, retinol-binding protein 4 (RBP4), omentin, and vaspin (2–7). Adipokines play important roles in energy homeostasis; their circulating levels are often dysregulated in conditions of obesity and/or diabetes (1). Adiponectin, the most highly expressed and intensely studied adipokine, has insulin-sensitizing, anti-inflammatory, and antiatherogenic properties (8). Allelic polymorphisms and reduced plasma adiponectin levels are tightly linked to insulin resistance and type 2 diabetes (8). A large body of evidence implicates adiponectin as a major insulin-sensitizing adipokine as well as an important biomarker and therapeutic target for obesity-associated metabolic diseases (1, 8). However, variable and relatively mild metabolic dysfunctions in adiponectin knock-out mice suggest the existence of additional metabolic regulators sharing overlapping function with adiponectin (9–12). Our efforts to identify such mediators produced by adipose tissue have led to the discovery of a highly conserved family of secreted proteins, designated as C1q/TNF-related proteins 1–10 (CTRP1 to -10) (13–15). Both CTRPs4 and adiponectin are part of the expanding C1q/TNF superfamily of proteins (16).
Most CTRPs are expressed by adipose tissue and circulate in plasma (14, 15). Their circulating levels vary according to sex, genetic background, and metabolic state of the animals (14, 15). All CTRPs form trimers as their basic structural unit; some are assembled into hexameric and high molecular weight oligomeric complexes that may have distinct biological and signaling properties (14, 15). Additionally, CTRPs form combinatorial associations, representing a potential mechanism to generate functionally distinct ligands with altered receptor specificity and/or function (14, 15, 17). Although the metabolic functions of adiponectin are well characterized, the physiological processes regulated by CTRPs are only beginning to be appreciated. Recombinant protein administration or adenovirus-mediated overexpression of some of these CTRPs reduces blood glucose in mice (14, 15). Apart from these studies, little is known about the biological significance and modes of action of CTRPs.
The present study aimed to characterize the metabolic function of CTRP3, a secreted protein expressed predominantly by adipose tissue in mice (14). Previous in vitro studies have shown that CTRP3 (also known as CORS26/cartducin) (18) stimulates chondrogenic precursor cell proliferation (19), promotes angiogenesis (20), and is overexpressed in the osteosarcoma cell line (21). In primary monocytes derived from healthy but not type 2 diabetic humans, recombinant human CTRP3 reduces proinflammatory cytokine IL-6 and TNF-α secretion in response to lipopolysaccharide stimulation, suggesting an anti-inflammatory role (22). Beyond these in vitro studies, the biological relevance and function of CTRP3 in vivo remain unknown.
Here we show that recombinant mouse CTRP3 administration lowered blood glucose in both normal and insulin-resistant ob/ob mice, an effect linked to activation of the Akt signaling pathway and a marked suppression of gluconeogenic gene expression in mouse liver. Consistent with its effect in mice, CTRP3 acts independently of insulin to reduce glucose output in rat H4IIE hepatocytes by suppressing the expression of glucose-6-phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK), two key enzymes involved in gluconeogenesis. Further, we identified two human CTRP3 isoforms that circulate in plasma. The two human proteins differ in size and glycosylation and form hetero-oligomers that appear to protect the longer isoform from proteolytic cleavage. Recombinant human CTRP3 also potently reduced gluconeogenesis in hepatocytes by suppressing the expression of gluconeogenic enzymes. This study represents the first functional characterization of CTRP3 in vivo and establishes the biological relevance of CTRP3 as a metabolic regulator of glucose homeostasis.
EXPERIMENTAL PROCEDURES
Animals
C57BL/6J and leptin-deficient obese (ob/ob) mice were purchased from the Jackson Laboratory and were allowed to acclimatize to the animal facility for at least 1 week. Laboratory mice consumed standard chow diet (no. 5001, Lab Diet, St. Louis, MO) and water ad libitum and were housed in polycarbonate cages on a 12-h light/dark photocycle. To model diet-induced obesity, 4-week-old C57BL/6 mice were placed on a high fat diet (60 kcal% fat; D12492) or an isocaloric low fat diet (10 kcal% fat; D12450B) purchased from Research Diets Inc. (New Brunswick, NJ). All experiments were approved by the Animal Care and Use Committee at Johns Hopkins University School of Medicine.
Mouse and Human Serum
Mouse serum samples were harvested by tail bleeding after overnight fast and separated using Microvette® CB 300 (Sarstedt). Serum samples were prepared according to the manufacturer's instructions for individual assay or diluted 1:20 in SDS loading buffer (50 mm Tris-HCl, pH 7.4, 2% (w/v) SDS, 6% (w/v) glycerol, 1% (v/v) 2-mercaptoethanol, and 0.01% (w/v) bromphenol blue) and subjected to Western blot analysis. Pooled normal human serum samples (Innovative Research) were diluted 1:20 in SDS loading buffer and subjected to Western blot analysis. An equivalent of 1 μl of serum was used to assess the presence of CTRP3A and CTRP3B.
Antibodies
Mouse monoclonal anti-FLAG M2 antibody was obtained from Sigma, and rat monoclonal anti-HA (clone 3F10) antibody was obtained from Roche Applied Science. Rabbit anti-CTRP3 antibody was generated as described previously (14). Goat polyclonal CTRP3/C1qTNF3 antibody was obtained from R&D Systems. Rabbit antibodies that recognize phospho-AKT (Thr-308), phospho-Erk1/2 (Thr-202/Tyr-204), phospho-AMPKα (Thr-172), phospho-GSK3β (Ser-9), total Akt, total AMPKα, and total Erk1/2 were obtained from Cell Signaling Technology.
Recombinant Protein
pCDNA3.1 expression constructs encoding C-terminal FLAG-tagged mouse CTRP3, human CTRP3A, or human CTRP3B were used in transient transfections to generate recombinant proteins. HEK 293 cells (Grip-TiteTM cells, Invitrogen) were cultured in DMEM containing 10% (v/v) bovine calf serum supplemented with 100 μg/ml penicillin and 100 μg/ml streptomycin. Transfections were performed in HEK 293 cells using Lipofectamine (Invitrogen) or calcium phosphate methods. At 24 h post-transfection, media were replaced with serum-free Opti-MEM media (Invitrogen) supplemented with vitamin C (0.1 mg/ml). Supernatants were collected three times, every 48 h, pooled, and purified using an anti-FLAG affinity gel according to the manufacturer's protocol (Sigma) and then eluted with 150 μg/ml FLAG peptide (Sigma). Purified proteins were dialyzed against 20 mm HEPES buffer (pH 8.0) containing 135 mm NaCl in a 10 kDa cut-off Slide-A-Lyzer dialysis cassette.
Recombinant Protein Injection
Food was removed in the morning 2 h prior to recombinant protein injection; drinking water was supplied for the duration of the experiment. Recombinant mouse CTRP3 (2 μg/g body weight) or the equivalent volume of vehicle buffer (20 mm HEPES (pH 8) and 135 mm NaCl) was injected intraperitoneally into 8–10-week-old C57BL/6 mice (n = 8). Blood glucose levels were measured at time 0 (prior to protein injection) and at the indicated time postinjection using the BD logicTM glucometer (BD Pharmingen). During the experiment, ∼100 μl of blood was collected from the tail tip for serum analysis. The experiment was repeated three times, using 6–8 new mice per experiment and different batches of recombinant protein; the glucose-lowering effects of CTRP3 were comparable across replicates. In addition, a third round of protein injection (2 μg/g body weight) was carried out with a different batch of mice and recombinant protein, and the livers were harvested and prepared for RNA and protein analysis 90 min postinjection. Furthermore, separate cohorts of mice were injected with different batches of recombinant CTRP3 at 1 μg/g (n = 6) and 4 μg/g (n = 6) to evaluate the glucose-lowering effect of CTRP3 at different doses.
