

Abstract
Heart failure is a chronic progressive disorder where frequent and recurrent hospitalizations are associated with high mortality and morbidity. The incidence and the prevalence of this disease will increase with the increase in the number of the aging population of the United States. Understanding the molecular pathology and pathophysiology of this disease will uncover novel targets and therapies that can restore the function or attenuate the damage of malfunctioning cardiomyocytes by gene therapy that becomes an interesting and a promising field for the treatment of heart failure as well as other diseases in the future. Of equal importance is developing vectors and delivery methods that can efficiently transduce the majority of the cardiomyocytes, that can offer a long term expression and that can escape the host immune response. Recombinant adeno-associated virus vectors have the potential to become a promising novel therapeutic vehicles for molecular medicine in the future.
1. Introduction
Cardiovascular disease is a major cause of death in both men and women in the United States, and is the major cause of death throughout the world. According to the center of disease and prevention (CDC), around 5.8 million people in the United States have heart failure and about 670,000 people are diagnosed with it each year. One in five people who have heart failure die within one year from diagnosis. Heart failure was a contributing cause of 282,754 deaths in 2006. In 2010, heart failure will cost the United States 39.2 billion dollars (1–2). The most common causes of heart failure are coronary artery disease, high blood pressure, and diabetes. The increase in heart failure incidence is partly due to the aging demographics of the United States population, as the rates of new and recurrent heart failure events increase substantially with age. Our better understanding of the molecular pathology in heart failure enable us to discover novel molecular therapeutic targets via gene therapy that will exceed the era of beta- blockers and ACE inhibitors, especially that the incidence of heart failure is the highest in the aging population who are not candidates for heart transplantation. The well-circumscribed geography of the heart makes it an attractive target that benefits from a wealth of clinical interventional experience. A variety of animal models of heart failure exist that reasonably reflect the human condition which enable us to examine potential gene-based therapies for heart failure before their transition into the clinical setting (3–5). Three elements are required for the clinical success of gene therapy. First, a vector or a packaging system is needed that can contain and deliver the genetic material of interest. Features of the vector determine the range of host cells that can be transduced, as well as the efficiency, level, and duration of transgene expression. Of note, only a few of the currently available vectors achieve efficient, high-level transgene expression in postmitotic cells, such as cardiomyocytes. These include recombinant adenoviruses (see below), adeno-associated viruses, and possibly lentivirus (6–8). Second, a delivery method is required to efficiently transduce the largest amount of cardiac cells. This is very important as in certain disease states, such as heart failure, we need to target large amount of cardiac cells in order to achieve a desirable outcome. Finally, an appropriate gene to be expressed in a particular clinical setting must be identified.
2. Mechanisms of Cardiac Contraction and Relaxation
2.a. Ultrastructure of the Contractile Cardiomyocyte
The major function of myocardial muscle cells is to execute the cardiac contraction-relaxation cycle. The contractile proteins of the heart lie within these myocytes. The sarcolemma of the myocyte invaginates to form an extensive tubular network, the T tubules, that extend the extracellular space into the interior of the cell. Immediately beneath the sarcolemma and in between the myofibrils, which are the contractile unit of the cell, are many mitochondria known as the subsarcolemmal and intermyofibrillar mitochondria (IMFM) respectively, the main function of which is to generate energy in the form of adenosine triphosphate (ATP) needed to maintain the heart contractile function and the associated ion gradients. The IMFM are found at very close proximity with the junctional sarcoplasmic reticulum and the T tubules and are connected to the sarcoplasmic reticulum (SR) by tethering complexes (9–10). Beside their role in energy production lately they are considered organelles of high calcium domain that play a role in calcium cycling in the cardiomyocyte. It is hypothesized that calcium flux in and out of the mitochondria and the intensity of such, plays critical role in the regulation of energy production (11–12). Of the other organelles, the sarcoplasmic reticulum (SR) is the most important that extends between the T tubules and the IMFM. It is the place where posttranslational modification of nascent proteins takes place and is considered the organelle of high calcium domain where calcium, bound to calsequestrin, is being stored and play a major and an essential role in the contraction and relaxation of the cardiomyocyte (13–17). The calcium release channels, the Ryanodine receptors (RYR), are found in the expanded parts of the SR that lie in very close apposition to the T tubules forming the subsarcolemmal cisternae or the junctional SR (JSR) and play a role in the release of calcium from the SR to the cytoplasm during contraction. The second part of the SR, the longitudinal or network SR consists of ramifying tubules and is involved with the uptake of calcium that initiates relaxation (18–21). This uptake is achieved by the ATP requiring pump known as the sarcoendoplasmic reticulum Ca ATPase (SERCA 2a), which increases its activity in response to beta-adrenergic stimulation. Calcium taken up into the SR is then stored at high concentration in a number of storage proteins, such as calsequestrin, calnexin and calreticulin, before being released again in response to the next depolarization wave figure 1.
Figure 1.
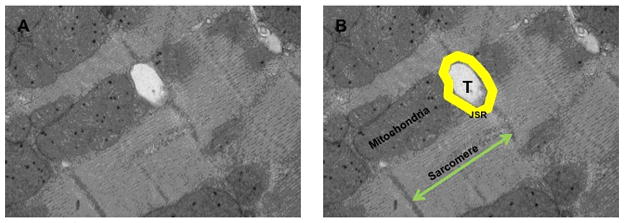
A. Electron microscopy image showing the ultrustructural architecture of the cardiomyocyte. B. is a schematic illustration of the T-tubules (T), junctional sarcoplasmic reticulum (JSR). mitochondria and sarcomere. Note mat these structures are found in close proximity and are considered organelles of high calcium domain in the cardiomyocyte.
2.b. Calcium Ion Fluxes in Cardiac Contraction-Relaxation Cycle
Calcium has a crucial role in regulating the contraction and relaxation phases of the cardiac cycle. The generally accepted hypothesis is based on the crucial role of calcium induced calcium release from the SR. Relatively small amounts of calcium ions, the trigger calcium ions, enter and leave the cardiomyocyte during each cardiac cycle, whereas much larger amounts move in and out of the SR. Each wave of depolarization traveling down the T tubules activates the L-type calcium channels (LTCC) that allow calcium entry into the cell. This calcium will trigger further release of calcium from the SR via the RYR receptors a phenomenon known as the calcium induced calcium release. The released calcium binds to troponin C, which with inactivation of troponin I via its phosphorylation and alteration of tropomyosin alignment, allow the interaction between actin and myosin leading to the contraction of the cardiomyocyte. The increase of cytosolic calcium that triggers contraction comes to an end as the wave of excitation passes. This turn-off of release is not as well understood as control of calcium release from the SR, but is important in preventing cytosolic calcium overload with serious consequences, such as arrhythmias and impaired contraction-relaxation cycles (22–24). It is speculated that the increase in cytosolic calcium ion concentration will activate the calcium uptake pump of the SR and hence will initiate the relaxation phase of the cardiac cycle. During relaxation phase, 25% of the cytosolic calcium ions are pumped into the extracellular space by sodium-calcium exchanger (NCX-1) while 75% of the calcium ions are taken up into the SR by the calcium pump SERCA 2a, which constitutes about 90% of the protein component of the SR. It straddles the SR in such a way that part of it protrudes into the cytosol. For each mole of ATP hydrolyzed by this enzyme two calcium ions are taken up and accumulate into the SR. The activity of SERCA 2a is inhibited by phospholamban, a protein that lies in close proximity to SERCA 2a. During diastole there is inhibition of phospholamban by its phosphorylation at two different sites; one is activated by PKA in response to beta-adrenergic stimulation and the other is activated by calcium ions and calmodulin, thus promoting and enhancing the activity of SERCA 2a (22–24). Of the other proteins that affect the function of SERCA 2a is the protein phosphatase 1 (PP1) and its inhibitor-1 (I-1). PP1 is a serine/threonine phosphatase that is localized to the SR and is inhibited by I-1 which becomes active upon phosphorylation of threonine-35 of phospholamban protein by PKA. This results in inhibition of PP1 and therefore enhanced PKA-mediated phosphorylation of phospholamban, leading to amplification of the β-adrenergic response in the heart. This phenomenon is of high importance as loading the SR with calcium will allow subsequent contraction in the next cycle and what is of more importance is that upon beta-adrenergic stimulation of the cardiomyocyte, the activity of SERCA 2a is enhanced as mentioned above allowing a faster relaxation, a higher calcium ion load into the SR and subsequently a more vigorous contraction in the subsequent cardiac cycle. Hence, beta-adrenergic stimulation of the cardiomyocyte has a lusitropic as well as an inotropic effect through enhancing the activity of SERCA 2a. Hence, SERCA 2a plays a critical, an essential and a central role by being the core of calcium cycling in the cardiomyocyte figure 2.
Figure 2. Calcium Ion Flux during the contraction-relaxation phase of the cardiac cycle.
