

Abstract
Climate warming is predicted to increase the frequency of invasions by pathogens and to cause the large-scale redistribution of native host species, with dramatic consequences on the health of domesticated and wild populations of plants and animals. The study of historic range shifts in response to climate change, such as during interglacial cycles, can help in the prediction of the routes and dynamics of infectious diseases during the impending ecosystem changes. Here we studied the population structure in Europe of two Microbotryum species causing anther smut disease on the plants Silene latifolia and Silene dioica. Clustering analyses revealed the existence of genetically distinct groups for the pathogen on S. latifolia, providing a clear-cut example of European phylogeography reflecting recolonization from southern refugia after glaciation. The pathogen genetic structure was congruent with the genetic structure of its host species S. latifolia, suggesting dependence of the migration pathway of the anther smut fungus on its host. The fungus, however, appeared to have persisted in more numerous and smaller refugia than its host and to have experienced fewer events of large-scale dispersal. The anther smut pathogen on S. dioica also showed a strong phylogeographic structure that might be related to more northern glacial refugia. Differences in host ecology probably played a role in these differences in the pathogen population structure. Very high selfing rates were inferred in both fungal species, explaining the low levels of admixture between the genetic clusters. The systems studied here indicate that migration patterns caused by climate change can be expected to include pathogen invasions that follow the redistribution of their host species at continental scales, but also that the recolonization by pathogens is not simply a mirror of their hosts, even for obligate biotrophs, and that the ecology of hosts and pathogen mating systems likely affects recolonization patterns.
Author Summary
Global change is expected to cause the large-scale redistribution of species, including pathogens that threaten the health of domestic populations and natural ecosystems. Predicting the dynamics of invasive pathogens is therefore a major challenge for the 21st century ecologists and epidemiologists. Because past climatic events have shaped the current distribution of species, the study of migration of pathogens during glaciation cycles provides insights into the constraints upon pathogen spread. We studied the structure of European populations of two pathogenic fungal species infecting two species of wild plants. The pathogen with the host's broadest ecological tolerance has followed the postglacial colonisation pathway of its host from the major refugia in southern Europe, although the pathogen likely persisted in more fragmented refugia. The other pathogen showed less clear-cut genetic patterns and some evidence of possible northern refugia, in agreement with the host's more fragmented distribution and ecological preference. These results indicate that pathogen invasions are likely to follow large-scale migration events of their host species in response to climate change, but also that the recolonization by pathogens is not simply a mirror of their hosts, and that the correlation between the colonization patterns of hosts and pathogens depends on host ecology.
Introduction
Understanding the dynamics of emerging infectious diseases and their routes to colonize new geographic regions is a major challenge for ecologists in an effort to prevent negative impacts upon human, domestic, and natural populations. Because pathogens causing emerging diseases have not coevolved with the host or the ecosystem in which they emerged, they may be more likely to pose a threat to biodiversity through biomass loss and extinction of host species than those responsible for endemic diseases [1]. In recent years, concerns about emerging diseases have been increasing in light of the first evidence of a current period of global climate change. Indeed, the increase of average temperatures in many areas of the world is thought to promote the expansion of exotic pathogens [2]. In particular, invasion by fungal pathogens is a major concern in agricultural and biodiversity management, as they infect many crops and wild plants [3], [4]. About 30% of emerging infectious diseases of plants are caused by fungi, and the change of environmental conditions is thought to be the major driver of fungal invasions [1] and disease outbreaks [5]. Fungal disease outbreaks in humans have also been suggested to be linked to climate change [6], [7]. Warmer and wetter conditions favour the growth and transmission of fungal pathogens, and host shifts often occur in conjunction with episodes of global climate change [8]. Based on current trends, emerging infectious diseases caused by fungal pathogens are likely to increase in the near future, with significant severe ecological, economic and social consequences.
One way to predict the invasion routes and dynamics of emerging infectious diseases in response to current climate warming is to study the past migrations of pathogens and their hosts during historic periods of climate changes. During the last glacial maximum, the Arctic ice sheet extended into a large part of Europe and limited the survival of most organisms into Southern Mediterranean refugia (i.e., Iberia, Italy and the Balkans [9], [10]). As the climate warmed and the ice sheet retreated, many species that had persisted inside the glacial refugia experienced massive migrations into the newly available temperate territories [11]. Such processes are expected to lead to a strong, large-scale geographic structure of genetic variation, consistent with what is observed in many widespread European plants (e.g. [12]–[17]). Genetic differences between spreading populations are likely to result from both natural selection and stochastic processes in small populations. Successive founder events during the process of range expansion are expected to lead to a loss of variation and further divergence between lineages derived from different refugia [9], [18]–[21].
Few studies have investigated whether the population structures of pathogens have also been impacted by the glaciations in Europe (but see e.g. [22]–[25]). In the case of host-pathogen systems, comparative phylogeography can also provide insights into host and pathogen co-evolutionary histories and identify causal factors determining their combined distributions [26]–[28]. In fact, pathogen populations are often more differentiated than their hosts, and the study of pathogens can complement or improve our knowledge on the host population genetic structure [29]–[37]. The extent to which the phylogeographic structure of pathogen populations mirrors that of the host depends on the degree of specificity and the obligate nature of pathogenic interaction [38]. A significant co-structure between the populations of the host and the pathogen suggests that the distribution and migration of the host impose a major constraint on the distribution of the pathogen [8]. On the other hand, the absence of congruence in population structure is consistent with independent host and pathogen colonization routes. In addition to pathogen specialization, the hosts' niche breadth and demographic characteristics may affect the persistence of disease and opportunities for host range expansion during large-scale migrations that follow climate change [39]–[41]. Thus, an approach that integrates knowledge of host and pathogen biology is essential to many theoretical and applied issues related to disease emergence in response to climate change.
Microbotryum violaceum sensu lato is a species complex of basidiomycete fungi responsible for anther smut disease in many plants in the Caryophyllaceae. These fungi are obligate pathogens that sterilize their hosts. Infected plants contain fungal teliospores in place of the pollen and female structures do not mature; female plants in dioecious species also develop spore-bearing anthers. Teliospores are transmitted from diseased to healthy plants mostly by insects that normally serve as pollinators. Therefore, the dispersal routes of the host's pollen and of the pathogen's spores are constrained by the same vectors. Plants also disperse by seed, while the fungus is not vertically transmitted, resulting in higher genetic differentiation in the pathogen than in the host [42]. The sibling species encompassed in Microbotryum violaceum sensu lato [43]–[49] show strong host specificity [45], [47], [50]. The most widely studied species are Microbotryum lychnis-dioicae [51] (called MvSl in [46] and hereafter) and M. silenes-dioicae [51] (called MvSd in [46] and hereafter), which infect respectively Silene latifolia (white campion) and S. dioica (red campion). These two closely related host-pathogen systems are interesting models for studying the combined demographic histories of pathogens and their hosts because (i) populations of this pathogen are more differentiated than those of its hosts [42], (ii) the fungus is completely dependent on its hosts and the same vectors disperse the fungus and the host pollen, (iii) these Microbotryum species are highly host specific in the field ([52]), (iv) Silene and Microbotryum species have similar generation times (one per year [53], [54]), (v) there has been no attempt to control the disease because it affects plants without economic interest, and (vi) Silene and Microbotryum are model organisms for a variety of topics in ecology and evolution, with therefore numerous studies available on their life-history and ecology [55].
Recent studies on the phylogeographic history of the two host plants showed genetic evidence of post-glacial recolonization from Mediterranean refugia. In S. latifolia, analyses of chloroplast DNA (cpDNA) polymorphism showed clearly structured haplotype variation in Europe, with haplotypes from Eastern and Western Europe forming divergent groups descended from haplotypes currently distributed in southern Europe, and in particular from the Iberian and Balkan Peninsulas [56]. The phylogeography of S. dioica in Europe has been less well studied, although the pattern of cpDNA polymorphism was also suggestive of post-glacial recolonisation from multiple refugia [57], [58]. A goal of the current study was therefore to determine the extent to which population structure of Microbotryum species parasitizing S. latifolia and S. dioica showed similar patterns of post-glacial history. We also investigated whether life history differences between the two host species constrained the distributions of the pathogens within the host migration pathways. S. latifolia and S. dioica differ with respect to their ecologies, which might strongly impact the genetic structure and diversity of their specialized pathogens. S. latifolia has an extensive range and occurs in most of Europe, as well as in Middle Asia and the Steppe area of south Siberia [59]. This plant is found mainly in open areas, such as hedgerows and in arable fields, therefore often experiencing extinction-recolonisation events in frequently disturbed habitats. In contrast, the distribution of S. dioica covers mainly Central, Northern, and Western Europe [59], but not the Mediterranean regions. This plant is found in meadows, cliffs, moist forest, and mown pastures at higher elevations, preferring colder and more humid habitats than S. latifolia, and experiencing more stable population dynamics [59]–[62]. It has been suggested that these differences in host life history have affected the distribution of genetic diversity at a small geographical scale in the pathogen species, with lower microsatellite variation and higher differentiation among populations in the anther smut fungus parasitizing S. latifolia than in the fungus parasitizing S. dioica [63]. It is likely that the phylogeographic structure of the European populations of the two pathogen species will also be influenced by differences in the dynamics of the host-pathogen systems.