Cell Culture and Gluconeogenesis Assay
Experiments were performed on rat hepatoma cell line H4IIE (obtained from Fredric Wondisford's laboratory at Johns Hopkins University), maintained in Dulbecco's modified Eagle's medium containing 5% newborn calf serum (Gemini Bio-product). For experimentation, cells were grown to near confluence in 24-well tissue culture plates (BD Biosciences) and incubated overnight with 500 nm dexamethasone and 0.1 mm 8-CTP-cAMP (Dex-cAMP), as described by Gocorko et al. (23). Cells were then stimulated with vehicle control or recombinant protein (5 μg/ml) and harvested at the indicated times for immunoblot and RNA analysis. For gluconeogenesis assays, H4IIE hepatoma cells were incubated overnight with recombinant CTRP3 (5 μg/ml) before transferring to glucose production buffer (glucose-free Dulbecco's modified essential medium, pH 7.4, containing 20 mm sodium lactate (Sigma) and 2 mm sodium pyruvate without phenol red) for an additional 5 h. At the end of this incubation, 0.2 ml of medium was taken to measure the glucose concentration using the Amplex Red glucose assay kit (Invitrogen). Glucose production was normalized to total protein concentration as measured by the Bradford method to correct for potential variation in cell number.
Glucose Uptake Assay
Glucose uptake experiments were performed on mouse 3T3-L1 adipocytes and rat L6 myotubes (ATCC). 3T3-L1 fibroblasts were cultured and differentiated as described previously (15). Rat L6 cells were maintained in DMEM (1 g/liter glucose) containing 10% (v/v) fetal bovine serum (Gemini Bio-Product). Myotubes were obtained by allowing undifferentiated myocytes to reach confluence and then switching to media containing 2% (v/v) fetal bovine serum for 7 days, with medium changes every second day. Myotubes were cultured for 2 h in serum-free DMEM containing 0.2% bovine serum albumin with CTRP3 (5 μg/ml) or vehicle buffer. Insulin was added during the last 30 min of incubation. Myotubes were washed twice in prewarmed PBS and then placed in HEPES-buffered saline solution (25 mm HEPES, pH 7.4, 120 mm NaCl, 5 mm KCl, 1.25 mm MgSO4, 1.2 mm CaCl2, 1.3 KH2PO4, and 0.5% BSA) containing 0.5 μCi/ml 2-deoxy-d-[14C]glucose (MP Biomedicals) and incubated for 10 min. The reactions were terminated by washing twice in ice-cold PBS. Cells were incubated 5 min with 1% Triton X-100, and the lysates were transferred to scintillation vials containing Ecoscint scintillation fluid (National Diagnostics) and then allowed to sit at room temperature for 1 h before being counted with a Beckman Coulter counter (model LS6000SC).
Blood Chemistry Analysis
Blood glucose was measured using a glucometer (BD Pharmingen). Serum insulin, glucagon, and adiponectin concentrations were measured using the Multiplex assay kit (Millipore). Serum leptin concentrations were measured using a leptin ELISA kit (Millipore). Serum non-esterified fatty acid and triglycerides were measured using assay kits from Wako.
Expression Profiling of CTRP3A and CTRP3B in Human Tissues
A semiquantitative PCR approach was used to evaluate the tissue distribution of CTRP3A and CTRP3B transcripts in human multiple tissue cDNA panels (Clontech). For human CTRP3, the following primer pair was used: forward primer, 5′-GAATCATGCTTTGGAGGCAGCTC-3′; reverse primer, 5′-CAGCTACAAGTCTTCCCCAAAGTGG-3′. 35-cycle PCRs were carried out using the Pfx polymerase (Invitrogen). For control human GAPDH, the following primer pair was used: forward primer, 5′-TGAAGGTCGGAGTCAACGGATTTGGT-3′; reverse primer, 5′-CATGTGGGCCATGAGGTCCACCAC-3′.
Identification and Cloning of Human CTRP3A and CTRP3B
Two isoforms of human CTRP3 mRNA were observed in human tissues. The two PCR products corresponding to alternative splicing of the CTRP3 gene were separated in 1% agarose gel, extracted from the gel, and cloned into the pCR2.1 TOPO vector (Invitrogen). The entire cDNA insert corresponding to CTRP3A and CTRP3B were sequenced, and DNA sequences were deposited into the NCBI GenBankTM data base with accession numbers ABY86416 and ABY86417, respectively.
Site-directed Mutagenesis
A PCR-based method was used to carry out site-directed mutagenesis using a kit from Stratagene. pCDNA3.1 expression vector encoding CTRP3A or CTRP3B was used as a PCR template. To generate the hCTRP3B (N70A) mutant, the following primer pair was used: forward primer, 5′-AACTGGGACTGTGGATGCTAACACTTCTACAGACC-3′; reverse primer, 5′-GGTCTGTAGAAGTGTTAGCATCCACAGTCCCAGTT-3′. To generate CTRP3A (C39A,C42A,C43A) and CTRP3B (C112A,C115A,C116A) Cys mutants, the following primer pair was used: forward primer, 5′-CCGGAGGACTACCCCCAGACGCCAGTAAGGCTGCTCATGGAGACTACAGCTTTCG-3′; reverse primer, 5′-CGAAAGCTGTAGTCTCCATGAGCAGCCTTACTGGCGTCTGGGGGTAGTCCTCCGG-3′. All constructs were verified by DNA sequencing.
Quantitative Real-time-PCR Analysis
For quantitative real-time-PCR analysis, RNAs were isolated from tissues (e.g. liver) or cell lines (e.g. H4IIE hepatocytes) using TRIzol® (Invitrogen) and reverse transcribed using Superscript II RNase H-reverse transcriptase (Invitrogen) according to the manufacturer's protocol. PCR primers used in the quantitative real-time-PCR analysis were as follows: G6Pase forward, 5′-CGACTCGCTATCTCCAAGTGA-3′; G6Pase reverse, 5′-GTTGAACCAGTCTCCGACCA-3′; PEPCK forward, 5′-CTGCATAACGGTCTGGACTTC-3′; PEPCK reverse, 5′-CAGCAACTGCCCGTACTCC-3′; 18 S RNA forward, 5′-GCAATTATTCCCCATGAACG-3′; 18 S RNA reverse, 5′-GGCCTCACTAAACCATCCAA-3′. Analyses were performed on an Applied Biosystems Prism 7000 sequence detection system. Samples were analyzed in 25-μl reactions according to the standard protocol provided in the SYBR® Green PCR Master Mix (Applied Biosystems). All expression levels were normalized to the corresponding 18 S RNA levels.