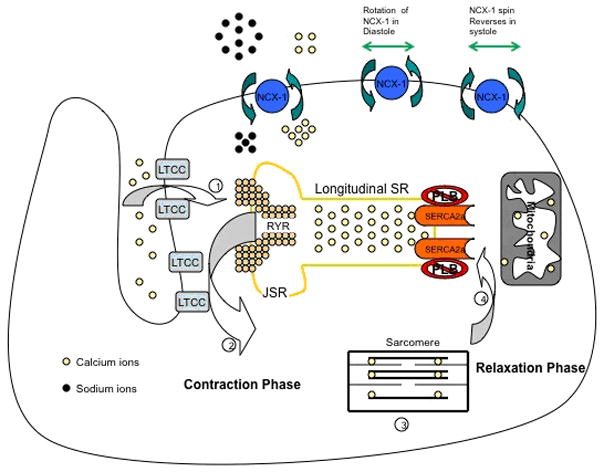
As the depolarization wave reaches the L-type calcium channels, calcium enters the cardiomyocyte triggering release of ca from the SR. Calcium binds to troponin C leading to contraction of the Cardiomyocyte. The Increase in intracellular ca activates SERCA 2a that pumps Ca into the SR so that relaxation takes place and the cell is ready for the next cardiac cycle.
3. Pathophysiology of Heart Failure
Heart failure is a clinical syndrome where the heart fails to meet the metabolic demand of the other organs. As a compensatory mechanism, there is activation of the neurohormonal and renin angiotensin aldosterone system (RAAS) followed by remodeling of the left ventricle and alterations in the left ventricle geometry(25). At the level of cardiomyocyte, there is alteration in the excitation contraction coupling, myosin heavy chain gene expression, beta-adrenergic desensitization, and hypertrophy. The cardiac myocyte death takes the form of necrotic, apoptotic and autophagic cell death. On the level of the extracellular matrix, there is enhanced matrix degradation and marked increase in interstitial fibrosis as well as perivascular fibrosis. In this review we will focus on changes in gene expression of the calcium handling proteins.
3.a. Changes of calcium handling proteins at the level of sarcoplasmic reticulum
As stated above SERCA 2a is the core of Ca cycling and plays a central role in the excitation-contraction coupling. The levels of SERCA 2a mRNA and protein expression are reduced in animal models as well as in myocardium samples obtained from patients with end stage heart failure. This reduction in the abundance and or functional activity in SERCA 2a could explain the slow decay of the calcium ion transients and the reduced sarcoplasmic reticulum calcium ion storage and release that has been observed in the failing cardiomyocytes. We have shown that SERCA 2a overexpression improves the contractile performance and restores cardiac function in the failing heart in rodent animal and large animal models of heart failure (26–31). Also overexpression of SERCA 2a improves myocardial performance in the senescent heart (32). At the same time there is decrease in phospholamban phosphorylation in the failing heart leading to further inhibition in SERCA 2a activity. The RYR receptors are hyperphosphorylated by protein kinase A. This hyperphosphorylation increases the open probability of the RYR receptors causing a persistent leak of calcium from the SR figure 3.
Figure 3.
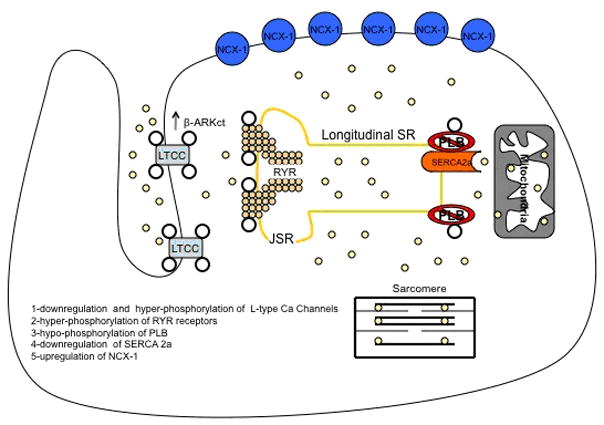
changes in calcium handling proteins in heart failure. There is (1) downregulation and hyperphosphorylation of LTCC by β-ARKct, (2) hyperphosphorylation of RYR receptors and increase calcium leak from the JSR into the cytoplasm. (3) hypo-Phosphorylation of PLB, (4) downregulation of SERCA2a and depletion of SR Ca stores. (5) upregulation of NCX-1 receptors.
3.b. Changes of calcium handling proteins at the level of the plasma membrane
The L-type calcium channel opens when the membrane depolarizes during the upstroke of the cardiac action potential. The subsequent calcium influx helps to produce the plateau phase of the action potential. Studies have shown that there is a reduction in the mRNA and protein levels and an increased level of phosphorylation of the L-type calcium channels in heart failure (33). Both of these abnormalities would reduce both the magnitude and homogeneity of the inward calcium current and thus adversely affect SR calcium release. The mRNA and protein levels of the Na-Ca exchanger have been reported to be increased in HF. This calcium entry via reverse-mode Na-Ca exchanger activity contributes to abnormal calcium handling in HF figure 3.
4. Viral Vectors for Gene Therapy
Gene delivery systems are classified into two main categories, the non-viral and the recombinant viral vectors (34–35). In this review we will focus on viral vectors. Briefly, non-viral vectors include plasmid DNA, liposome-DNA complexes and polymer-DNA complexes (36). The advantages of non-viral vectors include the ease of vector production, the reduced limitation on the expression cassette size and the relatively minimal biosafety risks. Their limitations include low transfection efficiency and transient effect due to their intracellular degradation. This makes them more suitable for transient, short lived expression of particular genes in certain disease states, such as angiopathies where transient expression of angiogenic factors by a modest number of gene modified cells is sufficient to obtain a desired phenotypic effect. On the other hand, certain cardiovascular disease states, such as heart failure require widespread and sustained transgene expression in order to achieve the desired effect. For these disease states viral vectors are the most suitable, as they offer a relatively high gene transfer efficiency and long term transgene expression. Their limitations include the limited packaging capacity, inconsistencies in bioactivity and purity between vector stocks, and biosafety risks. Moreover, the presence of neutralizing antibodies against viral vectors makes it more challenging and poses the demand to engineer or design a viral vector that can escape the immune response and at the same time that can transduce the largest number of cardiac cells. Viral vectors most commonly used in cardiovascular disease states are adenoviral vectors, adeno-associated virus vectors and lentivirus vectors.
4.a Adenoviral Vectors
Recombinant adenoviral vectors can transduce all major cardiac cell types both in vitro and in vivo. It is the most commonly used vector in preclinical gene therapy models and in clinical cardiovascular gene therapy protocols due to the ease of their production and the broad cell tropism, particularly within the cardiovascular system (37). However, in vivo, the pattern of myocardial transduction reflects the method of vector delivery. Direct myocardial injection results in intense transduction at the site of injection (38), whereas intracoronary delivery results in more widespread transduction in the distribution of the injected vessel (39). A number of maneuvers have been shown to enhance adenoviral myocardial transduction efficiency during intracoronary injection. Those maneuvers include physical approaches, such as increase in transcoronary pressure gradient (40) or the application of ultrasound energy to disperse circulating vector as it traverses the myocardium (41–42). Chemical approaches include the use of vasodilatory agents that facilitate vector passage from the vascular lumen to the myocardium (43–45). Translation of this vector system from research to clinical settings is going to be challenging for a number of reasons as the adenovirus vectors can evoke an intense immune and inflammatory reaction, beside the presence of neutralizing antibodies and the de novo development of these antibodies precludes readministration of the same vector serotype. Regarding their biosafety, a number of approaches have been pursued. These include the production of adenoviral vectors with reduced amounts of viral parental genes. The gutted adenoviral vectors are devoid of almost all of the viral parental genes. Another approach to reduce vector associated immunological responses is to modify vector tropism by engineering fiber coat proteins. This avoids transduction of unintended cells, such as antigen presenting cells, and increases gene transfer efficiency to target cells.