We therefore determined the population structures of the Microbotryum species infecting S. latifolia and S. dioica using microsatellite markers in order to address the following specific questions: (i) Do the phylogeographical structures of the Microbotryum species show signatures of post-glacial recolonisation of Europe, and in particular from Southern refugia? (ii) do the population structures of the two pathogens differ from each other, and if so, are these differences consistent with expectations based upon known ecological differences of their plant hosts? (iii) are the phylogeographic patterns of Microbotryum species comparable at a continental scale to those of their respective hosts?
Results
Genetic polymorphism in Microbotryum lychnidis-dioicae (MvSl) and M. silenes-dioicae (MvSd)
Among Microbotryum samples collected on S. latifolia and on S. doica across Europe (Figure S1), analysis of variation at 11 microsatellite markers revealed that both pathogen species displayed much lower levels of heterozygosity than expected under Hardy-Weinberg Equilibrium (HWE). The dataset included 701 MvSl individuals from 187 localities and 342 MvSd individuals from 68 localities, where hybrids and cross-species disease transmission between MvSl and MvSd identified in a previous study [52] were removed from the dataset. Descriptive statistics on the polymorphism of MvSd and MvSl and on deviations from HWE are shown in Table 1 and additional details are given in Text S1 and Tables S1 and S2. MvSl for instance exhibited only 3% of heterozygous genotypes while 73% were expected under HWE, which is consistent with the high selfing rates previously reported in Microbotryum. One marker, SL19, showed extreme FIS values in MvSl, between −1 and 1, and was almost fixed in the heterozygous state in MvSd (Table 1). Analyses were therefore performed with and without this marker for subsequent analyses, but the results were highly similar.
Table 1. Summary statistics on the 11 microsatellite loci in Microbotryum lychnidis-dioicae (MvSl) and Microbotryum silenes-dioicae (MvSd).
| Locus | Repeat | Microbotryum lychnidis-dioicae (MvSl) | Microbotryum silenes-dioicae (MvSd) | ||||||||
| N | Range | A | Ho/He | FIS | N | Range | A | Ho/He | FIS | ||
| E14 | (AG)15 | 670 | 7–23 | 11 | 0.03/0.74 | 0.86*** | 329 | 9–15 | 4 | 0.02/0.31 | 0.93*** |
| E17 | (AG)20 | 649 | 2–17 | 15 | 0.01/0.86 | 0.90*** | 326 | 1–5 | 5 | 0.01/0.76 | 0.98*** |
| E18 | (AG)15 | 535 | 4–25 | 20 | 0.01/0.88 | 0.98*** | 273 | 9–24 | 14 | 0.00/0.89 | 0.99*** |
| SL5 | (CT)18 | 678 | 11–22 | 12 | 0.00/0.82 | 0.98*** | 329 | 12–18 | 7 | 0.00/0.82 | 0.99*** |
| SL8 | (GA)15 | 534 | 1–10 | 10 | 0.01/0.63 | 0.96*** | 303 | 1–5 | 3 | 0.02/0.15 | 0.84*** |
| SL9 | (CT)9 | 683 | 3–14 | 12 | 0.04/0.83 | 0.89*** | 339 | 6–10 | 5 | 0.02/0.68 | 0.98*** |
| SL12 | (GT)10 | 680 | 1–7 | 7 | 0.03/0.64 | 0.73*** | 314 | 3–6 | 4 | 0.06/0.53 | 0.89*** |
| SL19 | (AAC)3AAA(AAC)12 | 662 | 1–9 | 9 | 0.10/0.84 | 0.29*** | 321 | 1–7 | 7 | 0.86/0.63 | −0.36+++ |
| SVG5 | (TG)8 | 695 | 2–9 | 7 | 0.09/0.68 | 0.59*** | 318 | 4–9 | 5 | 0.09/0.56 | 0.83*** |
| SVG8 | (GT)12 | 679 | 9–25 | 15 | 0.02/0.87 | 0.89*** | 337 | 9–23 | 10 | 0.04/0.82 | 0.95*** |
| SVG14 | (CTC)2(TTC)10 | 682 | 6–18 | 7 | 0.02/0.24 | 0.63*** | 318 | 14–22 | 6 | 0.05/0.70 | 0.92*** |
| all | 649.7 (±58.2) | 11.4 (±4.1) | 0.03/0.73 | 318.8 (±18.4) | 6.4 (±3.2) | 0.11/0.62 | |||||
Repeat: Motifs and repeat numbers of the microsatellites in the isolated clones that served to develop the markers. N: number of samples successfully genotyped; range: allelic size range in number of repeats; A: number of alleles observed; Ho: observed heterozygosity and He the expected heterozygosity (or overall genetic diversity in Nei 1987). Symbols (***) and (+++) show significant (p<0.001) deficit or excess in heterozygosity compared to Hardy-Weinberg expectations. A more detailed interpretation of the genetic polymorphism in MvSl and MvSd is provided in Text S1.
Genetic structure and genetic diversity in M. lychnidis-dioicae (MvSl)
For MvSl, multiple estimates of the genetic structure at the European scale showed the existence of at least three to five strongly supported clusters, i.e. populations genetically differentiated from each other. We used the model-based Bayesian clustering algorithms implemented in STRUCTURE, InStruct and TESS. The program STRUCTURE assumes a model with K clusters, each of which being characterized by a set of allelic frequencies. Assuming HWE and linkage equilibrium among loci within clusters, the program estimates allelic frequencies in each cluster and the proportion of ancestry from the different clusters in each individual. The program InStruct is an extension of the approach implemented in STRUCTURE, relaxing the assumption of HWE within clusters. InStruct instead jointly estimates selfing rates and individual membership on the basis of selfing rates, and is therefore well suited to selfing organisms such as Microbotryum. TESS is another extension of STRUCTURE, incorporating a spatial component into the clustering algorithm, so that geographically closer individuals are a priori more likely to belong to the same cluster. This may help revealing subtle geographical structure [64]. We attempted to identify the number of clusters (K) that best described the population structure using (1) the probability of the data under the considered value of K, i.e. Ln(Pr(X|K)), and its rate of change when increasing K; and (2) the Deviation Index Criterion (DIC), i.e. a model-complexity penalized measure of how well the model fits the data.
For MvSl, the programs STRUCTURE and InStruct showed that values of DIC decreased and LnP(X|K) increased from K = 1 to 10 (Figures S2a–c), indicating that increasing the number of clusters continuously improved the fit of the model to the data. However, the variation in LnP(X|K) showed a marked break at K = 5 in STRUCTURE analyses, with a much weaker increase of probability with increasing K afterwards (Figures S2a and b). The inclusion of space in the clustering modelling, as implemented in TESS 2.3, resulted in minimal DIC values at K = 5 and K = 6 (Figure S2d). Increasing K above 5 may therefore add little information for understanding large-scale population structure in Europe, although it would likely reveal a genuine population structure, relevant at smaller scale.
The admixture proportion (α) between clusters was low, as shown by the mean α = 0.033±0.000 over all the runs between K = 2 to 15 (10 replicates for each K) in the STRUCTURE analysis. This indicates that most of the genotypes are drawn from a single cluster, with little admixture among clusters (see Figure S3 for K = 5). There is therefore almost complete lack of gene flow among clusters.
Replicates conducted for each of the three algorithms showed dominant and minor modal solutions for membership probabilities (Figure S4). However, the dominant clustering solutions recovered from the three analyses (InStruct, STRUCTURE and TESS) were highly similar (see Figure S3 for K = 5). The three methods were therefore congruent in their inference of the population structure of MvSl in Europe. The differences between the dominant and minor modal solutions most often corresponded to a genetic structure appearing at higher K values (Figure S4). For instance, the Italian cluster was assigned to the Eastern group at K = 2 in the dominant solution, and to the Western group in other simulations.