Gel Filtration Chromatographic Analysis
Supernatants (500 μl) from transfected HEK 293 cells containing FLAG-tagged mouse CTRP3, human CTRP3A, or human CTRP3B were sequentially loaded onto an AKTA FPLC and fractionated through an HR 10/30 Superdex 200 column (GE Healthcare) in PBS. The collected fractions were subjected to Western blot analysis using the anti-FLAG or the anti-CTRP3 antibodies.
Co-immunoprecipitation
C-terminal FLAG or HA-tagged CTRP3A, CTRP3B, and their corresponding Cys mutants were transfected individually or together into HEK 293 cells as described above. An aliquot of the collected supernatants (250 μl) combined with 500 μl of immunoprecipitation buffer (150 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, and 1% Triton X-100) was subjected to immunoprecipitation using either 10 μl of EZviewTM anti-FLAG affinity gel (Sigma) or anti-HA affinity matrix (Roche Applied Science). Samples were rotated for 4 h at 4 °C, washed four times with IP buffer, and subjected to protein immunoblot analysis.
Protein Immunoblot Analysis
Mouse liver protein homogenates and H4IIE cell lysates were prepared in tissue protein extraction buffer (Thermo Scientific) supplemented with protease inhibitors (Roche Applied Science) and phosphatase inhibitors (Sigma). All samples were boiled for 4 min at 100 °C after the addition of an equal amount of 2× SDS loading buffer. The proteins were separated by electrophoresis (NuPAGE 10% or 4–12% BisTris gel, Invitrogen) according to the manufacturer's instructions. The gels were blotted onto nitrocellulose membranes (Bio-Rad) and stained with Ponceau S red (Sigma) to confirm equal loading and transferring of proteins to the membrane in each lane. Secondary antibodies were conjugated to horseradish peroxidase, and the signals were developed by chemiluminescence (GE Healthcare). The signals were imaged and quantified using FluorChem Q multiImage III (AlphaInnotech). Values were expressed as arbitrary units. The sizes of the proteins were verified by using the standard prestained SeeBlue® Plus 2 molecular weight markers (Invitrogen).
Statistical Analyses
Data from blood glucose measurements, after recombinant CTRP3 injection, were analyzed using a repeated measures analysis of variance followed by Tukey post hoc analysis. All remaining statistical analyses were performed using a one-way analysis of variance. Statistical analyses were performed using GraphPad Prism 5 statistical software. Statistical significance was accepted at p < 0.05. All data are given as mean ± S.E.
RESULTS
Inverse Relationship between Circulating CTRP3 and Leptin Levels
To address whether circulating levels of CTRP3 are regulated by the metabolic states of the animal, we fed normal C57BL/6 male mice with a high fat diet or an isocaloric-matched low fat diet for 9 or 12 weeks. Although no differences were observed in the circulating levels of CTRP3 when mice were fed a high or low fat diet for 9 weeks (Fig. 1A), we observed a significant decrease in the serum levels of CTRP3 in mice fed a high fat diet for 12 weeks (Fig. 1B). The diet-induced obese mice had much higher levels of circulating leptin, in proportion to their increased adiposity, compared with mice fed a low fat diet (Fig. 1C). Conversely, circulating levels of CTRP3 were elevated in 8- or 12-week-old leptin-deficient ob/ob mice (Fig. 1, D and E), consistent with increased CTRP3 mRNA levels found in adipose tissue of ob/ob mice (14). Moreover, older (12-week-old) ob/ob mice had higher serum CTRP3 levels compared with younger (8-week-old) mice. Further, serum CTRP3 levels were significantly elevated in mice after an overnight fast, in which leptin levels are known to be low (24), compared with ad libitum fed or fasted/refed mice (Fig. 1F). Together, these data indicate an inverse relationship between circulating CTRP3 and leptin levels. Using known amounts of purified recombinant mouse CTRP3 as our standard in immunoblot analysis, we determined the serum CTRP3 concentration to be ∼1 ± 0.3 μg/ml. Given that CTRP3 forms trimers and higher order multimers (14), the effective circulating concentration is ∼0.1–0.3 μg/ml or less. Precise measurement of CTRP3 concentration in serum requires the development of a CTRP3-specific ELISA assay, which is currently unavailable.
FIGURE 1.

Circulating levels of CTRP3. The CTRP3 immunoblot of sera from mice on either a high fat or low fat diet shows no change after 9 weeks (A) but significant reduction of CTRP3 after 12 weeks (B). Mice on a high fat diet had significantly elevated leptin levels (C). The immunoblot of sera from leptin-deficient obese (ob/ob) mice showed increased concentration of CTRP3 at 8 weeks (D) and 12 weeks (E) of age, compared with the lean phenotype. F, immunoblot of sera for circulating CTRP3 levels in fasted, refed, and ad libitum-fed mice. Each bar represents the mean ± S.E. (error bars) (n = 4–8). *, p < 0.05.
Recombinant Protein Administration Reduces Blood Glucose Levels
To address the potential metabolic function of CTRP3, we purified recombinant protein from the supernatant of HEK 293 mammalian cells overexpressing CTRP3 (Fig. 2A) and evaluated its in vivo effect when administered to mice. Intraperitoneal injection of recombinant CTRP3 (2 μg/g body weight) into normal C57BL/6 mice resulted in a 3-fold elevation of circulating CTRP3 levels within 2 h (Fig. 2B). The rise in serum CTRP3 levels led to a significant reduction in blood glucose levels compared with vehicle-injected animals (Fig. 2C), and this effect was sustained over a 7-h time period. Maximum reduction in blood glucose was observed 3–4 h after protein administration. Importantly, CTRP3 administration lowered blood glucose without altering insulin, glucagon, non-esterified free fatty acids (NEFA), leptin, or adiponectin levels in C57BL/6 mice (Fig. 2, E–I). To investigate whether CTRP3 has similar glucose-lowering effects in an insulin-resistant mouse model, we injected recombinant CTRP3 into ob/ob mice. Leptin-deficient obese (ob/ob) mice are hyperphagic and display hyperglycemia and pronounced hyperinsulinemia (2). As in normal C57BL/6 mice, injection of recombinant CTRP3 (2 μg/g body weight) into these mice led to a significant decrease in blood glucose levels 3 h postinjection (Fig. 2D). The reduction in blood glucose lasted for 2 h in ob/ob mice, compared with the more sustained effect seen in normal C57BL/6 mice (Fig. 2, C and D). Administration of different doses (1, 2, and 4 μg/g) and batches of recombinant CTRP3 in over 20 C57BL/6 mice demonstrated a consistent glucose-lowering effect compared with vehicle injection, with maximum glucose lowering achieved at 4 μg/g body weight (data not shown). The modest glucose-lowering effect (∼20%) of CTRP3 at 2 μg/g body weight is comparable with the reductions in blood glucose achieved by recombinant adiponectin injection into C57BL/6 mice (25). Importantly, a closely related protein family member, CTRP5, did not lower blood glucose levels when injected into C57BL/6 mice at 2 μg/g body weight, although the protein was produced and purified in an identical manner (14).
FIGURE 2.

CTRP3 administration decreases blood glucose in mice. Injection of mice with 2 μg/g body weight of purified recombinant CTRP3 (A) increased circulating CTRP3 levels 3-fold (B) and was sufficient to lower blood glucose in normal C57BL/6 (C) and obese and diabetic (ob/ob) mice (D) without altering serum insulin, glucagon, non-esterified fatty acids (NEFA), leptin, or adiponectin levels (E–I). Each bar represents the mean ± S.E. (error bars) (n = 8). *, p < 0.05.