4.b. Recombinant Adeno-Associated Virus vector
Recombinant adeno-associated virus (rAAV) vectors belong to the family of the dependant parvovirus AAV type 2 (46–47), a naturally occuring, non-pathogenic, non- integrating and non-replicating virus that depends on a helper virus for replication (48–49). When wild type AAV infects a cell in the absence of a helper virus, the AAV persists in a latent form within the cell until superinfection with a helper virus results in productive replication of the AAV along with the helper. The wtAAV is a 4.7 kb single-stranded molecule comprised of a single open-reading frame (ORF) encoding the four replication (Rep) proteins and a single ORF for the three structural capsid (cap) proteins (50). These protein coding regions are flanked on either side by paired 145 nucleotide inverted terminal repeat (ITR) sequences. The ITRs are the critical elements directing replication, viral packaging and host cell integration of the DNA sequences that they flank. The majority of integration events occur in a specific site, called AAVS1, in the q arm of the human chromosome 19 (51–52). There are 12 different AAV serotypes, each one of them has a different tissue tropism (53). AAV1 has been shown to transduce cardiac and skeletal muscle efficiently. Recently, AAV8 and AAV9 have shown strong tropism toward cardiac tissue which makes them suitable for gene therapy in the cardiovascular system (54–57). rAAV were produced in the mid 1980’s by creating ITR deleted helper plasmids to supply rep and cap function and as a result the property of site-specific integration into chromosome 19q is lost in rAAV vectors, which lack the AAV Rep78 and Rep68 proteins (58). rAAV2 vector plasmids were constructed in which ITR sequences flanked the therapeutic gene of interest, along with an appropriate promoter and a polyadenylation signal. This vector has a number of clinically favorable attributes over the adenoviral vector. First, it lacks parental agent pathogenicity and vector related cytotoxicity and as a result does not induce a host immune response. rAAV vectors have been used to deliver genes to over 200 study subjects by various routes of administration for potential treatment of genetic disorders such as cystic fibrosis, haemophilia, hereditary emphysema due to α1-antitrypsin deficiency and other neurologic disorders such as Canavan disease, Batten disease, Parkinson and Alzheimer disease without any significant safety concerns (59–60). Recently intra-articular injection of rAAV2-TNFR:Fc in 15 subjects with inflammatory arthritis was well tolerated without any side effects and had a favourable outcome in reducing the degree of inflammation comparable to systemic administration of TNF-α inhibitors (61). Second, it has the capacity for stable long term gene expression through random genomic integration and or stable extra-chromosomal (episomal) sequences maintenance in non-dividing or slowly dividing transduced cells such as the striated muscles and the cardiac myocytes (46–47, 62–63). Large animal models of single-administration of rAAV2 injection into the muscle (64–65) and the liver have demonstrated persistent expression for more than 3 years. Expression in primate lung and canine retina have persisted 4 and 6 months, respectively. In one trial administration of rAAV2 expressing the transgene for factor IX intramuscularly, achieved long term expression for more than 4 years. However, when the same dose of the same vector was administered through the hepatic artery to hemophilia patients, only short term expression of factor IX was observed (66). Results from human trials suggest that repeated delivery might be required, which heightens the interest in developing alternative serotypes and understanding their potential for cross- neutralization. Studies evaluating onset, peak and persistence of expression with nonAAV2 serotypes in vivo are at an early stage (67). A pseudotype AAV2/1 vector has been designed to effectively transduce cardiac myocytes. AAV2/1 vector has the capsid of AAV1 vector that efficiently transduces cardiac myocytes, while its ITRs are derived from the AAV2 vector as they have been used in many previous clinical trials and their safety profile is established. The time from vector administration to peak of therapeutic protein levels with rAAV2 vectors in vivo is much longer than with most other viral or non-viral gene therapeutics. The onset of transgenic protein expression is often detectable within two weeks of treatment. The plateau of expression occurs 4–8 weeks post viral injection into the muscle (64–65). The delay of peak expression is likely due to the requirement for input single-stranded rAAV transgene conversion to a double-stranded molecule that can serve as a template for DNA transcription within the target cell (68–69). One strategy to overcome this is to design a double-stranded template when delivering a small gene (70). Major limitations of the rAAV vector systems include the production of high titer vector stocks of consistent purity and bioactivity, a limited packaging capacity of 4.8 kb and the potential for pre-existing neutralizing antibodies in human populations.
4.c Lentivirus Vectors
Lentivirus vectors transduce mitotically quiescent cells through genomic integration, particularly within the cardiovascular system. Unfortunately, they are less clinically interesting for the fear regarding the pothogenicity of the parental virus which is based on the human immunodeficiency virus type 1 (HIV-1) (71). In order to become suitable for clinical use, vector modifications are needed such as deletion of all accessory proteins from the packaging system, separation of packaging elements into multiple plasmids and the use of a chimeric 5’-long term repeat (LTR) and a self inactivating 3’-LTR in the vector plasmid (72–73). Modifications to the vector backbone to optimize performance include the incorporation of a central polypurine tract (cPPT) and a posttranscriptional regulatory element PRE) to enhance nuclear import and mRNA translation respectively (74). These vector modifications have the potential to improve in vivo transduction efficiency of the myocardium (75). The strengths of this system include the ability to confer long term stable transgene expression and an increased packaging capacity compared with rAAV. However the major limitation of this vector system relates to its high biosafety risk.
5. Vector Delivery System
As noted above various cardiovascular diseases require a transduction threshold beyond which a desirable effect is achieved after gene therapy. Developing delivery methods that can provide us with the highest transduction efficiency to the largest number of cardiomyocytes when targeting molecular pathways involved in the pathophysiology of heart failure become very important. A number of delivery methods exist, each one of which has different transduction efficiency and hence will be suitable for specific therapies. Also the mode of delivery differs between rodents or small animals and large animals in vivo. In small animals the mode of delivery includes direct intramyocardial injection, tail vein injection and a catheter based technique that we have developed (40). In this approach, a catheter is inserted in the left ventricular apex and is advanced beyond the aortic valve. A high concentration of adenoviral vector is infused through the catheter while the aorta and the pulmonary artery are clamped simultaneously distal to the tip of the catheter for a period of approximately 30 sec. This method achieves grossly homogeneous transduction of cardiac myocytes throughout the left and the right ventricles of the heart and hence can produce dramatic transgene specific physiological effects in vivo (40). Maurice et al (76) used this technique to express β2 receptors in rabbit hearts; however, they only clamped the aorta and achieved predominantly epicardial gene expression. By clamping the pulmonary artery and the aorta simultaneously, we reduced left ventricle preload and as a consequence the left ventricle end-diastolic pressure which allows perfusion of the virus at relatively low pressure and the endocardium can be efficiently transduced. On the other hand, tail vein delivery, offers a high transduction efficiency of cardiac myocytes in small animals by certain rAAV serotypes, such as rAAV8-9 serotypes (77). Tail vein injection is applied to alter the expression of different genes in animal models of pressure overload hypertrophy and heart failure, where a second survival surgery would not be feasible in such sick animals.
The mode of gene delivery in large animal models include catheter based myocardial delivery, pericardial delivery and surgical delivery. Percutaneous catheter based gene delivery to the myocardium in vivo can be achieved by the intracoronary route, by endocardial delivery or by retrograde infusion of the coronary veins. Intracoronary delivery using a slow infusion as compared to bolus infusion is superior and offers on average 50% transduction efficiency which is affected by a number of factors such as animal species, pharmacological agents used to permeabilize vasculature and vector related titer variability. In contrast, percutaneous endocardial delivery offers focal gene delivery to the myocardium and is suitable for therapies involving angiogenesis (78) and to a lesser extent to focal arrhythmia therapy (79). Retrograde infusion of vector via the coronary veins constitutes a novel catheter based technique for myocardial gene delivery and can result in gene transfer efficiencies comparable to intracoronary delivery (80). It has advantage over the intracoronary method as it bypasses diseased arteries that are likely to impede vector delivery, which makes it a clinically attractive mode of gene delivery. More recently, a percutaneous and clinically applicable catheter based gene delivery method was developed that allowed selective antegrade myocardial gene transfer with concomitant specific coronary vein blockade and a high gene expression in targeted myocardium (81).
Pericardial delivery offers a low transduction efficiency that is mainly distributed to the epicardium. The transduction efficiency can be improved by the co-administration of proteolytic enzymes that disrupts the pericardial cellular and extracellular barriers to the myocardium (82). Surgical delivery is the most invasive where direct injection of vector into the myocardium can be achieved via the transthoracic or subxiphisternal approaches. This approach is mostly applied in small animals rather than large animals (38). The transduction efficiency with this method is mainly restricted to the local myocardium.
A novel technique, the V-Focus system, is a minimally invasive procedure that enables a closed circuit to be percutaneously established between the coronary arteries and the coronary sinus, hence allowing efficient vector delivery by isolating the coronary circulation from the general circulation, with minimal damage to the heart (83). The advantage of this technique is that it allows high transduction efficiency to the myocardium with minimal systemic vector delivery. Only 10% of the perfused blood escapes the closed circuit through the thebesian veins. Adequate oxygenation of the myocardium is maintained in part by circulating the perfusate through an extracorporeal membrane oxygenation system (ECMO). Once the circuit is established, the vector is directly injected into the circuit and circulates through the myocardium for approximately 10 min (83). This technique has allowed a widespread transduction of SERCA 2a in an ovine model of heart failure with a substantial increase in contractility both with adenoviral and rAAV gene transfer.
Surgical gene transfer methods have also been tried. Animals have been placed on bypass and antegrade injection of vector has been performed with high efficiency of gene transfer (84). More recently, investigator used a complete surgical isolation of the heart in situ with retrograde (through the coronary venous circulation) administration of both vector and endothelial permeabilizing agents to increase myocyte transduction efficiency (85)
This technique involves clamping both vena cavae and all pulmonary veins. On cardiopulmonary bypass, the aorta and pulmonary artery are crossclamped, and the heart isolated. Viral solution is then infused retrograde into the coronary sinus and recirculated for a total of 30 minutes. Even though this novel cardiac surgical technique of cardiac isolation and retrograde delivery of vector through the coronary sinus results in efficient myocyte transduction in an adult large animal in vivo, its translation to patients with severe heart failure may be limited (85).