Figure 1 shows the maps of mean membership probabilities per locality for MvSl genotypes from the InStruct analysis for K = 2 to 5. At K = 2, the analyses revealed a clear West-East partitioning. Simulation of a third cluster separated the Italian genotypes from the Eastern group. At K = 4, the Western cluster was subdivided into two clusters, one with a more northern distribution (blue, called hereafter Northwestern and abbreviated as Nwestern) and the other more to the south (yellow, called hereafter Southwestern and abbreviated as Swestern). At K = 5, the Eastern group splitted into two clusters, one bordering the Balkan peninsula (red, called hereafter the Balkan cluster) and one spreading toward Eastern Europe and Russia (purple, called hereafter the Eastern cluster). When increasing K, further clusters were identified, without evidence of admixture, and corresponding to more local geographical regions: for instance the UK became isolated, and then the most eastern part of Europe (Figure S4).
Figure 1. Maps of mean membership probabilities per localities from the InStruct analysis for Microbotryum lychnidis-dioicae (MvSl) assuming 2 to 5 clusters.

We also applied a Principal Component Analysis (PCA) on the microsatellite allele frequencies, which is a multivariate approach that does not rely on any model assumptions. It instead transforms a number of possibly correlated variables into a smaller number of uncorrelated components, the first principal components accounting for as much variability in the data as possible. The PCA fully recovered the population structure inferred by the three Bayesian clustering methods, as shown by the first four PCs, which explained 35% of the total variance in allelic frequencies (Figures S5 and S6).
The clusters displayed large and significant differences in allelic frequencies (global FST = 0.38, 95% CI: [0.29–0.46], P<0.001 for all pairs of clusters). The Nwestern and Swestern clusters showed the lowest differentiation (FST = 0.24) while the Balkan and Swestern clusters were the most different (FST = 0.47). The clusters also differed significantly with respect to their genetic diversities (Table 2), with the Italian cluster displaying significantly higher gene diversity (He) than the Balkan, Eastern and Nwestern clusters (Wilcoxon Signed Rank (WSR) tests, P = 0.008, P = 0.033, and P = 0.026, respectively, Table 2). The Balkan cluster exhibited a significantly lower allelic richness (number of alleles controlling for differences in sample size) than the Eastern and Italian clusters (P = 0.016 and P = 0.003, respectively), while the others had intermediate values.
Table 2. Genetic polymorphism and spatial pattern within each cluster of Microbotryum lychnidis-dioicae (MvSl) and Microbotryum silenes-dioicae (MvSd).
| Microbotryum lychnidis-dioicae (MvSl) | Microbotryum silenes-dioicae (MvSd) | ||||||
| Cluster | NWest_B (K3) | SWest_J (K5) | Italian (K4) | Balkan (K1) | Eastern (K2) | K1 | K2 |
| Microsatellite polymorphism | |||||||
| N | 130 | 147 | 132 | 93 | 84 | 219 | 122 |
| Ar ± SE | 3.6±0.6 | 3.8±0.7 | 5.0±0.5 | 3.3±0.5 | 4.4±0.6 | 4.5±0.9 | 4.6±0.6 |
| pAr ± SE | 0.58±0.25 | 0.60±0.20 | 1.17±0.29 | 0.47±0.18 | 0.89±0.35 | 1.43±0.42 | 1.57±0.42 |
| He | 0.46±0.26 | 0.45±0.32 | 0.63±0.13 | 0.35±0.27 | 0.43±0.28 | 0.48±0.30 | 0.55±0.18 |
| FIS | 0.85 | 0.90 | 0.95 | 0.93 | 0.96 | 0.74/0.91 | 0.86/0.96 |
| Selfing rate | 0.93 | 0.95 | 0.94 | 0.91 | 0.92 | 0.92 | 0.94 |
| Spatial pattern | |||||||
| Ln(distF(1)) | 3.53 | 4.32 | 3.06 | 2.17 | 6.52 | 6.43 | 2.24 |
| nF(1) | 245 | 285 | 655 | 16 | 176 | 1931 | 1082 |
| Sp | 0.17 | 0.02 | 0.06 | 0.29 | 0.01 | 0.06 | 0.15 |
| b | −0.070 | −0.013 | −0.037 | −0.113 | −0.004 | −0.037 | −0.089 |
| P-value | <0.001 | 0.115 | 0.004 | <0.001 | 0.407 | <0.001 | <0.001 |
N: within-cluster samples size; Ar: Allelic richness; pAr: private allelic richness; He: expected heterozygosity; Selfing rate: as inferred by InStruct; the first value of FIS for MvSd is when including the marker SL19 and the second value without this marker; Ln(distF(1)) and nF(1): logarithm of the mean geographic distance between genotypes of the first distance class and number of pairs considered in this class; spatial Sp statistic within cluster; b, regression slope between the kinship coefficient and the logarithm of the geographic distance; Mantel test's P-value (H0: bobs = bexp; H1 = bobs<bexp).
Within clusters, significant isolation by distance (IBD) was detected in the Balkan, Nwestern, and Italian clusters (Table 2), indicating that genetic differentiation increased with geographic distance in these clusters. No significant IBD was detected within the Swestern and Eastern clusters (Table 2). The level of spatial structure was quantified by the Sp statistic, which accounts for variation in sampling intensities; high values of Sp are indicative of low population density and/or limited dispersal [65]. Sp values were close to 0 within the Italian, Swestern, and Eastern clusters, but were much higher within the Balkan and Nwestern clusters (Table 2).
Within-cluster selfing rates estimated from InStruct analyses were extremely high (s = 0.91±0.03 on average), in agreement with previous studies and with the high FIS values within clusters (Table 2).
A European spatial map of genetic diversity was generated by aggregating geographically close samples together on a grid, considering only grid cells where the sample size was higher than 4. The interpolated values of allelic richness showed that genetic diversity increased in the southward direction, with the highest value observed in the Italian and Iberian peninsulas and the lowest values in northern Europe (Figure 2). Such a latitudinal trend was confirmed by the highly significant negative correlation observed between latitude and allelic richness (r = −0.57, P<0.0001); no significant correlation was found between longitude and allelic richness (r = 0.036, P = 0.892). High genetic diversities were also observed in the northern half of France and along a longitudinal line separating the Eastern and Western parts of Europe (Figure 2).
Figure 2. Map of allelic richness overall loci.

Samples have been aggregated on a grid in order to make this calculation on a minimum sample size of at least 4 genotypes. Localities where the sample size was below this threshold were not considered.
We analyzed the relationships among clusters using neighbour-joining population trees, respectively based on Nei's DA distance, shared allele distance DSA, Chord's distance and Goldstein's (δµ)2 distance. As the different trees provided similar topologies, only the tree based on Nei's DA distance is presented (Figure 3). The trees suggested that the Eastern groups would have diverged first, followed by the Italian cluster and then by the Western groups. The Eastern and Western group would then have further split into two clusters each. Rough estimate of separation time between clusters can be deduced from distances between clusters assuming that the divergence between the two species occurred 400,000 yr BP [52] and assuming clocklike evolution of microsatellite markers [66]. The separation of the 5 clusters can thus be roughly estimated to have occurred between 200,000 and 350,000 yr BP (Figure 3).
Figure 3. Microsatellite distance-based Neighbour-Joining trees on intraspecific clusters for both Microbotryum lychnidis-dioicae (MvSl) and M. silenes-dioicae (MvSd).

The root of the trees was placed on the branch separating the two species. The bootstrap values above 50% are shown.
Genetic structure, genetic diversity and selfing rates in M. silenes-dioicae (MvSd)
In MvSd, multiple estimates of the genetic structure at the European scale provided confidence in existence of several distinct clusters. As for MvSl, the three Bayesian clustering analyses (InStruct, STRUCTURE and TESS) all indicated that DIC decreased and LnP(X|K) increased with increasing K (Figure S7). Again, genotype assignment probabilities were always very high, with very little admixture among clusters (mean α = 0.030±0.002 over the 140 runs from K = 2 to 15 simulated clusters; Figure S8), and were similar for the three algorithms used (data not shown). The spatial distributions of the two clusters identified at K = 2 appeared highly intermingled, with however a slight West-East trend of separation. Further clusters differentiated as K increased, but without any obvious large-scale geographical pattern (Figure 4). Similar genetic partitioning was recovered using PCA (Figure S9). The first PC accounted for 20% of the variance in the allelic frequencies and clearly separated genotypes into the same two groups as those identified using Bayesian clustering approaches at K = 2 (Figure S9). The differences in allelic frequencies between them were high, with a FST value of 0.34 (95% CI: 0.17–0.49). The successive PCs accounted for less than 11% of the total variance in allelic frequencies each and revealed the same clusters of genotypes as those observed in Bayesian clustering analyses (Figure S9).
Figure 4. Maps of mean membership probabilities per locality from the InStruct analysis for Microbotryum silenes-dioicae (MvSd), assuming 2 to 5 clusters.