CTRP3 Suppresses Gluconeogenic Gene Expression in Mouse Liver
Blood glucose levels are affected by the rate of glucose uptake in the peripheral tissues and by hepatic glucose output (26). In the postprandial state, insulin secreted from the pancreatic β-cells is largely responsible for glucose disposal in the muscle and adipose tissue. In the postadsorptive state, in which insulin levels are low, blood glucose levels are largely controlled by the rate of hepatic glucose output. All of the recombinant protein injection experiments (Fig. 2) were performed in the morning (around 10 a.m.) with food removed 2 h prior to protein administration. Under this condition, insulin levels are low, and thus the glucose-lowering effects we observed with CTRP3 injection are probably due to the suppression of hepatic glucose output. To address whether CTRP3 regulates hepatic gluconeogenesis, livers were harvested 90 min after administration with vehicle or 2 μg/g body weight of recombinant protein. Quantitative real-time PCR analysis revealed a profound suppression (∼80%) of two key gluconeogenic enzymes, G6Pase and PEPCK, in the liver of CTRP3-administered mice relative to vehicle-treated controls (Fig. 3A). The PKB/Akt signaling pathway plays a major role in controlling hepatic glucose output (27–29); thus, CTRP3 may activate this signaling pathway in the liver to regulate gluconeogenic enzyme expression. Indeed, recombinant CTRP3 induces phosphorylation and activation of the Akt and ERK1/2 signaling pathway in mouse liver (Fig. 3, B and C). These effects are specific; we observed no changes in AMPKα phosphorylation (Fig. 3, B and C).
FIGURE 3.

CTRP3 administration suppresses hepatic gluconeogenic gene expression. Injection of mice with 2 μg/g body weight of recombinant CTRP3 suppressed the expression of gluconeogenic genes PEPCK and G6Pase in mouse liver, as assessed by quantitative real-time PCR analysis (A). Administration of recombinant CTRP3 to mice increased Akt and Erk1/2 phosphorylation in liver with no change in AMPKα phosphorylation (B and C). Each bar represents the mean ± S.E. (error bars) (n = 5). *, p < 0.05.
CTRP3 Acts Directly and Independently of Insulin to Regulate Glucose Metabolism in Hepatocytes
Rat hepatoma cells, H4IIE, are a well established hepatocyte cell line used in studying the transcriptional control of gluconeogenesis (30, 31). Using H4IIE cells, we established a direct effect of CTRP3 in regulating glucose production. Treatment of H4IIE hepatoma cells with recombinant CTRP3 significantly attenuated gluconeogenesis (Fig. 4A). Further, CTRP3 acts independently of insulin to suppress de novo glucose production (Fig. 4B); increasing the concentration of insulin did not alter the magnitude of CTRP3 in suppressing gluconeogenesis. Reduction of glucose output in H4IIE hepatoma cells by CTRP3 treatment was due to suppressed expression of gluconeogenic enzymes G6Pase and PEPCK (Fig. 4C). As with its effects on mouse liver, CTRP3 induced the phosphorylation and activation of Akt in hepatoma cells (Fig. 4D). Further, GSK3-β, a downstream target of Akt, was also phosphorylated following CTRP3 treatment (Fig. 4E). In contrast, we observed no changes in the phosphorylation of AMPK (Fig. 4F). Besides liver, muscle and adipose tissue play important roles in controlling whole-body glucose metabolism (32, 33). To investigate whether CTRP3 effects are specific to liver cells, we treated mouse 3T3-L1 adipocytes and rat L6 myotubes, representing the adipocyte and muscle cells, respectively, with recombinant CTRP3 (34, 35). With or without insulin, CTRP3 treatment did not affect the Akt signaling pathway (data not shown) or glucose uptake in these cells (Fig. 4, G and H).
FIGURE 4.

CTRP3 acts directly on liver cells, independent of insulin, to suppress gluconeogenic gene expression and glucose production. Gluconeogenesis was reduced in rat H4IIE hepatoma cells treated with recombinant CTRP3 (n = 6) for 16 h (A); this effect was independent of insulin concentration (n = 6) (B). Recombinant CTRP3 suppressed the expression of gluconeogenic genes PEPCK and G6Pase in hepatoma cells (n = 6) (C). Additionally, CTRP3 treatment increased Akt (D) and GSK3-β (E) phosphorylation but not AMPKα (F) in hepatoma cells. Treatment with recombinant CTRP3 in the absence or presence of insulin (10 nm) resulted in no change in glucose uptake in 3T3-L1 adipocytes (G) and rat L6 myotubes (H). In all experiments, cells were treated with 5 μg/ml recombinant CTRP3. All in vitro signaling experiments have been repeated at least twice, and comparable results were obtained. Each bar represents the mean ± S.E. (error bars). *, p < 0.05.
Humans Express Two CTRP3 Isoforms
In mice, CTRP3 is predominantly expressed by adipose tissue (14). In humans, CTRP3 is more broadly expressed, with transcript detectable in pancreas, small intestine, colon, and adipose tissue (Fig. 5A). Brain, kidney, thymus, and ovary also express low levels of CTRP3, but none of the resting or activated immune cells express detectable levels of CTRP3 mRNA. Although CTRP3 is overexpressed in some cancer cells (21), we did not detect CTRP3 expression in human cancer xenografts (Fig. 5A). Two specific PCR products were amplified from human tissues (Fig. 5A, arrows). Cloning, sequencing, and genomic mapping revealed that the two PCR products derived from alternative splicing of the CTRP3 gene (Fig. 5B). The first product was designated as CTRP3A, and the longer splice variant was designated as CTRP3B. CTRP3B has a larger exon 1 due to the retention of 219 nucleotides from the intron 1 sequence. The deduced CTRP3B cDNA encodes a protein with an extra 73 amino acids in the N terminus, which contains a highly conserved N-linked glycosylation site (Fig. 5B, circled, and supplemental Fig. S2). Human CTRP3A protein sequence is highly conserved throughout vertebrate evolution, with amino acid identity ranging from 78 to 99% (supplemental Fig. S1). The extended N terminus of CTRP3B is also highly conserved, with amino acid identity ranging from 38 to 100% between different vertebrate species (supplemental Fig. S2). Using the anti-CTRP3 antibody, both CTRP3A and CTRP3B can be detected in pooled normal human sera (Fig. 5C). The apparent molecular weights of these two proteins on SDS-PAGE immunoblot are consistent with their predicted molecular mass. Unlike human CTRP3, CTRP3A is the predominant circulating form found in mouse serum (Fig. 5C).
FIGURE 5.

Identification of a novel splice variant of CTRP3 in human tissues. A semiquantitative PCR analysis was carried out to evaluate the expression of CTRP3 and control GAPDH in human tissues (A). Two splice variants were identified and designated as CTRP3A and CTRP3B. B, organization of the CTRP3A and CTRP3B genes and proteins. The human CTRP3A gene is 25.3 kb in size, consists of six exons and five introns, and is located on chromosome 5p13. Exons 1, 2, 3, 4, 5, and 6 of the CTRP3A gene are 171, 112, 155, 130, 100, and 2,885 bp in size, respectively. The size of each intron is also indicated. Exon 1B (gray square) contains a 219-nucleotide sequence found in CTRP3B cDNA, coding for an extra 73 amino acid residues. A potential N-linked glycosylation site is circled. The consensus splice donors are shown in italic type. C, pooled human and mouse sera were subjected to immunoblot analysis using the anti-CTRP3 antibody.