6. Targets for Gene Therapy
Our understanding of the molecular pathways involved in the pathophysiology of heart failure, enable us to target by gene therapy multiple pathways at different levels in the cardiomyocyte. Those molecular targets mainly exist at the level of the myofibrils, the endoplasmic reticulum and the mitochondria, as these organelles lie in very close proximity and are essential for the proper function of the cardiomyocyte. At the level of the plasma membrane, prolonged β-adrenergic stimulation leads to β-adrenergic receptor (βAR) downregulation, increase in βAR kinase (βARK) activity and increased inhibitory effect of G-protein α-subunit (GαI) function (86–91). Several gene-based approaches have been reported to successfully restore this signaling defect and as a result rescue the heart failure phenotype. Apoptotic and autophagic cell death are the main forms of cell death in heart failure that lead to loss of cardiomyocyte. Gene therapy plays a role in altering those forms of cell death and promotes cardiomyocyte survival. In this review we will focus on the role of gene therapy in restoring the homeostasis of the calcium handling proteins at the level of the endoplasmic reticulum and its anti-apoptotic effect figure 4.
Figure 4.
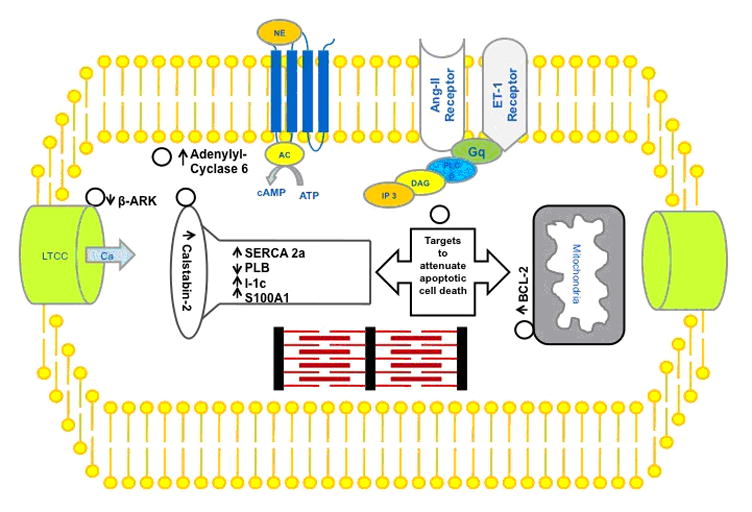
Potential gene therapy targets for the treatment of heart failure.
6.a The Calcium Handling Proteins
Several calcium handling protein defects have been described in the context of heart failure. There is decrease in SERCA2a protein level as well as decrease in phosphorylation of phospholamban (PLN), which promotes further SERCA2a inhibition. Restoring PLN to SERCA2a ratio has a favorable effect in improving cardiac function. Transgenic mice where overexpression of SERCA2a or PLN ablation has been reported to result in favorable changes in cardiac haemodynamics and to prevent dilated cardiomyopathy in cross-bred genetically predisposed mice respectively. Adenovirus gene mediated transfer of SERCA2a restored calcium transient morphology in cardiomyocytes isolated from failing human hearts (92). Transcoronary delivery of the same adenoviral vector in vivo in a pressure overload rat heart failure model restored SERCA 2a level, improved cardiac hemodynamics and increased animal survival (93). Over expression of SERCA 2a in heart failure restored the end systolic pressure-volume relationship, increased the stroke volume and decreased the end diastolic volume. The survival of those animals improved from 9% to 64% (93).
Beside overexpression of SERCA2a, restoring SERCA2a to PLN ratio can be achieved by ablation of PLN function via either antisense or a dominant negative approach. Adenovirus mediated ablation of PLN by antisense gene transfer restored calcium transients and contractility of cardiomyocytes isolated from failing human hearts. It has been reported that transcoronary delivery of recombinant adeno-associated virus encoding a dominant negative form of PLN has improved calcium handling and cardiac contractility and retarted the progression to heart failure (94–95). However, the discovery of a PLN dominant negative mutation as a cause of human dilated cardiomyopathy made this approach less interesting. Moreover, there are inconsistencies in the ability of PLN knockout mice to rescue heart failure phenotype in cross-bred genetic heart failure models.
Another critical target in the calcium handling proteins involves protein phosphatase inhibitor 1 (I–1). Two classes of serine-threonine phosphatases, type 1 and type 2, have been shown to regulate cardiac contractile performance. The type 1 phosphatase (PP1) is localized to the SR membranes and is regulated by two heat and acid stable proteins, inhibitor 1 and 2. Inhibitor 1 becomes active on phosphorylation of threonine 35 by PKA (96–97). This result in inhibition of PP-1and therefore enhanced PKA mediated protein phosphorylation leading to amplification of the β-agonist response in the heart (98–99). PP1 activity increases in heart failure which worsens cardiac function as shown by Dr. Krania’s group. Studies performed by Dr. Kranias group have shown that overexpression of the catalytic subunit of PP1 in murine was associated with depressed cardiac function, dilated cardiomyopathy and premature mortality consistent with heart failure (96). Adenoviral-mediated expression of a constitutively active I-1 protein (I-1t35d) in myocytes isolated from human failing hearts, we were able to decrease PP1 activity and enhance contractile function in the presence or absence of β-agonist (96). In rats with pressure overload induced heart failure, overexpression of I-1t35d induced an improvement in contractility. Transgenic expression of a constitutively active I-1 enhanced PLN phosphorylation, augmented cardiac contractility and attenuated heart failure development on aortic constriction. Moreover, we found that active I-1 gene transfer results in marked restoration of contractility in rats with pressure overload induced heart failure (100). Beside that PKCα was found to be a critical regulator of contractility through its direct inhibitory effects on I-1(99).
S100A1, another protein target, is a low molecular weight calcium cycling protein and is abundantly expressed in myocardium with multiple intracellular targets (101). The expression of S100A1 is reduced in heart failure and its ovrexpression by gene transfer induces an increase in SERCA 2a and RYR activities, a decrease in [Na]I, a decrease in NCX and restoration of creatine phosphate resulting in improved intracellular calcium handling and overall enhanced systolic and diastolic function in rats following cryo-infarction (102–104).
6.b Anti-apoptotic effect of gene therapy
As mentioned above, cell death in heart failure takes the form of oncotic, apoptotic and autophagic cell death. Apoptotic and autophagic cell death are forms of programmed cell death that are activated when the cellular reparative mechanisms fail to maintain cellular homeostatsis and integrity as a result of continuous cellular insult. Apoptotic cascades are activated by two major organelles in the cardiomyocyte; the endoplasmic reticulum and the mitochondria. Each one of these organelles can activate apoptotic pathways on its own which is believed to take place when the damage to these organelles tends to be irreversible. This results in loss in cardiomyocytes and as a consequence worsening of cardiac function. Gene therapy plays a role in preventing or attenuating the apoptotic activity. It has been shown that adenovirus mediated overexpression of BCL-2 in rat cardiomyocytes, significantly decreased apoptosis by inhibiting the activation of p53 oncogene. Similarly, overexpression of AKT and phosphatidyl inositol 3 kinase 1 (PI3K1) decreased apoptosis respectively. In small animal models of ischemia-reperfusion injury, adenovirus mediated gene transfer of AKT significantly reduced the number of apoptoic cells in the region of injury, limited infarct size and preserved cardiac function. In another study, intracoronary delivery of adenoviral vector encoding p35, a caspase-3 inhibitor, prevented the onset of heart failure. IL-6 is a cytokine that signals cell survival and has an anti-apoptotic effect through its interaction with glycoprotein 130 receptor. Cardiac restricted ablation of this receptor has been shown to markedlyincrease the rate of apoptosis after pressure overload of the heart. In the future, further understanding of molecular targets of apoptosis and autophagy pathways, may constitute novel targets by gene therapy for the treatment of heart failure.
6.c. Homing of stem cells
It has been well established that damaged and inflamed tissues send out signals to attract stem cells to the injured tissue, and many of these signals have been identified, including stromal derived factor 1 (SDF-1). There has been growing evidence that SDF-1:CXCR4 (its receptor) axis plays an important role in regulating myocardial repair following ischemic injury. SDF-1 has been shown to be increased following myocardial ischemia and a number of studies have shown that enhancing SDF1 levels around the infarct improves myocardial remodeling following infarction. Currently the strategy of delivering SDF1 naked DNA by percutaneous measures to the peri-infarct area is being exploited clinically (105–108).
7. Clinical Trials
There are currently a number of trials ongoing or in the planning stages targeting various pathways for rescuing heart failure. The targets that have been taken forward towards clinical trials include SERCA2a, adenylyl cyclase type 6, and SDF-1.