The two clusters identified at K = 2 represented the only structure with a large-scale geographical pattern. The FST values between sites where the sampling was higher or equal to 10 samples showed that a very high level of genetic differentiation between clusters was observed, even in the regions where populations from different clusters were intermingled (Figure S10). Within clusters, there was no significant IBD, i.e. no significant increase in FST with geographic distance (Mantel test, P = 0.418). The two clusters showed a spatial genetic structure of a similar level to that observed in MvSl, with Sp values of 0.06 and 0.15 for the clusters 1 and 2, respectively (Table 2).
Selfing rates within each of the two clusters, inferred from Instruct, were high and similar to those in MvSl (mean selfing rates at K = 2: 0.93±0.01), consistent with the high FIS values (Table 2). The two clusters of MvSd showed genetic diversities comparable to those observed in MvSl (Table 2), and did not differ significantly from each other with respect to gene diversity (WSR test, P = 0.424) and allelic richness (WSR test, P = 0.594). In contrast to MvSl, spatially interpolated values of allelic richness increased northwards (Figure 2), although the correlation with latitude was significant only at a marginal level (r = 0.43, P = 0.073). The correlation with longitude was not significant (r = −0.008, P = 0.975).
Discussion
Mating system and local population structure
Very high selfing rates were inferred in both Microbotryum species (s = 0.91 for MvSl and s = 0.93 for MvSd), in agreement with the high deficits in heterozygotes (Text S1, Table 1). Microbotryum species are in fact known to have a selfing mating system [67], [68], but the estimations of selfing rates in natural populations inferred here are more precise and even higher than previously thought [67]. These high selfing rates appear to result both from an intrinsic preference for intra-tetrad mating [69] and from lack of outcrossing opportunities when the spores are deposited on a new host plant. The lack of outcrossing opportunity is supported by the observation that selfing rates under choice experiments (when given the opportunity to self or outcross on plants) are lower (ca. 0.70 [70]) than those inferred here in natural populations.
The marker SL19 showed extreme FIS values in MvSl and was almost fixed in the heterozygous state in MvSd. This was not particularly surprising given that Microbotryum species undergo mostly intra-tetrad mating, which can lead to an excess in heterozygosity in regions of the genome near the centromeres and on the sex chromosomes carrying the mating type locus [69], [71]. Because the mating type segregates at the first meiosis division, intra-tetrad mating automatically restores heterozygosity in all regions linked to centromeres and linked to the mating type locus, as they also segregate at the first meiotic division [69], [71].
In addition to selfing, the study of local genetic structure and diversity for both Microbotryum species across Europe revealed patterns consistent with the dynamics of a metapopulation [72]. In particular, we observed very low genetic diversity within demes, and strong differences in allele frequencies (high FST values) between demes (see text S1). Metapopulation dynamics involve frequent extinctions and recolonizations, thus creating strong genetic drift in local populations. In addition, selfing reduces the local effective population size and the frequency of gene exchange between individuals and populations, which reinforces the effects of genetic drift upon allelic frequencies [72], [73]. These results are consistent with previous population genetics and demographic studies conducted at more local scales on Microbotryum species infecting S. latifolia and S. dioica, also showing patterns consistent with metapopulation dynamics [42], [74]–[77].
Genetic structure shaped by multiple glacial refugia
From a biogeographic point of view, Europe is a large peninsula with an East-West orientation, delimited in the south by a strong barrier, the Mediterranean Sea. During glaciation epochs, many species likely went through alternating contractions and expansions of range, involving extinctions of northern populations when the ice-sheet extended southward, and spread of the southern populations from different refugial areas as the glaciation receded. Such colonization processes were likely characterized by recurrent bottlenecks that would have led to lower diversity in the northern populations compared with the southern refugia [21]. The idea that refugia were localized in three areas (Iberia, Italy, Balkans) in Europe is now well-established [10], [11], [15], [17], although increasing evidence suggest that northern and eastern refugia also existed [78]–[84].
Microbotryum lychnidis-dioicae (MvSl)
In MvSl, the strong phylogeographic structure observed at the European scale was composed of at least three genetic clusters with distributions strikingly similar to the major glacial refugia commonly recognized to have existed in Europe for many plant and animal temperate species (e.g., [14]). This pattern suggests that the pathogen likely colonized Northern Europe from at least the three main Mediterranean refugia (Iberian, Italian and Balkan).
The scenario may have been more complex, however, as the Eastern and Western clusters each further split into two groups, with divergence times of the five clusters roughly estimated between 200,000 and 350,000 yr BP. One of the eastern clusters was located north of the Balkans (mainly in Hungary and Czech Republic) and the other from Germany eastward. This pattern is consistent with colonization from distinct refugia located in the Balkans and further East in Eurasia, following a similar scenario as those reported in some animals, such as the bear Ursus actor [10], the vole Myodes glareolus [79], [80], some plant species [78], [85], [86], and also in some pathogens (e.g., [22]–[25]).
The two clusters identified in Western Europe had more diffuse geographic distributions, with a slight longitudinal partitioning. One of the clusters was distributed more towards the West of France while the second was more present in east-central France and in Eastern UK. Such a pattern may be due to the pathogen having survived in distinct regional refugia in Western Europe, from which they would have expanded their range over France and UK. Such a hypothesis is consistent with recent findings that the main glacial refugia in Europe were probably not composed of a single population, but instead could have been structured into several local refugia more or less isolated from one another (see the concept of “refugia within refugia” [87]–[91]).
While the north-south gradient in genetic diversity can be taken as a sign of range expansion from southern glacial refugia, a band of high genetic diversity was observed north of Italy and extending into Germany, as well as a hotspot of diversity in the centre of France. These areas of high of genetic diversity likely come from the colonization history of Europe by the different genetic clusters, establishing suture zones where genetic clusters meet and become intermingled. Such a pattern has been observed previously in a comparative approach of the history of colonization of 22 widespread and co-distributed European trees and shrubs [92], where hotspots of genetic diversity in the colonised ranges were found to be the result of mixed colonization from genetically isolated eastern and western European refugia [92]. The high genetic diversity found in MvSl in the Iberian peninsula also likely results from the co-occurrence of genotypes from four clusters.
Previous studies indicated that the phylogeographic pattern of the host plant S. latifolia similarly showed genetic evidence of post-glacial recolonization from Mediterranean refugia [56]. Analyses of cpDNA haplotypes revealed clear biogeographic structure in Europe, with haplotypes from Eastern and Western Europe forming divergent groups descending from haplotypes currently distributed in Iberian and Balkan Peninsulas [56]. The phylogeographic patterns in the plant S. latifolia and in its anther smut pathogen therefore seem to be congruent. In particular, the Eastern and Western clades identified in the host could correspond to the Eastern and Western genetic clusters in the pathogen MvSl. The pathogen however seems to display a finer genetic structure than its host, with particularly clear genetic evidence of an Italian glacial refugium for the pathogen, but not for the host plant (see figure 4 in [56]). More pronounced geographic structure is in fact expected in anther smut pathogens compared to their hosts, as has been observed at smaller scales [42]. This can be explained by the following observations: 1) the distribution of the pathogen is necessarily embedded within the range of its host, 2) anther smut pathogens are dispersed by the same vectors as the pollen of the plants, without being dispersed by seeds, so that their dispersal ability is lower than that of their host plants [42]. It is therefore likely that MvSl persisted in more fragmented refugia compared with its host. This highlights the potential use of pathogens as proxies for understanding host past migrations and distributions: the finding of distinct clusters in Italy and the Balkan in MvSl reveals that S. latifolia persisted in both these refugia during last glaciations, which was not obvious based solely on our current knowledge of the plant's phylogeograpy. However, statistically explicit comparative analyses linking the host and pathogen genetic polymorphisms, using comparable genetic markers, would be required to draw firm conclusions regarding correlations between the biogeographic structure of S. latifolia and MvSl (e.g. [93]).
Microbotryum silenes-dioicae (MvSd)
The anther smut pathogen MvSd also exhibited a strong genetic structure, albeit with biogeographic patterns more difficult to interpret. The two main clusters had largely intermingled distributions, with an estimated time of divergence of the same order of magnitude as observed for MvSl. The distinct clusters in MvSd could correspond to genetic groups having diverged in distinct southern refugia during the glaciations, similar to MvSl, although the locations of the putative refugia are more difficult to identify. This may be due to the restricted distribution of MvSd, constrained by ecological specificities of the host and disease: the plant S. dioica is very rare in Mediterranean regions, and even more so the disease (we did not find anther smut symptoms on any S. dioica plant in the Pyrenees despite several years of searching). On the other hand, given the more northern current distribution of the plant S. dioica compared to S. latifolia, one can alternatively speculate that its tolerance to cold temperatures [62] might have allowed the host and the disease to remain in more northern refugia, as suggested for other species adapted to cold environments [84]. This could provide an explanation of the marginally significant increase in allelic richness with latitude in MvSd, although we cannot rule out that this pattern resulted from the co-occurrence of a greater number of different clusters in the north.