Human CTRP3 Isoforms Differ in Size and Glycosylation
Both CTRP3A and CTRP3B were robustly detected in the conditioned media of transfected HEK 293 cells (Fig. 6A). The presence of an extended N terminus in CTRP3B is reflected in its greater apparent molecular mass on immunoblot (∼40 kDa), as compared with CTRP3A (∼32 kDa), allowing unambiguous separation and identification of the two proteins. Two migrating bands were observed for CTRP3B, suggesting the presence of carbohydrate moieties. Indeed, N-glycanase (peptide:N-glycosidase F (PNGaseF)) treatment shifted the apparent molecular weight of CTRP3B on immunoblot, confirming the presence of N-linked glycans (Fig. 6A). The predicted site of glycosylation is the highly conserved Asn-70, located in the extended N terminus of CTRP3B (Fig. 5B and supplemental Fig. S2). When mutated to Ala, the N70A mutant migrated as a single band on immunoblot, in contrast to the two bands observed for WT protein, confirming Asn-70 as the attachment site for glycans (Fig. 6B).
FIGURE 6.

Human CTRP3A forms hetero-oligomers with CTRP3B, protecting CTRP3B from proteolytic cleavage. A, immunoblot analysis was carried out on conditioned media containing FLAG-tagged CTRP3A or CTRP3B incubated with (+) or without (−) peptide:N-glycosidase F (PNGaseF) to determine the presence of N-linked glycans. B, conditioned media containing FLAG-tagged wild-type (WT) and mutant (N70A) CTRP3B were subjected to immunoblot analysis. C, supernatant and cell lysate containing epitope-tagged CTRP3A and/or CTRP3B were subjected to immunoblot analysis. D, secreted epitope-tagged CTRP3A and/or CTRP3B were immunoprecipitated (IP) with the anti-FLAG affinity gel and immunoblotted (IB) with the anti-HA antibody. E, size exclusion chromatographic (FPLC) analysis of secreted epitope-tagged CTRP3A and/or CTRP3B. Fractions 10–27 were subjected to immunoblot analysis (panels 1–4). Additionally, fractions 10–27 were immunoprecipitated with the anti-FLAG affinity gel and immunoblotted with the anti-HA antibody (panel 5). The indicated molecular weight markers (669, 440, 232, and 158 kDa) correspond to the peak elution fraction of molecular standard thyroglobulin, ferritin, catalase, and aldolase, respectively. FPLC fractions that correspond to adiponectin trimers, hexamers, and high molecular weight oligomers are indicated. F, co-expressed proteins (lane 4) or a mixture of separately expressed protein (lane 3) were immunoprecipitated with the anti-FLAG affinity gel and immunoblotted with the anti-HA antibody (top). Replicate blots were probed for the presence of epitope-tagged input proteins. The cysteine mutant of CTRP3A and/or CTRP3B (lanes 5–7) were immunoprecipitated with the anti-FLAG affinity gel and immunoblotted with the anti-HA antibody (top). Replicate blots were probed for the presence of epitope-tagged input proteins.
Physical Association with CTRP3A Protects CTRP3B from Proteolytic Cleavage
When expressed alone in HEK 293 cells, FLAG- or HA-tagged human CTRP3B was subjected to proteolytic cleavage, as revealed by multiple migrating bands on immunoblot with apparent molecular weights smaller than that of the full-length protein (Fig. 6C, lanes 3 and 10). In contrast, no proteolytic cleavage was observed for CTRP3A when expressed in HEK 293 cells. However, when co-expressed, CTRP3A appears to protect CTRP3B from proteolytic cleavage (Fig. 6C, lanes 6 and 11), suggesting physical association between CTRP3A and CTRP3B. Indeed, secreted epitope-tagged CTRP3A can be reciprocally co-immunoprecipitated with CTRP3B from cells expressing both proteins (Fig. 6D, lanes 5 and 6). This physical association is specific; other related proteins, such as adiponectin and CTRP9, did not co-immunoprecipitate with CTRP3 (Fig. 6D, lane 7 and 8). As with mouse CTRP3 (14), human CTRP3A and CTRP3B form higher order structures corresponding to the presumed trimeric, hexameric, and high molecular weight oligomeric forms (Fig. 6E, panels 1 and 2). Using gel filtration followed by immunoprecipitation, CTRP3B co-immunoprecipitated with CTRP3A in most FPLC fractions, confirming the formation of heterotrimeric and hetero-oligomeric complexes of CTRP3A and CTRP3B (Fig. 6E, panel 5).
CTRP3A-CTRP3B Complexes Form during Biosynthesis and Do Not Require N-terminal Cys Residues
Human tissues that express CTRP3A mRNA invariably express CTRP3B. The co-regulated expression of both transcripts in the same tissue suggests that CTRP3A-CTRP3B complexes formed during biosynthesis. Indeed, CTRP3A interacted with CTRP3B only when they were co-expressed in the same cell (Fig. 6F, lane 4); mixing supernatants containing CTRP3A and CTRP3B expressed separately did not result in co-immunoprecipitation (Fig. 6F, lane 3). Three Cys residues are located in the N termini of CTRP3A (Cys-39, -42, and -43) and CTRP3B (Cys-112, -115, and -116); these Cys residues can potentially participate in interdisulfide bond formation to covalently link CTRP3A to CTRP3B. Mutations of all three N-terminal Cys residues did not disrupt physical complex formation; secreted Cys mutant of CTRP3A can still co-immunoprecipitate with the Cys mutant of CTRP3B (Fig. 6F, lane 7), indicating that disulfide bond formation between CTRP3A and CTRP3B is not required for physical association.
Human CTRP3 Suppresses Gluconeogenesis in Hepatocytes
Despite 95% amino identity between full-length human and mouse CTRP3, we investigated whether human CTRP3 retains a similar biological function. As with mouse CTRP3, treatment of H4IIE hepatoma cells with purified recombinant human CTRP3A or CTRP3B significantly attenuated gluconeogenesis in these cells (Fig. 7A). The reduction in de novo glucose production in hepatoma cells is also due to the suppression of gluconeogenic enzymes G6Pase and PEPCK expression (Fig. 7B).
FIGURE 7.