The first clinical trial of gene therapy in patients with HF was launched in the United States in 2007 (27–28) . CUPID (Calcium Up-Regulation by Percutaneous Administration of Gene Therapy in Cardiac Disease) is a multicenter trial designed to evaluate the safety profile and the biological effects of gene transfer of the SERCA2a cDNA by delivering a recombinant AAV1 (AAV1/SERCA2a) in patients with advanced HF. Participants in this trial were administered a single intracoronary infusion of AAV1/SERCA2a in an open-label approach (27–28). Cohorts 1, 2, 3, and 4 of 3 patients each, received sequentially a single escalating dose of AAV1/SERCA2a. The infusion was spread over multiple coronary arteries in an attempt to provide homogeneous myocardial exposure. 6 to 12 month follow-up of these patients showed an acceptable safety profile (27–28). None of the serious adverse events reported was considered to be secondary to the study drug. Improvement was detected in several patients, reflected by symptomatic (5 patients), functional (4 patients), biomarker (2 patients) and LV function/remodeling (6 patients) parameters. Although this was a phase 1 study involving a small number of patients, early results found that AAV1.SERCA2a treatment conferred quantitative biological benefit in every patient without pre-existing Nabs. The results of the phase 1 study demonstrated an acceptable safety profile, and improvements at 6 months across symptomatic, functional, biomarker and LV function/remodeling parameters. During this preliminary trial, two patients who failed to improve had pre-existing anti-AAV1 neutralizing antibodies (NAb) underscoring the importance of this immunologic mechanism in blocking AAV delivery to the cardiomyocyte after intracoronary infusion. In the phase 2 trial (which was reported at the European Society of Cardiology Heart Failure Meeting on May 30, 2010 in Berlin), 39 patients with advanced HF, as determined by NYHA class III/IV, VO2 max ≤ 20 mL/kg/min and left ventricular (LV) ejection fraction ≤ 35%, were randomized to receive intracoronary adeno-associated virus 1 (AAV1) mediated SERCA2a gene delivery (in one of 3 doses (low dose − 6 × 1011 DRP, middle dose − 3 × 1012 DRP and high dose − 1 × 1013 DRP) versus placebo. Patient’s symptoms (NYHA class, Minnesota Living With Heart Failure Questionnaire [MLWHFQ]), functional status (6 minute walk test [6MWT] and VO2 max), NT-proBNP levels and echocardiographic measures were evaluated over 6 months. Treatment success was determined by examining concordant trends in the above endpoints for group- and patient-based comparisons, as well as clinical outcomes. The AAV1.SERCA2a high-dose group met the pre-specified criteria for success at the group and individual patient levels. AAV1.SERCA2a treated patients, versus placebo, demonstrated improvement or stabilization in NYHA class, MLWHFQ, 6MWT, VO2 max, NT-proBNP levels, and LV end-systolic volumes. Significant increases in time to adjudicated CV events, and a decreased frequency of CV events per patient were observed on AAV1.SERCA2a. No increases in adverse events, disease-related events, laboratory abnormalities or arrhythmias were observed in AAV1.SERCA2a treated patients compared to placebo.
Two other clinical trials targeting SERCA2a are currently enrolling patients. The first trial is in patients with advanced heart failure having received left ventricular assist devices at least one month prior to treatment and who will receive either AAV6.SERCA2a or saline. This trial is being conducted in the United Kingdom. A second Phase 2 monocenter double blind randomized placebo-controled, parallel study will be held in the Institut of Cardiology Pitié-Salpêtrière, Paris, France with the primary objective to investigate the impact of AAV6-CMV-SERCA2a on cardiac remodeling parameters in patients with severe heart failure.
In a separate clinical study adenovirus-5 encoding human adenylyl cyclase type 6 (is being delivered through intracoronary injection to patients with congestive heart failure. Intracoronary delivery of Ad5.hAC6 or PBS in 3:1 randomization with dose escalation, The patients will be randomized in a dose dependent fashion starting at 3.2 × 10^9 viral particles to 3.2 × 10^12 viral particles in 6 dose groups using a 3:1 randomization fashion with PBS (buffered saline being used for control). The trial is currently enrolling patients.
An additional trial is examining the effects of injecting SDF-1 directly into the myocardium of patients with ischemic heart disease. An open label dose escalation study to evaluate the safety of a single escalating dose of SDF-1 administered by endomyocardial injection to cohorts of adults with ischemic heart failure is currently enrolling patients. SDF-1 naked DNA will be injected directly into the myocardium as a single dose at multiple sites through a percutaneous, left ventricular approach using a needle injection catheter.
8. Conclusion and Future Directions
The application of gene therapy was limited initially for the treatment of monogenic diseases. However, the identification of specific disruption in signaling pathways in various disease states has extended the application of gene therapy as a potential treatment in molecular medicine. Regarding the molecular pathology in heart failure, a wealth of novel targets have been uncovered and have increased the interest in gene therapy as a novel therapy for the treatment of heart failure and other cardiovascular diseases. Although improvements regarding vector delivery, safety and bioengineering have been established, it is reasonable to view current vectors, mainly rAAV, as parent compounds, the development of which should stimulate a rationale and broad generation of derivative drugs. Among these derivatives will be vectors based on each of the AAV serotypes, vectors targeted to specific cell types via modified capsid sequences, capsid ligands or tissue specific promoters. Potential gene-based therapies for heart failure can be validated in realistic animal models and, when appropriate, rigorously analyzed in clinical trials for effects on both physiological and clinical end-points. Based on a foundation of rigorous science and a growing understanding of heart failure pathogenesis, there is reason for cautious optimism for the future.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. Executive summary: heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010 Feb 23;121(7):948–54. doi: 10.1161/CIRCULATIONAHA.109.192666. [DOI] [PubMed] [Google Scholar]
- 2.Lloyd-Jones D, Adams RJ, Brown TM, Carnethon M, Dai S, De Simone G, et al. Heart disease and stroke statistics--2010 update: a report from the American Heart Association. Circulation. 2010 Feb 23;121(7):e46–e215. doi: 10.1161/CIRCULATIONAHA.109.192667. [DOI] [PubMed] [Google Scholar]
- 3.Mitchell GF, Pfeffer JM, Pfeffer MA. The transition to failure in the spontaneously hypertensive rat. Am J Hypertens. 1997 May;10(5 Pt 2):120S–6S. [PubMed] [Google Scholar]
- 4.Pfeffer JM, Pfeffer MA, Fletcher PJ, Braunwald E. Progressive ventricular remodeling in rat with myocardial infarction. Am J Physiol. 1991 May;260(5 Pt 2):H1406–14. doi: 10.1152/ajpheart.1991.260.5.H1406. [DOI] [PubMed] [Google Scholar]
- 5.Takewa Y, Chemaly ER, Takaki M, Liang LF, Jin H, Karakikes I, et al. Mechanical work and energetic analysis of eccentric cardiac remodeling in a volume overload heart failure in rats. Am J Physiol Heart Circ Physiol. 2009 Apr;296(4):H1117–24. doi: 10.1152/ajpheart.01120.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosenzweig A. Vectors for cardiovascular gene therapy. J Mol Cell Cardiol. 2003 Jul;35(7):731–3. doi: 10.1016/s0022-2828(03)00144-5. [DOI] [PubMed] [Google Scholar]
- 7.Sakoda T, Kasahara N, Hamamori Y, Kedes L. A high-titer lentiviral production system mediates efficient transduction of differentiated cells including beating cardiac myocytes. J Mol Cell Cardiol. 1999 Nov;31(11):2037–47. doi: 10.1006/jmcc.1999.1035. [DOI] [PubMed] [Google Scholar]
- 8.Svensson EC, Marshall DJ, Woodard K, Lin H, Jiang F, Chu L, et al. Efficient and stable transduction of cardiomyocytes after intramyocardial injection or intracoronary perfusion with recombinant adeno-associated virus vectors. Circulation. 1999 Jan 19;99(2):201–5. doi: 10.1161/01.cir.99.2.201. [DOI] [PubMed] [Google Scholar]