The phylogeography of the host plant S. dioica based on cpDNA RFLP similarly indicated the existence of genetically distinct groups, more or less longitudinally separated, albeit with large overlap in their ranges [58]. The distribution of the common cpDNA haplotypes was suggested to result from a post-glacial expansion of S. dioica across Europe from multiple southern refugia [57], [58]. However the absence of sampling from Mediterranean peninsulas in prior studies prevents any definitive conclusion regarding the number and location of these refugia. In addition, the geographic distribution of the shared haplotypes in S. dioica and S. latifolia was consistent with a history of hybridization and introgression events, making it difficult to assess whether the present distribution of these haplotypes resulted from the recolonization history of S. dioica or S. latifolia [58].
Lack of gene flow among clusters and lack of IBD
A striking pattern observed in both Microbotryum species was the low level of admixture among genetic clusters (≤3%), suggesting almost complete lack of gene flow, despite the existence of contact zones. Such low levels of gene flow among clusters are likely influenced by the very high selfing rates in Microbotryum. High selfing rates have been invoked to explain reproductive isolation between sympatric Microbotryum species [67], [94], and there could be a similar effect in keeping the genetic clusters distinct within species. The high selfing rates also explain why increasing the number of clusters in Bayesian analyses always increased the explanatory value in describing the population genetic structure, without the appearance of admixed clusters, even for very high K values: this is because each diploid individual mostly reproduces with itself and therefore the smallest ‘panmictic unit’ may indeed be the individual. In selfing species, the genetic structure extends to a much finer scale than in outcrossing species [32], [95]. The lack of gene flow among clusters may result in addition to metapopulation dynamics and rapid expansion during post-glacial recolonization. A theoretical study [96] indeed showed that rapid growth in population size after founding events resulted in gene frequency divergence that is resistant to decay by gene exchange.
Inference for climate change and emerging infectious diseases
Large-scale congruence between the pathogens' phylogeographic patterns and those of their respective hosts indicates that their glacial refugia and migration pathways during recolonization have been similar. While this may be expected for obligate pathogens like Microbotryum species, highly dependent on their hosts for survival and using the same dispersal vectors, we interestingly found that the pathogens likely subsisted during glaciations in a more fragmented distribution, with their genetic diversity divided among a higher number of smaller refugia. Moreover, the extent of large-scale dispersal across Europe after recolonization was less for the pathogen than for its host: in particular, the clusters were much more clumped in MvSl than in S. latifolia, and footprints of refugia appeared in MvSl that were absent in S. latifolia, such as the Italian peninsula. Our findings thus indicate that vector-borne, obligate pathogens may colonize new areas following climate warming with some delay compared to their hosts, and to a lesser extent. The invasive potential of pathogens following climate change is therefore likely to depend on the obligate nature of the interactions with their host and on the dispersal modes, as could be expected.
However, once the original host and its fungal pathogen invade a geographic region, the pathogen poses a risk of emerging as an infectious disease on new host species found in that area. For instance, MvSl was introduced in the United States some time after its host plant S. latifolia, and has remained in a much more restricted geographic area [97]. Cross-species disease transmission was nevertheless documented in the United States to another non-native species, S. vulgaris, that is otherwise free of anther smut disease in the continent [98]. In Europe, we have previously detected rare events of cross-species disease transmission between S. dioica and S. latifolia and of hybridization between MvSl and MvSd that occurred after secondary contact [52]. Host shifts are frequent in fungal pathogens [4], [99], [100] in particular in Microbotryum, where co-phylogenetic analyses showed that speciation events were most often associated with host shifts [47]. This suggests that climate warming may cause emerging infectious diseases, by resulting in contacts between different potential hosts that were allopatric, even when the intrinsic dispersal capacity of the pathogens is limited and their migration pathways are constrained by those of their hosts. Climate warming can also bring into contact differentiated populations from the same species, promoting introgression between previously geographically isolated populations, which can have important and unpredictable evolutionary consequences. We showed in the present study that secondary contact between genetically differentiated clusters happened after the glaciations in MvSl, and that the highly selfing mating system was here important in preventing introgression.
The substantive contrast in phylogeography for the anther smut fungi on S. latifolia and S. dioica, which may be attributed to differences in the hosts' ecology, is also relevant for predicting the fate of infectious diseases following global warming. The redistribution of pathogens under warmer climatic conditions should indeed be highly dependent on the hosts' ecological preferences and adaptive potentials, in particular regarding the temperature and competition in new ecological communities [39]–[41].
Conclusion
In this study, we showed that high selfing rates and metapopulation dynamics in two plant pathogenic fungi had strong impact on their genetic diversity and structure. At the scale of the species' distribution ranges, the population structures in the two fungal species were quite different, likely due to differences in the ecological preferences of the two host-pathogen systems. The broadly distributed S. latifolia and its anther smut pathogen have kept clear genetic footprints of postglacial colonization from the major southern European refugia. The pathogens showed striking evidence for more numerous and localized refugia than their hosts. On the other hand, the ecological preference of the plant S. dioica for wetter and colder habitats [62] probably led to a more restricted and more northern distribution of the plant and its anther smut pathogen, and may have induced a drastic contraction of their distribution ranges with the post-glacial warming and the fragmentation of suitable habitat conditions. The European genetic structures of the anther smut fungi seem to match those of their respective hosts, with even a finer genetic structure, so that the geographic distribution of genetic variation in the pathogens may be useful to draw inferences about host phylogeography.
Beyond the interest of our study for understanding the dynamics of diseases under climate warming and the impact of host life histories on the genetic structure of pathogens, our study illustrates several important points to take into account when performing clustering genetic analyses, which are still often poorly recognized. First, several K values are often interesting to consider in clustering analyses, and it may be non-heuristic to search for a “single optimal” number of clusters. As long as increasing K does not lead to admixed clusters, the new clusters revealed by increasing K probably reveal a genuine genetic structure that may be interesting to investigate. This appears especially true in selfing species, for which the smallest panmictic cluster may be the individual.
Materials and Methods
Teliospore collection and populations
The individuals of Microbotryum analyzed in this study were collected as diploid teliospores from 187 localities on S. latifolia (n = 701) and 68 localities on S. dioica (n = 342) across Europe (Figure S10) and stored in silica gel (see Figure S1 for a detailed description of the sampling). DNA from teliospores of one flower per diseased plant was extracted for genetic analyses. Multiple infections by different genotypes are frequent in the Silene-Microbotryum system, but teliospores within a single flower originate from a single diploid individual [101].
Microsatellite genotyping
DNA was extracted as described in [77]. Teliospores were genotyped using 11 microsatellite markers following the protocol of [77] (Table 1). Among the 11 microsatellite loci used, E14, E17, E18 were described in [102], SL8, SL9, SL12, SL19, SVG5, SVG8, SVG14 described in [103], and SL5 was described in [104].
Data analyses
Sample handling
Hybridization between MvSl and MvSd has been suggested previously [105]. However, we showed recently in a study investigating the divergence process between the two species using the same dataset as in the present work that hybridization was very rare in natural populations [52]. Therefore, we investigated here the genetic polymorphism and population structure in MvSl and MvSd separately, after having removed the few inter-specific hybrids and cross-disease species transmissions from the datasets (see [52] for more details).
Descriptive statistics
The within-species genetic polymorphism at each locus was quantified using the allelic richness (Ar), the observed and unbiased expected heterozygosities (Ho and He), and the fixation index (FIS). These statistics were calculated using FSTAT 2.9.3.2 [106]. Departure from Hardy-Weinberg expectations was tested using exact tests implemented in GENEPOP 4.0 [107], [108].
Linkage disequilibrium (LD) among loci was quantified using the correlation coefficient (r2) calculated in Genetix 4.05 [109]. In order to assess the impact of mating system on the genetic structure, we calculated r2 within sampled localities that contained at least 10 genotypes, i.e. 15 out of 187 localities for MvSl (n = 210, Figure S11) and 13 out of 68 localities for MvSd (n = 197, Figure S12). Genotypic disequilibrium was tested using permutation test implemented in FSTAT 2.9.3.2. The nominal P-value of 0.05 was adjusted for multiple comparisons using a Bonferroni correction (i.e. 6.1×10−5, based on 82,500 permutations).