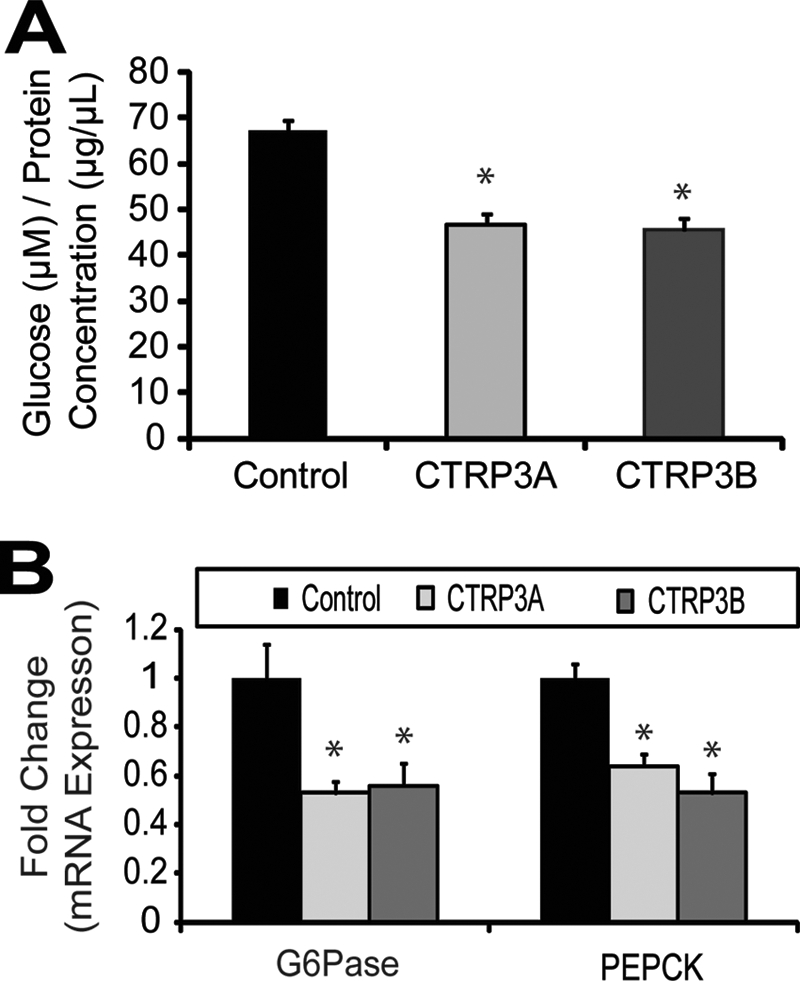
Human CTRP3A or CTRP3B suppresses gluconeogenesis in vitro. Glucose production was reduced in rat H4IIE hepatoma cells treated with 5 μg/ml recombinant human CTRP3A or CTRP3B for 16 h (A). Human CTRP3 treatment also suppressed the expression of gluconeogenic genes PEPCK and G6Pase (B). Each bar represents the mean ± S.E. (error bars) (n = 5). *, p < 0.05.
DISCUSSION
A 2-fold increase in plasma levels of adiponectin is sufficient to ameliorate many metabolic abnormalities associated with insulin resistance and/or diabetes in various animal models (25, 36–39). Despite its antidiabetic, anti-inflammatory, and antiatherogenic properties, adiponectin knock-out mice show surprisingly mild phenotypes, exhibiting varying degrees of insulin resistance when challenged with a high fat diet (9, 11, 12). These findings suggest the existence of secreted factors or mechanisms that can compensate for the absence of adiponectin. Indeed, increased leptin sensitivity in adiponectin knock-out mice partially accounts for the mild metabolic phenotypes (40). Additionally, the highly conserved family of adiponectin paralogs we recently identified may have overlapping functions, preventing a more severe phenotype in adiponectin knock-out mice (13–15, 41). However, limited data exist concerning the biological functions of CTRP proteins in the context of metabolism. Here we provide functional evidence establishing CTRP3 as a novel adipokine that regulates hepatic glucose metabolism.
A modest 3-fold increase in plasma CTRP3 levels by recombinant protein administration is sufficient to activate the Akt signaling pathway in liver, suppress hepatic gluconeogenic enzyme expression, and lower blood glucose levels in both normal and insulin-resistant ob/ob mice, without altering insulin, glucagon, or adiponectin levels. Glucose lowering appears to be a direct effect of CTRP3; in cultured rat hepatoma cells, we demonstrate that recombinant CTRP3 acts directly and independently of insulin to reduce glucose output by suppressing the expression of gluconeogenic genes G6Pase and PEPCK. Importantly, rat hepatocytes treated with a saturating dose of insulin continue to respond to CTRP3 treatment with a further decrease in gluconeogenesis, indicating that the biological pathway activated by CTRP3 to suppress glucose output is parallel to the insulin signaling pathway. This property of CTRP3 may be exploited in a therapeutic setting to treat diabetes, a metabolic condition characterized by severe hyperinsulinemia and profound insulin resistance.
Although adiponectin and CTRP3 lower blood glucose levels when injected into mice, major differences were noted between these proteins. First, although both proteins circulate in blood, the plasma concentration of adiponectin (10–30 μg/ml) is at least 10-fold higher than that of CTRP3 (precise measurement of CTRP3 concentrations in the blood requires the development of sensitive CTRP3-specific ELISA).
Second, circulating adiponectin levels are consistently decreased in obese and/or diabetic humans and rodents (1, 8), partly accounting for the decrease in systemic insulin sensitivity. In contrast, an inverse relationship appears to exist between circulating leptin and CTRP3 levels. During fasting, when leptin levels are low, serum CTRP3 levels increase. In diet-induced obese mice, in which serum leptin levels are high, CTRP3 levels decrease. Conversely, serum CTRP3 levels are elevated in leptin-deficient ob/ob mice. The mechanisms (direct or indirect) by which leptin affects circulating levels of CTRP3 remain unknown.
Third, adiponectin is an insulin sensitizer that potentiates insulin action in liver (25, 36). In the absence of insulin, recombinant adiponectin has little effect in suppressing gluconeogenesis in primary rat hepatocytes (25). In contrast, CTRP3 acts independently of insulin to suppress gluconeogenic gene expression and glucose output. In the absence of insulin, CTRP3 can still activate the Akt signaling pathway, suppress the expression of gluconeogenic enzymes G6Pase and PEPCK, and decrease glucose production in rat hepatoma cells.
Fourth, adiponectin exerts its whole-body metabolic effects mainly by modulating the AMPK signaling pathway in muscle, liver, and the hypothalamus (36, 42, 43). These effects are attenuated by the use of a dominant negative AMPK (36) or a specific AMPK inhibitor (44). In contrast, CTRP3 activates the Akt signaling pathway in mouse liver and cultured hepatocytes without any significant effect on the AMPK pathway. It is known that Akt activation in liver potently suppresses hepatic gluconeogenesis (27–29) by phosphorylating the transcription factor Foxo1. Phosphorylated Foxo1 is thus excluded from the nucleus, preventing the transcription of gluconeogenic genes (45).
Fifth, CTRP3 activates the Erk1/2 signaling pathway in the mouse liver; in contrast, adiponectin in general has little effect on the same pathway. The biological significance of Erk1/2 activation by CTRP3 in the liver is unknown. However, Erk1/2 has been shown to regulate transcription factors, such as Foxo1 (46), which may link Erk1/2 signaling either directly or indirectly with the transcriptional suppression of hepatic glucose output.
Sixth, adiponectin potentiates the action of insulin to suppress glucose output in liver (25) and promotes glucose uptake and fatty acid oxidation in muscle (36, 42, 47). Although CTRP3 suppresses gluconeogenesis in vitro and in vivo, it has no significant effect on the Akt signaling pathway and glucose uptake in 3T3-L1 adipocytes or L6 myotubes. The potential metabolic effect of CTRP3 on mouse muscle remains to be determined. Although CTRP3 and adiponectin belong to the C1q/TNF superfamily (16, 48) and share a modest 31% amino acid identity at the conserved C-terminal globular domain (13), our studies indicate clearly that adiponectin and CTRP3 are functionally distinct adipokines.