- 9.Giorgi C, De Stefani D, Bononi A, Rizzuto R, Pinton P. Structural and functional link between the mitochondrial network and the endoplasmic reticulum. Int J Biochem Cell Biol. 2009 Oct;41(10):1817–27. doi: 10.1016/j.biocel.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walter L, Hajnoczky G. Mitochondria and endoplasmic reticulum: the lethal interorganelle cross-talk. J Bioenerg Biomembr. 2005 Jun;37(3):191–206. doi: 10.1007/s10863-005-6600-x. [DOI] [PubMed] [Google Scholar]
- 11.Bell CJ, Bright NA, Rutter GA, Griffiths EJ. ATP regulation in adult rat cardiomyocytes: time-resolved decoding of rapid mitochondrial calcium spiking imaged with targeted photoproteins. J Biol Chem. 2006 Sep 22;281(38):28058–67. doi: 10.1074/jbc.M604540200. [DOI] [PubMed] [Google Scholar]
- 12.Murgia M, Giorgi C, Pinton P, Rizzuto R. Controlling metabolism and cell death: at the heart of mitochondrial calcium signalling. J Mol Cell Cardiol. 2009 Jun;46(6):781–8. doi: 10.1016/j.yjmcc.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Boncompagni S, Rossi AE, Micaroni M, Beznoussenko GV, Polishchuk RS, Dirksen RT, et al. Mitochondria are linked to calcium stores in striated muscle by developmentally regulated tethering structures. Mol Biol Cell. 2009 Feb;20(3):1058–67. doi: 10.1091/mbc.E08-07-0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hayashi T, Martone ME, Yu Z, Thor A, Doi M, Holst MJ, et al. Three-dimensional electron microscopy reveals new details of membrane systems for Ca2+ signaling in the heart. J Cell Sci. 2009 Apr 1;122(Pt 7):1005–13. doi: 10.1242/jcs.028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta. 2009 Nov;1787(11):1324–33. doi: 10.1016/j.bbabio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 16.Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: molecular determinants and functional consequences. Physiol Rev. 2006 Jan;86(1):369–408. doi: 10.1152/physrev.00004.2005. [DOI] [PubMed] [Google Scholar]
- 17.Szabadkai G, Duchen MR. Mitochondria: the hub of cellular Ca2+ signaling. Physiology (Bethesda) 2008 Apr;23:84–94. doi: 10.1152/physiol.00046.2007. [DOI] [PubMed] [Google Scholar]
- 18.Asghari P, Schulson M, Scriven DR, Martens G, Moore ED. Axial tubules of rat ventricular myocytes form multiple junctions with the sarcoplasmic reticulum. Biophys J. 2009 Jun 3;96(11):4651–60. doi: 10.1016/j.bpj.2009.02.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Di Maio A, Block BA. Ultrastructure of the sarcoplasmic reticulum in cardiac myocytes from Pacific bluefin tuna. Cell Tissue Res. 2008 Oct;334(1):121–34. doi: 10.1007/s00441-008-0669-6. [DOI] [PubMed] [Google Scholar]
- 20.Popov V, Medvedev NI, Davies HA, Stewart MG. Mitochondria form a filamentous reticular network in hippocampal dendrites but are present as discrete bodies in axons: a three-dimensional ultrastructural study. J Comp Neurol. 2005 Nov 7;492(1):50–65. doi: 10.1002/cne.20682. [DOI] [PubMed] [Google Scholar]
- 21.Staehelin LA, Kang BH. Nanoscale architecture of endoplasmic reticulum export sites and of Golgi membranes as determined by electron tomography. Plant Physiol. 2008 Aug;147(4):1454–68. doi: 10.1104/pp.108.120618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bers DM. Calcium fluxes involved in control of cardiac myocyte contraction. Circ Res. 2000 Aug 18;87(4):275–81. doi: 10.1161/01.res.87.4.275. [DOI] [PubMed] [Google Scholar]
- 23.Katz AM, Lorell BH. Regulation of cardiac contraction and relaxation. Circulation. 2000 Nov 14;102(20 Suppl 4):IV69–74. doi: 10.1161/01.cir.102.suppl_4.iv-69. [DOI] [PubMed] [Google Scholar]
- 24.Okada J, Sugiura S, Nishimura S, Hisada T. Three-dimensional simulation of calcium waves and contraction in cardiomyocytes using the finite element method. Am J Physiol Cell Physiol. 2005 Mar;288(3):C510–22. doi: 10.1152/ajpcell.00261.2004. [DOI] [PubMed] [Google Scholar]
- 25.Mann DL. Left ventricular size and shape: determinants of mechanical signal transduction pathways. Heart Fail Rev. 2005 Jun;10(2):95–100. doi: 10.1007/s10741-005-4636-y. [DOI] [PubMed] [Google Scholar]
- 26.Beeri R, Chaput M, Guerrero JL, Kawase Y, Yosefy C, Abedat S, et al. Gene Delivery of Sarcoplasmic Reticulum Calcium ATPase Inhibits Ventricular Remodeling in Ischemic Mitral Regurgitation. Circ Heart Fail. 2010 Jul 15; doi: 10.1161/CIRCHEARTFAILURE.109.891184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hajjar RJ, Zsebo K, Deckelbaum L, Thompson C, Rudy J, Yaroshinsky A, et al. Design of a phase 1/2 trial of intracoronary administration of AAV1/SERCA2a in patients with heart failure. J Card Fail. 2008 Jun;14(5):355–67. doi: 10.1016/j.cardfail.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 28.Jaski BE, Jessup ML, Mancini DM, Cappola TP, Pauly DF, Greenberg B, et al. Calcium upregulation by percutaneous administration of gene therapy in cardiac disease (CUPID Trial), a first-in-human phase 1/2 clinical trial. J Card Fail. 2009 Apr;15(3):171–81. doi: 10.1016/j.cardfail.2009.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang LF, et al. Targeted gene transfer increases contractility and decreases oxygen cost of contractility in normal rat hearts. Am J Physiol Heart Circ Physiol. 2007 May;292(5):H2356–63. doi: 10.1152/ajpheart.01310.2006. [DOI] [PubMed] [Google Scholar]
- 30.Sakata S, Lebeche D, Sakata N, Sakata Y, Chemaly ER, Liang LF, et al. Restoration of mechanical and energetic function in failing aortic-banded rat hearts by gene transfer of calcium cycling proteins. J Mol Cell Cardiol. 2007 Apr;42(4):852–61. doi: 10.1016/j.yjmcc.2007.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakata S, Lebeche D, Sakata Y, Sakata N, Chemaly ER, Liang L, et al. Transcoronary gene transfer of SERCA2a increases coronary blood flow and decreases cardiomyocyte size in a type 2 diabetic rat model. Am J Physiol Heart Circ Physiol. 2007 Feb;292(2):H1204–7. doi: 10.1152/ajpheart.00892.2006. [DOI] [PubMed] [Google Scholar]
- 32.Schmidt U, del Monte F, Miyamoto MI, Matsui T, Gwathmey JK, Rosenzweig A, et al. Restoration of diastolic function in senescent rat hearts through adenoviral gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase. Circulation. 2000 Feb 22;101(7):790–6. doi: 10.1161/01.cir.101.7.790. [DOI] [PubMed] [Google Scholar]
- 33.Bodi I, Mikala G, Koch SE, Akhter SA, Schwartz A. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 2005 Dec;115(12):3306–17. doi: 10.1172/JCI27167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kay MA, Glorioso JC, Naldini L. Viral vectors for gene therapy: the art of turning infectious agents into vehicles of therapeutics. Nat Med. 2001 Jan;7(1):33–40. doi: 10.1038/83324. [DOI] [PubMed] [Google Scholar]
- 35.Nishikawa M, Huang L. Nonviral vectors in the new millennium: delivery barriers in gene transfer. Hum Gene Ther. 2001 May 20;12(8):861–70. doi: 10.1089/104303401750195836. [DOI] [PubMed] [Google Scholar]
- 36.Felgner PL. Nonviral strategies for gene therapy. Sci Am. 1997 Jun;276(6):102–6. doi: 10.1038/scientificamerican0697-102. [DOI] [PubMed] [Google Scholar]
- 37.Isner JM. Myocardial gene therapy. Nature. 2002 Jan 10;415(6868):234–9. doi: 10.1038/415234a. [DOI] [PubMed] [Google Scholar]
- 38.Guzman RJ, Lemarchand P, Crystal RG, Epstein SE, Finkel T. Efficient and selective adenovirus-mediated gene transfer into vascular neointima. Circulation. 1993 Dec;88(6):2838–48. doi: 10.1161/01.cir.88.6.2838. [DOI] [PubMed] [Google Scholar]
- 39.Barr E, Carroll J, Kalynych AM, Tripathy SK, Kozarsky K, Wilson JM, et al. Efficient catheter-mediated gene transfer into the heart using replication-defective adenovirus. Gene Ther. 1994 Jan;1(1):51–8. [PubMed] [Google Scholar]
- 40.Hajjar RJ, Schmidt U, Matsui T, Guerrero JL, Lee KH, Gwathmey JK, et al. Modulation of ventricular function through gene transfer in vivo. Proc Natl Acad Sci U S A. 1998 Apr 28;95(9):5251–6. doi: 10.1073/pnas.95.9.5251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shohet RV, Chen S, Zhou YT, Wang Z, Meidell RS, Unger RH, et al. Echocardiographic destruction of albumin microbubbles directs gene delivery to the myocardium. Circulation. 2000 Jun 6;101(22):2554–6. doi: 10.1161/01.cir.101.22.2554. [DOI] [PubMed] [Google Scholar]