Population structure
We investigated population structure in MvSl and MvSd using two kinds of analyses: Bayesian model-based clustering and principal component analyses. We applied three variants of Bayesian model-based clustering algorithms. The first one, implemented in STRUCTURE 2.3 [110], [111], partitions multilocus genotypes into clusters while minimizing departure from Hardy Weinberg and linkage equilibrium (HWLE) among loci [110], [111]. The second one, implemented in the InStruct software, is a variant of the previous one specifically suited to selfing organisms [112], such as Microbotryum species (e.g. [69]–[71], [77]). InStruct relaxes the assumption of Hardy-Weinberg equilibrium through the calculation of the expected genotype frequencies on the basis of selfing rates (jointly inferred from the data) rather than as simple products of allele frequencies [112]. The third one implemented in TESS 2.3.1 [64], [113], [114] differs from the STRUCTURE algorithm in including spatially explicit prior distributions describing which sets of individuals are likely to have similar cluster membership [113]. In this approach, clusters correspond to spatially and genetically continuous units separated by small discontinuities that occur where genetic barriers are crossed. The incorporation of a spatial component into the clustering model has the potential to determine if clines provide a sensible description of the underlying pattern of variation [64].
For both STRUCTURE and TESS algorithms, we used a haploid setting because both species of Microbotryum are almost completely homozygous (Table 1). Run conditions for STRUCTURE analyses were as follows: we conducted a series of independent runs with different proposals for the number of clusters (K), testing all values from 1 to 15. Each run used 500,000 iterations after a burn-in of 250,000 iterations, using a model allowing for admixture and correlated allele frequencies. To ensure convergence of the MCMC, we performed 10 independent replicates for each value of K and checked the consistency of results visually and using the procedure implemented in the program CLUMPP 1.1.1 [115]. We used CLUMPP 1.1.1 to account for label switching. We also used CLUMPP 1.1.1 to compute with the Greedy algorithm the symmetric similarity coefficient between pairs of runs (100 random input sequences, G' statistic), in order to identify potential distinct modes among the results of independent replicate runs for each K value and to average individual assignment probabilities (q) over replicated runs showing a similar mode. Barplots were generated by the DISTRUCT program [116]. We attempted to identify the number(s) of clusters (K) that best explained the data using the probability of the data under the considered value of K, i.e. Ln(Pr(X|K)) where X are the data (Ln(D) in [117]) and its rate of change when increasing K.
InStruct run conditions were as follows: we used diploid data to jointly estimate the selfing rate within each clusters and the individual assignment probability (q) to each cluster [112]. We ran 10 independent replicates for each K, testing all values between 1 and 10. Each MCMC chain used 1,000,000 iterations after a burn-in of 200,000 iterations and a thinning interval of 50. The convergence of each run was tested using CLUMPP in a similar way to that described above for the STRUCTURE analysis, and barplots were produced using DISTRUCT. InStruct does not estimate Ln(Pr(X|K)), but it computes instead a Deviation Index Criterion (DIC), i.e. a model-complexity penalized measure of how well the model fits the data [118].
TESS run conditions were as follows: we used the conditional auto-regressive (CAR) Gaussian model of admixture with linear trend surface [64], and set the admixture parameter to α = 1 and the interaction parameter ρ = 0.6 as starting values and subsequently updated. The algorithm was run with a burn-in period of length 20,000 cycles, and estimation was performed using 30,000 additional cycles. We increased the maximal number of clusters from Kmax = 2 to Kmax = 15 (10 replicates for each value). For Kmax = 5, we performed 50 additional longer runs (burn-in 30×103, run-length 100×103, ρ = 0.6, α = 1), and we averaged the estimated admixture coefficients (Q matrix) over the 5 runs with the smallest values of the DIC (DIC = 14 819, s.d. = 9). To account for label switching among runs, we used the software CLUMPP version 1.1, whose greedy algorithm computed a symmetric similarity coefficient equal to 0.85 (100 random input sequences, G' statistic).
Finally, we applied a centred-normed Principal Component Analysis (PCA) on the microsatellite allele frequencies for each species to corroborate the clustering solution inferred by the three Bayesian clustering algorithms using a multivariate approach that does not rely on any model assumption. We conducted the analyses using ADEgenet [119] and ADE4 packages [120] in the R-environment [121].
We represented the relationships among clusters using neighbour-joining population trees, respectively based on Nei's DA distance [122], shared allele distance DSA [123], [124], Chord's distance [125] and Goldstein's (δµ)2 distance, the latter one assuming that microsatellite evolve following a Stepwise Mutation Model (SMM) model [126]. The computation of distance matrix and of bootstrapped distances was performed using PowerMarker [127], and the consensus tree was obtained and plotted using MEGA 4 [128]. As the different trees provided similar topologies, only the tree based on the Da distance of Nei et al. [122] is presented. This distance was found to perform comparatively well in estimation of population trees from microsatellite allele frequency data [129]. The root was placed between the two species.
Spatial pattern in genetic diversity
Allelic richness and private allelic richness of each genetic cluster or combination of clusters identified in the Bayesian clustering analysis were calculating using ADZE 1.0 [130]. The expected heterozygosity (He) was calculated using FSTAT 2.9.3. Variation in allelic richness across space was analysed by aggregating samples according to a grid system whose mesh size was chosen in order to have at least 4 samples per cell (132×139 km). This cut-off value was chosen as a trade-off between the sample size per cell and its spatial distribution. This led for MvSl to 66 cells, out of which 47 contained at least 4 samples, and for MvSd to 31 cells, out of which 19 had at least 4 samples. Spatial interpolation was performed using a thin plate spline method, with a smoothing parameter of λ = 0.005, as implemented in the R package ‘fields’ [121].
Isolation by distance (IBD) analyses
We used pairwise kinship coefficients between individuals (Fij) (Loiselle et al. 1995) to test for isolation by distance, as recommended for highly selfing species [65]. We calculated the average kinship coefficient for different intervals of distance ranges (10, 100, 500, 1500 and 5000 km), the slope of the correlation coefficient between individual kinship coefficients and the logarithm of geographic distance, and the significance of the slope (10,000 permutations of localities) using the software SPAGeDi 1.3 [131]. When the slope was significant, we calculated the ‘Sp’ statistic, which is the ratio
where 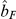 is the slope of the correlation coefficient and
is the slope of the correlation coefficient and 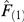 is the mean Fij between individuals belonging to a first distance interval that includes all pairs of neighbours (in our case, this distance interval corresponds to a local population). Under some assumptions, the Sp statistic is equal to the inverse of 4πDσ
2 (with D: effective population density and σ
2: average squared axial parent-offspring distance), which matches Wright's neighbourhood size, under the hypothesis of Gaussian dispersal functions. Therefore, high values of the Sp statistic are indicative of low population density or limited dispersal ability.
is the mean Fij between individuals belonging to a first distance interval that includes all pairs of neighbours (in our case, this distance interval corresponds to a local population). Under some assumptions, the Sp statistic is equal to the inverse of 4πDσ
2 (with D: effective population density and σ
2: average squared axial parent-offspring distance), which matches Wright's neighbourhood size, under the hypothesis of Gaussian dispersal functions. Therefore, high values of the Sp statistic are indicative of low population density or limited dispersal ability.
Supporting Information
Map of sampled localities for Microbotryum lychnidis-dioicae (MvSl, n = 701) and M. silenes-dioicae (MvSd, n = 342).
(0.68 MB TIF)
Estimated number of populations in Microbotryum lychnidis-dioicae (MvSl) from STRUCTURE (a and b), InStruct (c) and TESS (d) analyses. STRUCTURE analyses: (a) mean (± SD) probabilities of the data [LnPr(X|K)] over 10 Structure replicated runs plotted as a function of the putative number of clusters (K). (b) Mean variations of probabilities of the data (Δ(LnPr(X|K)) between successive K considered in STRUCTURE analyses. Deviation Index Criterion (DIC) for InStruct (c) and TESS (d) analyses.
(0.16 MB TIF)
Estimated population structure from the STRUCTURE, InStruct and TESS analyses assuming 5 clusters. Each individual is represented by a thin horizontal line divided into K coloured segments that represent the individual's estimated membership fractions in K clusters. Black lines separate individuals from different geographic areas labelled on the right. Each plot is based on the dominant clustering solution identified at that value of K.
(0.46 MB TIF)
Population structure inferred from InStruct analyses assuming 2 to 8 clusters. Each individual is represented by a thin vertical line divided into K coloured segments that represent the individual's estimated membership fractions in K clusters. Black lines separate individuals from different geographic areas. Several barplots are shown for each K and represent the distinct modal solutions observed.
(5.37 MB TIF)
Principal component analysis on microsatellite allelic frequencies of Microbotryum lychnidis-dioicae (MvSl). Scatter plots for the first four principal components are shown using a colour labelling of genotypes defined according to the membership probability to belong to the 5 identified clusters using Bayesian clustering analyses. Each genotype that received a probability above 0.7 was coloured according to the colour pattern used in Bayesian clustering, otherwise it was considered as admixed and coloured in orange.