Circulating human CTRP3 exists in two isoforms, encoded by alternatively spliced transcripts. Expression of the splice variants is co-regulated; human tissues that express CTRP3A invariably express CTRP3B, and vice versa. It appears that the highly conserved CTRP3B isoform was first acquired during tetrapod evolution; teleosts, such as zebrafish and fugu, do not have the CTRP3B-specific exon 1B in their sequenced genome.
The longer isoform, CTRP3B, has an extended N terminus with 73 extra amino acids. These extra sequences confer two novel biochemical properties to CTRP3B. First, there is a highly conserved Asn residue in the N-terminal extension that is glycosylated; mutation of Asn-70 to Ala abolishes N-glycosylation. In contrast, CTRP3A contains no N-linked glycans. It is well known that secreted proteins are often glycosylated; this post-translational modification tends to enhance the half-life of the protein in vivo or serves as a carbohydrate epitope recognized by the cell surface receptor. Glycosylated human CTRP3B appears not to have a longer half-life in vivo; in fact, we failed to detect epitope-tagged recombinant CTRP3B in the blood when injected intraperitoneally into mice (data not shown), suggesting that the recombinant protein either gets rapidly cleared from the circulatory system or alternatively, binds strongly to cell surface receptor(s) lining the blood vessels. In contrast, an equivalent amount of recombinant CTRP3A injected into mice can be detected in the blood 10 min postinjection and remains in the circulatory system for up to 6 h (data not shown). Second, CTRP3B, but not CTRP3A, is susceptible to proteolytic cleavage when expressed in vitro. Cleavage occurs inside the cell prior to protein secretion; the cleaved CTRP3B can be detected in both the cell lysate and supernatant of HEK 293 cells expressing the protein. Cleavage is not induced by the presence of either FLAG or HA epitope; similarly tagged CTRP3A is not cleaved.
When co-expressed, CTRP3A forms hetero-oligomers with CTRP3B; this physical association limits proteolytic cleavage of CTRP3B. This may be a reason both transcripts are co-expressed in human tissues. However, the in vivo biological significance and relevance of these in vitro observations remain to be elucidated. Despite differences in size, glycosylation, and susceptibility to cleavage, both recombinant CTRP3A and CTRP3B reduce glucose output in hepatocytes by suppressing the expression of gluconeogenic genes G6Pase and PEPCK.
In summary, we have provided the first line of in vitro and in vivo evidence to establish CTRP3 as a novel adipokine that regulates hepatic glucose metabolism. Future gain- and loss-of-function studies in different metabolic contexts will further clarify CTRP3 roles and its mechanisms in controlling whole-body energy balance.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grant DK084171 (to G. W. W.). This work was also supported in part by American Heart Association Grant SDG2260721 and Baltimore Diabetes Research and Training Center Grant P60DK079637.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- CTRP
- C1q/TNF-related protein
- G6Pase
- glucose-6-phosphatase
- PEPCK
- phosphoenolpyruvate carboxykinase
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- AMPK
- AMP-activated protein kinase.
REFERENCES
- 1.Scherer P. E. (2006) Diabetes 55, 1537–1545 [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y., Proenca R., Maffei M., Barone M., Leopold L., Friedman J. M. (1994) Nature 372, 425–432 [DOI] [PubMed] [Google Scholar]
- 3.Scherer P. E., Williams S., Fogliano M., Baldini G., Lodish H. F. (1995) J. Biol. Chem. 270, 26746–26749 [DOI] [PubMed] [Google Scholar]
- 4.Steppan C. M., Bailey S. T., Bhat S., Brown E. J., Banerjee R. R., Wright C. M., Patel H. R., Ahima R. S., Lazar M. A. (2001) Nature 409, 307–312 [DOI] [PubMed] [Google Scholar]
- 5.Yang Q., Graham T. E., Mody N., Preitner F., Peroni O. D., Zabolotny J. M., Kotani K., Quadro L., Kahn B. B. (2005) Nature 436, 356–362 [DOI] [PubMed] [Google Scholar]
- 6.Yang R. Z., Lee M. J., Hu H., Pray J., Wu H. B., Hansen B. C., Shuldiner A. R., Fried S. K., McLenithan J. C., Gong D. W. (2006) Am. J. Physiol. Endocrinol. Metab. 290, E1253–E1261 [DOI] [PubMed] [Google Scholar]
- 7.Hida K., Wada J., Eguchi J., Zhang H., Baba M., Seida A., Hashimoto I., Okada T., Yasuhara A., Nakatsuka A., Shikata K., Hourai S., Futami J., Watanabe E., Matsuki Y., Hiramatsu R., Akagi S., Makino H., Kanwar Y. S. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 10610–10615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadowaki T., Yamauchi T., Kubota N., Hara K., Ueki K., Tobe K. (2006) J. Clin. Invest. 116, 1784–1792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kubota N., Terauchi Y., Yamauchi T., Kubota T., Moroi M., Matsui J., Eto K., Yamashita T., Kamon J., Satoh H., Yano W., Froguel P., Nagai R., Kimura S., Kadowaki T., Noda T. (2002) J. Biol. Chem. 277, 25863–25866 [DOI] [PubMed] [Google Scholar]
- 10.Ma K., Cabrero A., Saha P. K., Kojima H., Li L., Chang B. H., Paul A., Chan L. (2002) J. Biol. Chem. 277, 34658–34661 [DOI] [PubMed] [Google Scholar]
- 11.Maeda N., Shimomura I., Kishida K., Nishizawa H., Matsuda M., Nagaretani H., Furuyama N., Kondo H., Takahashi M., Arita Y., Komuro R., Ouchi N., Kihara S., Tochino Y., Okutomi K., Horie M., Takeda S., Aoyama T., Funahashi T., Matsuzawa Y. (2002) Nat Med 8, 731–737 [DOI] [PubMed] [Google Scholar]
- 12.Nawrocki A. R., Rajala M. W., Tomas E., Pajvani U. B., Saha A. K., Trumbauer M. E., Pang Z., Chen A. S., Ruderman N. B., Chen H., Rossetti L., Scherer P. E. (2006) J. Biol. Chem. 281, 2654–2660 [DOI] [PubMed] [Google Scholar]