- 42.Beeri R, Guerrero JL, Supple G, Sullivan S, Levine RA, Hajjar RJ. New efficient catheter-based system for myocardial gene delivery. Circulation. 2002 Oct 1;106(14):1756–9. doi: 10.1161/01.cir.0000035240.92015.e4. [DOI] [PubMed] [Google Scholar]
- 43.Donahue JK, Kikkawa K, Johns DC, Marban E, Lawrence JH. Ultrarapid, highly efficient viral gene transfer to the heart. Proc Natl Acad Sci U S A. 1997 Apr 29;94(9):4664–8. doi: 10.1073/pnas.94.9.4664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Donahue JK, Kikkawa K, Thomas AD, Marban E, Lawrence JH. Acceleration of widespread adenoviral gene transfer to intact rabbit hearts by coronary perfusion with low calcium and serotonin. Gene Ther. 1998 May;5(5):630–4. doi: 10.1038/sj.gt.3300649. [DOI] [PubMed] [Google Scholar]
- 45.Nagata K, Marban E, Lawrence JH, Donahue JK. Phosphodiesterase inhibitor-mediated potentiation of adenovirus delivery to myocardium. J Mol Cell Cardiol. 2001 Mar;33(3):575–80. doi: 10.1006/jmcc.2000.1322. [DOI] [PubMed] [Google Scholar]
- 46.Monahan PE, Samulski RJ. AAV vectors: is clinical success on the horizon? Gene Ther. 2000 Jan;7(1):24–30. doi: 10.1038/sj.gt.3301109. [DOI] [PubMed] [Google Scholar]
- 47.Monahan PE, Samulski RJ, Tazelaar J, Xiao X, Nichols TC, Bellinger DA, et al. Direct intramuscular injection with recombinant AAV vectors results in sustained expression in a dog model of hemophilia. Gene Ther. 1998 Jan;5(1):40–9. doi: 10.1038/sj.gt.3300548. [DOI] [PubMed] [Google Scholar]
- 48.Flotte TR, Carter BJ. Adeno-associated virus vectors for gene therapy. Gene Ther. 1995 Aug;2(6):357–62. [PubMed] [Google Scholar]
- 49.Schnepp BC, Jensen RL, Chen CL, Johnson PR, Clark KR. Characterization of adeno-associated virus genomes isolated from human tissues. J Virol. 2005 Dec;79(23):14793–803. doi: 10.1128/JVI.79.23.14793-14803.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monahan PE, Jooss K, Sands MS. Safety of adeno-associated virus gene therapy vectors: a current evaluation. Expert Opin Drug Saf. 2002 May;1(1):79–91. doi: 10.1517/14740338.1.1.79. [DOI] [PubMed] [Google Scholar]
- 51.Dutheil N, Shi F, Dupressoir T, Linden RM. Adeno-associated virus site-specifically integrates into a muscle-specific DNA region. Proc Natl Acad Sci U S A. 2000 Apr 25;97(9):4862–6. doi: 10.1073/pnas.080079397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Young SM, Jr, Samulski RJ. Adeno-associated virus (AAV) site-specific recombination does not require a Rep-dependent origin of replication within the AAV terminal repeat. Proc Natl Acad Sci U S A. 2001 Nov 20;98(24):13525–30. doi: 10.1073/pnas.241508998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carter PJ, Samulski RJ. Adeno-associated viral vectors as gene delivery vehicles. Int J Mol Med. 2000 Jul;6(1):17–27. doi: 10.3892/ijmm.6.1.17. [DOI] [PubMed] [Google Scholar]
- 54.Inagaki K, Fuess S, Storm TA, Gibson GA, McTiernan CF, Kay MA, et al. Robust systemic transduction with AAV9 vectors in mice: efficient global cardiac gene transfer superior to that of AAV8. Mol Ther. 2006 Jul;14(1):45–53. doi: 10.1016/j.ymthe.2006.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pacak CA, Mah CS, Thattaliyath BD, Conlon TJ, Lewis MA, Cloutier DE, et al. Recombinant adeno-associated virus serotype 9 leads to preferential cardiac transduction in vivo. Circ Res. 2006 Aug 18;99(4):e3–9. doi: 10.1161/01.RES.0000237661.18885.f6. [DOI] [PubMed] [Google Scholar]
- 56.Wang Z, Zhu T, Qiao C, Zhou L, Wang B, Zhang J, et al. Adeno-associated virus serotype 8 efficiently delivers genes to muscle and heart. Nat Biotechnol. 2005 Mar;23(3):321–8. doi: 10.1038/nbt1073. [DOI] [PubMed] [Google Scholar]
- 57.Zhu T, Zhou L, Mori S, Wang Z, McTiernan CF, Qiao C, et al. Sustained whole-body functional rescue in congestive heart failure and muscular dystrophy hamsters by systemic gene transfer. Circulation. 2005 Oct 25;112(17):2650–9. doi: 10.1161/CIRCULATIONAHA.105.565598. [DOI] [PubMed] [Google Scholar]
- 58.Tratschin JD, Miller IL, Smith MG, Carter BJ. Adeno-associated virus vector for high-frequency integration, expression, and rescue of genes in mammalian cells. Mol Cell Biol. 1985 Nov;5(11):3251–60. doi: 10.1128/mcb.5.11.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Carter BJ. Adeno-associated virus vectors in clinical trials. Hum Gene Ther. 2005 May;16(5):541–50. doi: 10.1089/hum.2005.16.541. [DOI] [PubMed] [Google Scholar]
- 60.Flotte TR. Adeno-associated virus-based gene therapy for inherited disorders. Pediatr Res. 2005 Dec;58(6):1143–7. doi: 10.1203/01.pdr.0000189226.03684.fe. [DOI] [PubMed] [Google Scholar]
- 61.Mease PJ, Hobbs K, Chalmers A, El-Gabalawy H, Bookman A, Keystone E, et al. Local delivery of a recombinant adenoassociated vector containing a tumour necrosis factor alpha antagonist gene in inflammatory arthritis: a phase 1 dose-escalation safety and tolerability study. Ann Rheum Dis. 2009 Aug;68(8):1247–54. doi: 10.1136/ard.2008.089375. [DOI] [PubMed] [Google Scholar]
- 62.Monahan PE, Samulski RJ. Adeno-associated virus vectors for gene therapy: more pros than cons? Mol Med Today. 2000 Nov;6(11):433–40. doi: 10.1016/s1357-4310(00)01810-4. [DOI] [PubMed] [Google Scholar]
- 63.Schnepp BC, Clark KR, Klemanski DL, Pacak CA, Johnson PR. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J Virol. 2003 Mar;77(6):3495–504. doi: 10.1128/JVI.77.6.3495-3504.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Snyder RO, Miao C, Meuse L, Tubb J, Donahue BA, Lin HF, et al. Correction of hemophilia B in canine and murine models using recombinant adeno-associated viral vectors. Nat Med. 1999 Jan;5(1):64–70. doi: 10.1038/4751. [DOI] [PubMed] [Google Scholar]
- 65.Kessler PD, Podsakoff GM, Chen X, McQuiston SA, Colosi PC, Matelis LA, et al. Gene delivery to skeletal muscle results in sustained expression and systemic delivery of a therapeutic protein. Proc Natl Acad Sci U S A. 1996 Nov 26;93(24):14082–7. doi: 10.1073/pnas.93.24.14082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Manno CS, Pierce GF, Arruda VR, Glader B, Ragni M, Rasko JJ, et al. Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat Med. 2006 Mar;12(3):342–7. doi: 10.1038/nm1358. [DOI] [PubMed] [Google Scholar]
- 67.Rabinowitz JE, Rolling F, Li C, Conrath H, Xiao W, Xiao X, et al. Cross-packaging of a single adeno-associated virus (AAV) type 2 vector genome into multiple AAV serotypes enables transduction with broad specificity. J Virol. 2002 Jan;76(2):791–801. doi: 10.1128/JVI.76.2.791-801.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ferrari FK, Samulski T, Shenk T, Samulski RJ. Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol. 1996 May;70(5):3227–34. doi: 10.1128/jvi.70.5.3227-3234.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Malik AK, Monahan PE, Allen DL, Chen BG, Samulski RJ, Kurachi K. Kinetics of recombinant adeno-associated virus-mediated gene transfer. J Virol. 2000 Apr;74(8):3555–65. doi: 10.1128/jvi.74.8.3555-3565.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Nakai H, Storm TA, Kay MA. Recruitment of single-stranded recombinant adeno-associated virus vector genomes and intermolecular recombination are responsible for stable transduction of liver in vivo. J Virol. 2000 Oct;74(20):9451–63. doi: 10.1128/jvi.74.20.9451-9463.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Klages N, Zufferey R, Trono D. A stable system for the high-titer production of multiply attenuated lentiviral vectors. Mol Ther. 2000 Aug;2(2):170–6. doi: 10.1006/mthe.2000.0103. [DOI] [PubMed] [Google Scholar]
- 72.Galimi F, Noll M, Kanazawa Y, Lax T, Chen C, Grompe M, et al. Gene therapy of Fanconi anemia: preclinical efficacy using lentiviral vectors. Blood. 2002 Oct 15;100(8):2732–6. doi: 10.1182/blood-2002-04-1245. [DOI] [PubMed] [Google Scholar]
- 73.Galimi F, Verma IM. Opportunities for the use of lentiviral vectors in human gene therapy. Curr Top Microbiol Immunol. 2002;261:245–54. doi: 10.1007/978-3-642-56114-6_13. [DOI] [PubMed] [Google Scholar]
- 74.Sirven A, Pflumio F, Zennou V, Titeux M, Vainchenker W, Coulombel L, et al. The human immunodeficiency virus type-1 central DNA flap is a crucial determinant for lentiviral vector nuclear import and gene transduction of human hematopoietic stem cells. Blood. 2000 Dec 15;96(13):4103–10. [PubMed] [Google Scholar]
- 75.Bonci D, Cittadini A, Latronico MV, Borello U, Aycock JK, Drusco A, et al. 'Advanced' generation lentiviruses as efficient vectors for cardiomyocyte gene transduction in vitro and in vivo. Gene Ther. 2003 Apr;10(8):630–6. doi: 10.1038/sj.gt.3301936. [DOI] [PubMed] [Google Scholar]