(0.33 MB TIF)
Principal component analysis on microsatellite allelic frequencies of Microbotryum lychnidis-dioicae (MvSl). Maps for the first four principal components scores (PC1 to 4) are shown.
(0.97 MB TIF)
Principal component analysis on microsatellite allelic frequencies of Microbotryum silenes-dioicae (MvSd) for K = 2, 3 and 5. Scatter plots for the first four principal components are shown using a colour labelling of genotypes defined according to the membership probability to belong to the K identified clusters using Bayesian clustering analyses. Each genotype that received a probability above 0.7 was coloured according to the colour pattern used in Bayesian clustering, otherwise it was considered as admixed and coloured in orange.
(0.21 MB TIF)
Level of inter-sites differentiation, expressed a FST value, as a fonction of their geographic distance in Microbotryum silenes-dioicae (MvSd).
(0.05 MB TIF)
Mapped values overall loci for several descriptive statistics in Microbotryum lychnidis-dioicae (MvSl) population where the sample size was at least of 10 teliospores. The statistics shown include the sample size per locations (N), the mean allelic richness (Ar) per population, the mean observed and expected heterozygosity (Ho and He), the average number of fixed loci per populations (Nb. fixed loci), the overall loci inbreeding coefficient (FIS) per population, and the linkage disequilibrium across loci estimated using the correlation coefficient (r2) averaged across loci. We used the function quilt.plot in the R “Fields” package to plot the values on a map according to a grid system of 50×33 pixels. Locations that fall into the same grid box will have their values averaged.
(0.75 MB TIF)
Same as Figure S11, but for Microbotryum silenes-dioicae (MvSd). The locus SL19 was excluded in the calculation of Ho, He, FIS, to avoid bias related to its almost complete fixation in a heterozygous state (see the text).
(0.58 MB TIF)
Description of genetic polymorphism in Microbotryum lychnidis-dioicae (MvSl) and M. silenes-dioicae (MvSd).
(0.07 MB PDF)
Level of pairwise differentiation, estimated as FST value, among samples of M. lychnidis-dioicae where n was higher than 10 strains. All pairwise values were significantly different at p<0.0001.
(0.05 MB PDF)
Same as Table S1 but for Microbotryum silenes-dioicae (MvSd) populations. All pairwise values but one (*) were significantly different at p<0.0001.
(0.05 MB PDF)
Acknowledgments
We thank the following people for help with strain collection: Janis Antonovics, Jacqui Shykoff, Anne Thomas, Levente Kiss, Matthias Lutz, Martin Kemler, Irene Till, Jacqueline Giraud, Thierry Darras, Michel Bartoli, Julien Capelle, Jerome Enjalbert, Anders P Moller, Mickael Le Gac, Aurélie Garnier, Jane Lecomte, Lucie Salvaudon, Damien M de Vienne, Sarah Triki, Fabio Conti, Alex Widmer, Ulla Carlsson, Barbara Giles, Marcel Dorken, Giorgina Bernasoni, Dmitri Filatov, Elisabeth Fournier, Marc-André Selosse, Christian Raquin, Ilya Zakharov, Jean-Yves Hernet, Solenn Le Cadre, Sylvain Billiard, Lynda Delph, Martine Davoust, Robert Haicourt, Tunde Jankovics, Alexandra Pintye, Claire Garraud, Francois Rodolphe, Garbor Kovacs, Hans Lawton, Cvetomir Denchev, Roman Hobza, Michel Goslino, Laurent Penet, Rémy Relli, Arjen Biere, Gabor M. Kovacs, William Lévy, Amanda Gibson, Guillaume Delaunay, Bozo Frajman, Sophie Karrenberg, Thierry Hamon, Pierre-Henri Gouyon, Kalman Vanky, Valentine Métais, Honor Prentice, Anne Sophie Walker, Christine Dillman, Philippe Reignault, Guislaine Refrégier, Fabien Halkett, Sonja Yakovlev, Lorn Wolfe, Rémy Petit, Philippe Heeb, Jean-Philippe Rioult and the French Parc National des Pyrénées. We thank Stuart JE Baird and Filipe Santos for their help in data analyses.
Footnotes
The authors have declared that no competing interests exist.
TG acknowledges the grants ANR 06-BLAN-0201 and ANR 07-BDIV-003, ANR-09-0064-01, MCF a post-doctoral grant from the Ile de France Region. MEH acknowledges the grant NSF-DEB 0747222. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Anderson PK, Cunningham AA, Patel NG, Morales FJ, Epstein PR, et al. Emerging infectious diseases of plants: pathogen pollution, climate change and agrotechnology drivers. Trends Ecol Evol. 2004;19:535–544. doi: 10.1016/j.tree.2004.07.021. [DOI] [PubMed] [Google Scholar]
- 2.Harvell CD, Mitchell CE, Ward JR, Altizer S, Dobson AP, et al. Climate warming and disease risks for terrestrial and marine biota. Science. 2002;296:2158–2162. doi: 10.1126/science.1063699. [DOI] [PubMed] [Google Scholar]
- 3.Desprez-Loustau M, Robin C, Buee M, Courtecuisse R, Garbaye J, et al. The fungal dimension of biological invasions. Trends Ecol Evol. 2007;22:472–480. doi: 10.1016/j.tree.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 4.Giraud T, Gladieux P, Gavrilets S. Linking the emergence of fungal plant diseases with ecological speciation. Trends Ecol Evol. 2010;25:387–395. doi: 10.1016/j.tree.2010.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fisher MC, Koenig GL, White TJ, Taylor JW. Pathogenic clones versus environmentally driven population increase: Analysis of an epidemic of the human fungal pathogen Coccidioides immitis. J Clin Microbiol. 2000;38:807–813. doi: 10.1128/jcm.38.2.807-813.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kidd SE, Hagen F, Tscharke RL, Huynh M, Bartlett KH, et al. A rare genotype of Cryptococcus gattii caused the cryptococcosis outbreak on Vancouver Island (British Columbia, Canada). Proc Natl Acad Sci USA. 2004;101:17258–17263. doi: 10.1073/pnas.0402981101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Datta K, Bartlett KH, Baer R, Byrnes E, Galanis E, et al. Spread of Cryptococcus gattii into Pacific Northwest Region of the United States. Emerg Infect Dis. 2009;15:1185–1191. doi: 10.3201/eid1508.081384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brooks DR, Hoberg EP. How will global climate change affect parasite-host assemblages? Trends Parasitol. 2007;23:571–574. doi: 10.1016/j.pt.2007.08.016. [DOI] [PubMed] [Google Scholar]
- 9.Hewitt GM. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc. 1996;58:247–276. [Google Scholar]
- 10.Hewitt GM. Genetic consequences of climatic oscillations in the Quaternary. Phil Trans R Soc London Biol. 2004;359:183–195. doi: 10.1098/rstb.2003.1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hewitt G. Ice ages: their impact on species distributions and evolution. In: Rotschild L, Lister A, editors. Evolution on Planet Earth. Academic press; 2003. pp. 339–361. [Google Scholar]
- 12.King RA, Ferris C. Chloroplast DNA phylogeography of Alnus glutinosa (L.) Gaertn. Mol Ecol. 1998;7:1151–1161. [Google Scholar]
- 13.Taberlet P, Fumagalli L, Wust-Saucy AG, Cosson JF. Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol. 1998;7:453–464. doi: 10.1046/j.1365-294x.1998.00289.x. [DOI] [PubMed] [Google Scholar]
- 14.Hewitt G. The genetic legacy of quaternary ice ages. Nature. 2000;405:907–913. doi: 10.1038/35016000. [DOI] [PubMed] [Google Scholar]
- 15.Palme AE, Vendramin GG. Chloroplast DNA variation, postglacial recolonization and hybridization in hazel, Corylus avellana. Mol Ecol. 2002;11:1769–1780. doi: 10.1046/j.1365-294x.2002.01581.x. [DOI] [PubMed] [Google Scholar]
- 16.Petit RJ, Brewer S, Bordacs S, Burg K, Cheddadi R, et al. Identification of refugia and post-glacial colonisation routes of European white oaks based on chloroplast DNA and fossil pollen evidence. Forest Ecol Manag. 2002;156:49–74. [Google Scholar]