- 13.Wong G. W., Wang J., Hug C., Tsao T. S., Lodish H. F. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 10302–10307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong G. W., Krawczyk S. A., Kitidis-Mitrokostas C., Revett T., Gimeno R., Lodish H. F. (2008) Biochem. J. 416, 161–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong G. W., Krawczyk S. A., Kitidis-Mitrokostas C., Ge G., Spooner E., Hug C., Gimeno R., Lodish H. F. (2009) FASEB J. 23, 241–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kishore U., Gaboriaud C., Waters P., Shrive A. K., Greenhough T. J., Reid K. B., Sim R. B., Arlaud G. J. (2004) Trends Immunol. 25, 551–561 [DOI] [PubMed] [Google Scholar]
- 17.Peterson J. M., Wei Z., Wong G. W. (2009) Biochem. Biophys. Res. Commun. 388, 360–365 [DOI] [PubMed] [Google Scholar]
- 18.Maeda T., Abe M., Kurisu K., Jikko A., Furukawa S. (2001) J. Biol. Chem. 276, 3628–3634 [DOI] [PubMed] [Google Scholar]
- 19.Akiyama H., Furukawa S., Wakisaka S., Maeda T. (2006) FEBS J. 273, 2257–2263 [DOI] [PubMed] [Google Scholar]
- 20.Akiyama H., Furukawa S., Wakisaka S., Maeda T. (2007) Mol. Cell Biochem. 304, 243–248 [DOI] [PubMed] [Google Scholar]
- 21.Akiyama H., Furukawa S., Wakisaka S., Maeda T. (2009) Oncol. Rep. 21, 1477–1481 [DOI] [PubMed] [Google Scholar]
- 22.Weigert J., Neumeier M., Schäffler A., Fleck M., Schölmerich J., Schütz C., Buechler C. (2005) FEBS Lett. 579, 5565–5570 [DOI] [PubMed] [Google Scholar]
- 23.Govorko D., Logendra S., Wang Y., Esposito D., Komarnytsky S., Ribnicky D., Poulev A., Wang Z., Cefalu W. T., Raskin I. (2007) Am. J. Physiol. Endocrinol. Metab. 293, E1503–E1510 [DOI] [PubMed] [Google Scholar]
- 24.Ahima R. S., Prabakaran D., Mantzoros C., Qu D., Lowell B., Maratos-Flier E., Flier J. S. (1996) Nature 382, 250–252 [DOI] [PubMed] [Google Scholar]
- 25.Berg A. H., Combs T. P., Du X., Brownlee M., Scherer P. E. (2001) Nat. Med. 7, 947–953 [DOI] [PubMed] [Google Scholar]
- 26.Cherrington A. D. (1999) Diabetes 48, 1198–1214 [DOI] [PubMed] [Google Scholar]
- 27.Liao J., Barthel A., Nakatani K., Roth R. A. (1998) J. Biol. Chem. 273, 27320–27324 [DOI] [PubMed] [Google Scholar]
- 28.Ono H., Shimano H., Katagiri H., Yahagi N., Sakoda H., Onishi Y., Anai M., Ogihara T., Fujishiro M., Viana A. Y., Fukushima Y., Abe M., Shojima N., Kikuchi M., Yamada N., Oka Y., Asano T. (2003) Diabetes 52, 2905–2913 [DOI] [PubMed] [Google Scholar]
- 29.Miyake K., Ogawa W., Matsumoto M., Nakamura T., Sakaue H., Kasuga M. (2002) J. Clin. Invest. 110, 1483–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmoll D., Walker K. S., Alessi D. R., Grempler R., Burchell A., Guo S., Walther R., Unterman T. G. (2000) J. Biol. Chem. 275, 36324–36333 [DOI] [PubMed] [Google Scholar]
- 31.Perrot V., Rechler M. M. (2003) J. Biol. Chem. 278, 26111–26119 [DOI] [PubMed] [Google Scholar]
- 32.Rosen E. D., Spiegelman B. M. (2006) Nature 444, 847–853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zierler K. (1999) Am. J. Physiol. 276, E409–E426 [DOI] [PubMed] [Google Scholar]
- 34.Green H., Kehinde O. (1975) Cell 5, 19–27 [DOI] [PubMed] [Google Scholar]
- 35.Klip A., Li G., Logan W. J. (1984) Am. J. Physiol. 247, E291–E296 [DOI] [PubMed] [Google Scholar]
- 36.Yamauchi T., Kamon J., Minokoshi Y., Ito Y., Waki H., Uchida S., Yamashita S., Noda M., Kita S., Ueki K., Eto K., Akanuma Y., Froguel P., Foufelle F., Ferre P., Carling D., Kimura S., Nagai R., Kahn B. B., Kadowaki T. (2002) Nat. Med. 8, 1288–1295 [DOI] [PubMed] [Google Scholar]
- 37.Yamauchi T., Kamon J., Waki H., Imai Y., Shimozawa N., Hioki K., Uchida S., Ito Y., Takakuwa K., Matsui J., Takata M., Eto K., Terauchi Y., Komeda K., Tsunoda M., Murakami K., Ohnishi Y., Naitoh T., Yamamura K., Ueyama Y., Froguel P., Kimura S., Nagai R., Kadowaki T. (2003) J. Biol. Chem. 278, 2461–2468 [DOI] [PubMed] [Google Scholar]
- 38.Yamauchi T., Kamon J., Waki H., Terauchi Y., Kubota N., Hara K., Mori Y., Ide T., Murakami K., Tsuboyama-Kasaoka N., Ezaki O., Akanuma Y., Gavrilova O., Vinson C., Reitman M. L., Kagechika H., Shudo K., Yoda M., Nakano Y., Tobe K., Nagai R., Kimura S., Tomita M., Froguel P., Kadowaki T. (2001) Nat. Med. 7, 941–946 [DOI] [PubMed] [Google Scholar]
- 39.Satoh H., Nguyen M. T., Trujillo M., Imamura T., Usui I., Scherer P. E., Olefsky J. M. (2005) Diabetes 54, 1304–1313 [DOI] [PubMed] [Google Scholar]
- 40.Yano W., Kubota N., Itoh S., Kubota T., Awazawa M., Moroi M., Sugi K., Takamoto I., Ogata H., Tokuyama K., Noda T., Terauchi Y., Ueki K., Kadowaki T. (2008) Endocr. J. 55, 515–522 [DOI] [PubMed] [Google Scholar]
- 41.Davis K. E., Scherer P. E. (2008) Biochem. J. 416, e7–e9 [DOI] [PubMed] [Google Scholar]
- 42.Tomas E., Tsao T. S., Saha A. K., Murrey H. E., Zhang Cc, C., Itani S. I., Lodish H. F., Ruderman N. B. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 16309–16313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kubota N., Yano W., Kubota T., Yamauchi T., Itoh S., Kumagai H., Kozono H., Takamoto I., Okamoto S., Shiuchi T., Suzuki R., Satoh H., Tsuchida A., Moroi M., Sugi K., Noda T., Ebinuma H., Ueta Y., Kondo T., Araki E., Ezaki O., Nagai R., Tobe K., Terauchi Y., Ueki K., Minokoshi Y., Kadowaki T. (2007) Cell Metab. 6, 55–68 [DOI] [PubMed] [Google Scholar]
- 44.Yoon M. J., Lee G. Y., Chung J. J., Ahn Y. H., Hong S. H., Kim J. B. (2006) Diabetes 55, 2562–2570 [DOI] [PubMed] [Google Scholar]
- 45.Gross D. N., van den Heuvel A. P., Birnbaum M. J. (2008) Oncogene 27, 2320–2336 [DOI] [PubMed] [Google Scholar]
- 46.Asada S., Daitoku H., Matsuzaki H., Saito T., Sudo T., Mukai H., Iwashita S., Kako K., Kishi T., Kasuya Y., Fukamizu A. (2007) Cell. Signal. 19, 519–527 [DOI] [PubMed] [Google Scholar]
- 47.Fruebis J., Tsao T. S., Javorschi S., Ebbets-Reed D., Erickson M. R., Yen F. T., Bihain B. E., Lodish H. F. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 2005–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shapiro L., Scherer P. E. (1998) Curr. Biol. 8, 335–338 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.