- 76.Maurice JP, Hata JA, Shah AS, White DC, McDonald PH, Dolber PC, et al. Enhancement of cardiac function after adenoviral-mediated in vivo intracoronary beta2-adrenergic receptor gene delivery. J Clin Invest. 1999 Jul;104(1):21–9. doi: 10.1172/JCI6026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zincarelli C, Soltys S, Rengo G, Rabinowitz JE. Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther. 2008 Jun;16(6):1073–80. doi: 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 78.Koransky ML, Robbins RC, Blau HM. VEGF gene delivery for treatment of ischemic cardiovascular disease. Trends Cardiovasc Med. 2002 Apr;12(3):108–14. doi: 10.1016/s1050-1738(01)00158-x. [DOI] [PubMed] [Google Scholar]
- 79.Edelberg JM, Huang DT, Josephson ME, Rosenberg RD. Molecular enhancement of porcine cardiac chronotropy. Heart. 2001 Nov;86(5):559–62. doi: 10.1136/heart.86.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Boekstegers P, von Degenfeld G, Giehrl W, Heinrich D, Hullin R, Kupatt C, et al. Myocardial gene transfer by selective pressure-regulated retroinfusion of coronary veins. Gene Ther. 2000 Feb;7(3):232–40. doi: 10.1038/sj.gt.3301079. [DOI] [PubMed] [Google Scholar]
- 81.Hayase M, Del Monte F, Kawase Y, Macneill BD, McGregor J, Yoneyama R, et al. Catheter-based antegrade intracoronary viral gene delivery with coronary venous blockade. Am J Physiol Heart Circ Physiol. 2005 Jun;288(6):H2995–3000. doi: 10.1152/ajpheart.00703.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fromes Y, Salmon A, Wang X, Collin H, Rouche A, Hagege A, et al. Gene delivery to the myocardium by intrapericardial injection. Gene Ther. 1999 Apr;6(4):683–8. doi: 10.1038/sj.gt.3300853. [DOI] [PubMed] [Google Scholar]
- 83.Preovolos AC, Mennen MT, Bilney A, Mariani J, Kaye DM, Power JM. Development of a novel perfusion technique to allow targeted delivery of gene therapy-–the V-Focus system. J Extra Corpor Technol. 2006 Mar;38(1):51–2. [PMC free article] [PubMed] [Google Scholar]
- 84.Bridges CR, Burkman JM, Malekan R, Konig SM, Chen H, Yarnall CB, et al. Global cardiac-specific transgene expression using cardiopulmonary bypass with cardiac isolation. Ann Thorac Surg. 2002 Jun;73(6):1939–46. doi: 10.1016/s0003-4975(02)03509-9. [DOI] [PubMed] [Google Scholar]
- 85.Bridges CR, Gopal K, Holt DE, Yarnall C, Cole S, Anderson RB, et al. Efficient myocyte gene delivery with complete cardiac surgical isolation in situ. J Thorac Cardiovasc Surg. 2005 Nov;130(5):1364. doi: 10.1016/j.jtcvs.2005.07.035. [DOI] [PubMed] [Google Scholar]
- 86.Bristow MR. The beta-adrenergic receptor. Configuration, regulation, mechanism of action. Postgrad Med. 1988 Feb 29;Spec No:19–26. [PubMed] [Google Scholar]
- 87.Bristow MR. beta-adrenergic receptor blockade in chronic heart failure. Circulation. 2000 Feb 8;101(5):558–69. doi: 10.1161/01.cir.101.5.558. [DOI] [PubMed] [Google Scholar]
- 88.Bristow MR, Ginsburg R. Beta 2 receptors on myocardial cells in human ventricular myocardium. Am J Cardiol. 1986 Apr 25;57(12):3F–6F. doi: 10.1016/0002-9149(86)90880-5. [DOI] [PubMed] [Google Scholar]
- 89.Bristow MR, Kantrowitz NE, Ginsburg R, Fowler MB. Beta-adrenergic function in heart muscle disease and heart failure. J Mol Cell Cardiol. 1985 Jul;17( Suppl 2):41–52. doi: 10.1016/0022-2828(85)90007-0. [DOI] [PubMed] [Google Scholar]
- 90.Feldman DS, Carnes CA, Abraham WT, Bristow MR. Mechanisms of disease: beta-adrenergic receptors--alterations in signal transduction and pharmacogenomics in heart failure. Nat Clin Pract Cardiovasc Med. 2005 Sep;2(9):475–83. doi: 10.1038/ncpcardio0309. [DOI] [PubMed] [Google Scholar]
- 91.Feldman DS, Elton TS, Sun B, Martin MM, Ziolo MT. Mechanisms of disease: detrimental adrenergic signaling in acute decompensated heart failure. Nat Clin Pract Cardiovasc Med. 2008 Apr;5(4):208–18. doi: 10.1038/ncpcardio1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.del Monte F, Williams E, Lebeche D, Schmidt U, Rosenzweig A, Gwathmey JK, et al. Improvement in survival and cardiac metabolism after gene transfer of sarcoplasmic reticulum Ca(2+)-ATPase in a rat model of heart failure. Circulation. 2001 Sep 18;104(12):1424–9. doi: 10.1161/hc3601.095574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davia K, Bernobich E, Ranu HK, del Monte F, Terracciano CM, MacLeod KT, et al. SERCA2A overexpression decreases the incidence of aftercontractions in adult rabbit ventricular myocytes. J Mol Cell Cardiol. 2001 May;33(5):1005–15. doi: 10.1006/jmcc.2001.1368. [DOI] [PubMed] [Google Scholar]
- 94.Hoshijima M, Ikeda Y, Iwanaga Y, Minamisawa S, Date MO, Gu Y, et al. Chronic suppression of heart-failure progression by a pseudophosphorylated mutant of phospholamban via in vivo cardiac rAAV gene delivery. Nat Med. 2002 Aug;8(8):864–71. doi: 10.1038/nm739. [DOI] [PubMed] [Google Scholar]
- 95.Iwanaga Y, Hoshijima M, Gu Y, Iwatate M, Dieterle T, Ikeda Y, et al. Chronic phospholamban inhibition prevents progressive cardiac dysfunction and pathological remodeling after infarction in rats. J Clin Invest. 2004 Mar;113(5):727–36. doi: 10.1172/JCI18716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Carr AN, Schmidt AG, Suzuki Y, del Monte F, Sato Y, Lanner C, et al. Type 1 phosphatase, a negative regulator of cardiac function. Mol Cell Biol. 2002 Jun;22(12):4124–35. doi: 10.1128/MCB.22.12.4124-4135.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.El-Armouche A, Pamminger T, Ditz D, Zolk O, Eschenhagen T. Decreased protein and phosphorylation level of the protein phosphatase inhibitor-1 in failing human hearts. Cardiovasc Res. 2004 Jan 1;61(1):87–93. doi: 10.1016/j.cardiores.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 98.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, et al. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin–NFAT signaling. J Clin Invest. 2003 May;111(10):1475–86. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, et al. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004 Mar;10(3):248–54. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 100.Pathak A, del Monte F, Zhao W, Schultz JE, Lorenz JN, Bodi I, et al. Enhancement of cardiac function and suppression of heart failure progression by inhibition of protein phosphatase 1. Circ Res. 2005 Apr 15;96(7):756–66. doi: 10.1161/01.RES.0000161256.85833.fa. [DOI] [PubMed] [Google Scholar]
- 101.Donato R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int J Biochem Cell Biol. 2001 Jul;33(7):637–68. doi: 10.1016/s1357-2725(01)00046-2. [DOI] [PubMed] [Google Scholar]
- 102.Most P, Pleger ST, Volkers M, Heidt B, Boerries M, Weichenhan D, et al. Cardiac adenoviral S100A1 gene delivery rescues failing myocardium. J Clin Invest. 2004 Dec;114(11):1550–63. doi: 10.1172/JCI21454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Pleger ST, Most P, Heidt B, Voelkers M, Hata JA, Katus HA, et al. S100A1 gene transfer in myocardium. Eur J Med Res. 2006 Oct 27;11(10):418–22. [PubMed] [Google Scholar]
- 104.Pleger ST, Remppis A, Heidt B, Volkers M, Chuprun JK, Kuhn M, et al. S100A1 gene therapy preserves in vivo cardiac function after myocardial infarction. Mol Ther. 2005 Dec;12(6):1120–9. doi: 10.1016/j.ymthe.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 105.Agarwal U, Ghalayini W, Dong F, Weber K, Zou YR, Rabbany SY, et al. Role of Cardiac Myocyte CXCR4 Expression in Development and Left Ventricular Remodeling After Acute Myocardial Infarction. Circ Res. 2010 Jul 15; doi: 10.1161/CIRCRESAHA.110.223289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Penn MS. Importance of the SDF-1:CXCR4 axis in myocardial repair. Circ Res. 2009 May 22;104(10):1133–5. doi: 10.1161/CIRCRESAHA.109.198929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Rabbany SY, Pastore J, Yamamoto M, Miller T, Rafii S, Aras R, et al. Continuous delivery of stromal cell-derived factor-1 from alginate scaffolds accelerates wound healing. Cell Transplant. 2010;19(4):399–408. doi: 10.3727/096368909X481782. [DOI] [PubMed] [Google Scholar]
- 108.Segers VF, Lee RT. Protein Therapeutics for Cardiac Regeneration after Myocardial Infarction. J Cardiovasc Transl Res. 2010 Jul 7; doi: 10.1007/s12265-010-9207-5. [DOI] [PMC free article] [PubMed] [Google Scholar]