- 17.Francois O, Blum MGB, Jakobsson M, Rosenberg NA. Demographic history of European populations of Arabidopsis thaliana. PLoS Genet. 2008;4:e1000075. doi: 10.1371/journal.pgen.1000075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ibrahim KM, Nichols RA, Hewitt GM. Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity. 1996;77:282–291. [Google Scholar]
- 19.Petit RJ, Pineau E, Demesure B, Bacilieri R, Ducousso A, et al. Chloroplast DNA footprints of postglacial recolonization by oaks. Proc Natl Acad Sci USA. 1997;94:9996–10001. doi: 10.1073/pnas.94.18.9996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Widmer A, Lexer C. Glacial refugia: sanctuaries for allelic richness, but not for gene diversity. Trends Ecol Evol. 2001;16:267–269. doi: 10.1016/s0169-5347(01)02163-2. [DOI] [PubMed] [Google Scholar]
- 21.Excoffier L, Foll M, Petit RJ. Genetic Consequences of Range Expansions. Ann Rev Ecol Syst. 2008;40:481–501. [Google Scholar]
- 22.Nieberding CM, Durette-Desset MC, Vanderpoorten A, Casanova JC, Ribas A, et al. Geography and host biogeography matter for understanding the phylogeography of a parasite. Mol Phylogenet Evol. 2008;47:538–554. doi: 10.1016/j.ympev.2008.01.028. [DOI] [PubMed] [Google Scholar]
- 23.Rich KA, Thompson JN, Fernandez CC. Diverse historical processes shape deep phylogeographical divergence in the pollinating seed parasite Greya politella. Mol Ecol. 2008;17:2430–2448. doi: 10.1111/j.1365-294X.2008.03754.x. [DOI] [PubMed] [Google Scholar]
- 24.Amico GC, Nickrent DL. Population structure and phylogeography of the mistletoes Tristerix corymbosus and T. aphyllus (Loranthaceae) using chloroplast DNA sequence variation. Am J Bot. 2009;96:1571–1580. doi: 10.3732/ajb.0800302. [DOI] [PubMed] [Google Scholar]
- 25.Nakao M, Xiao N, Okamoto M, Yanagida T, Sako Y, et al. Geographic pattern of genetic variation in the fox tapeworm Echinococcus multilocularis. Parasitol Int. 2009;58:384–389. doi: 10.1016/j.parint.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 26.Thrall PH, Burdon JJ. The spatial scale of pathogen dispersal: Consequences for disease dynamics and persistence. Evol Ecol Res. 1999;1:681–701. [Google Scholar]
- 27.Grenfell BT, Pybus OG, Gog JR, Wood JLN, Daly JM, et al. Unifying the epidemiological and evolutionary dynamics of pathogens. Science. 2004;303:327–332. doi: 10.1126/science.1090727. [DOI] [PubMed] [Google Scholar]
- 28.Miura O, Torchin ME, Kuris AM, Hechinger RF, Chiba S. Introduced cryptic species of parasites exhibit different invasion pathways. Proc Natl Acad Sci USA. 2006;103:19818–19823. doi: 10.1073/pnas.0609603103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reed DL, Smith VS, Hammond SL, Rogers AR, Clayton DH. Genetic analysis of lice supports direct contact between modern and archaic humans. PLoS Biol. 2004;2:e340. doi: 10.1371/journal.pbio.0020340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whiteman NK, Parker PG. Using parasites to infer host population history: a new rationale for parasite conservation. Animal Conservation. 2005;8:175–181. [Google Scholar]
- 31.Wirth T, Meyer A, Achtman M. Deciphering host migrations and origins by means of their microbes. Mol Ecol. 2005;14:3289–3306. doi: 10.1111/j.1365-294X.2005.02687.x. [DOI] [PubMed] [Google Scholar]
- 32.Criscione CD, Cooper B, Blouin MS. Parasite genotypes identify source populations of migratory fish more accurately than fish genotypes. Ecology. 2006;87:823–828. doi: 10.1890/0012-9658(2006)87[823:pgispo]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 33.Nieberding CM, Olivieri I. Parasites: proxies for host genealogy and ecology? Trends Ecol Evol. 2007;22:156–165. doi: 10.1016/j.tree.2006.11.012. [DOI] [PubMed] [Google Scholar]
- 34.Whiteman NK, Kimball RT, Parker PG. Co-phylogeography and comparative population genetics of the threatened Galapagos hawk and three ectoparasite species: ecology shapes population histories within parasite communities. Mol Ecol. 2007;16:4759–4773. doi: 10.1111/j.1365-294X.2007.03512.x. [DOI] [PubMed] [Google Scholar]
- 35.Blakeslee AMH, Byers JE, Lesser MP. Solving cryptogenic histories using host and parasite molecular genetics: the resolution of Littorina littorea's North American origin. Mol Ecol. 2008;17:3684–3696. doi: 10.1111/j.1365-294X.2008.03865.x. [DOI] [PubMed] [Google Scholar]
- 36.Bruyndonckx N, Isabelle H, Philippe C, Gerald K. Spatio-temporal population genetic structure of the parasitic mite Spinturnix bechstein is shaped by its own demography and the social system of its bat host. Mol Ecol. 2009;18:3581–3592. doi: 10.1111/j.1365-294X.2009.04299.x. [DOI] [PubMed] [Google Scholar]
- 37.Bruyndonckx N, Biollaz F, Dubey S, Goudet J, Christe P. Mites as biological tags of their hosts. Mol Ecol. 2010;19:2770–2778. doi: 10.1111/j.1365-294X.2010.04699.x. [DOI] [PubMed] [Google Scholar]
- 38.Barrett LG, Thrall PH, Burdon JJ, Linde CC. Life history determines genetic structure and evolutionary potential of host-parasite interactions. Trends Ecol Evol. 2008;23:678–685. doi: 10.1016/j.tree.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hood ME, Mena-Alí JI, Gibson AK, Oxelman B, Giraud T, et al. Distribution of the anther-smut pathogen Microbotryum on species of the Caryophyllaceae. New Phytologist. 2010;187:217–229. doi: 10.1111/j.1469-8137.2010.03268.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fargette D, Konate G, Fauquet C, Muller E, Peterschmitt M, et al. Molecular ecology and emergence of tropical plant viruses. Ann Rev Phytopathol. 2006;44:235–260. doi: 10.1146/annurev.phyto.44.120705.104644. [DOI] [PubMed] [Google Scholar]
- 41.Wilson DJ, Falush D, McVean G. Germs, genomes and genealogies. Trends Ecol Evol. 2005;20:39–45. doi: 10.1016/j.tree.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 42.Delmotte F, Bucheli E, Shykoff JA. Host and parasite population structure in a natural plant-pathogen system. Heredity. 1999;82:300–308. doi: 10.1038/sj.hdy.6884850. [DOI] [PubMed] [Google Scholar]
- 43.Lutz M, Goker M, Piatek M, Kemler M, Begerow D, et al. Anther smuts of Caryophyllaceae: molecular characters indicate host-dependent species delimitation. Mycol Prog. 2005;4:225–238. [Google Scholar]
- 44.Kemler M, Goker M, Oberwinkler F, Begerow D. Implications of molecular characters for the phylogeny of the Microbotryaceae (Basidiomycota: Urediniomycetes). BMC Evol Biol. 2006;6:35. doi: 10.1186/1471-2148-6-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Le Gac M, Hood ME, Giraud T. Evolution of reproductive isolation within a parasitic fungal complex. Evolution. 2007;61:1781–1787. doi: 10.1111/j.1558-5646.2007.00144.x. [DOI] [PubMed] [Google Scholar]
- 46.Le Gac M, Hood ME, Fournier E, Giraud T. Phylogenetic evidence of host-specific cryptic species in the anther smut fungus. Evolution. 2007;61:15–26. doi: 10.1111/j.1558-5646.2007.00002.x. [DOI] [PubMed] [Google Scholar]
- 47.Refrégier G, Le Gac M, Jabbour F, Widmer A, Hood ME, et al. Cophylogeny of the anther smut fungi and their Caryophyllaceous hosts: Prevalence of host shifts and Importance of delimiting parasite species. BMC Evol Biol. 2008;8:100. doi: 10.1186/1471-2148-8-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sloan D, Giraud T, Hood M. Maximized virulence in a sterilizing pathogen: the anther-smut fungus and its co-evolved hosts. J Evol Biol. 2008;21:1544–1554. doi: 10.1111/j.1420-9101.2008.01604.x. [DOI] [PubMed] [Google Scholar]
- 49.de Vienne DM, Refrégier G, Hood M, Guigue A, Devier B, et al. Hybrid sterility and inviability in the parasitic fungal species complex Microbotryum. J Evol Biol. 2009;22:683–698. doi: 10.1111/j.1420-9101.2009.01702.x. [DOI] [PubMed] [Google Scholar]
- 50.de Vienne DM, Hood ME, Giraud T. Phylogenetic determinants of potential host shifts in fungal pathogens. J Evol Biol. 2009;22:2532–2541. doi: 10.1111/j.1420-9101.2009.01878.x. [DOI] [PubMed] [Google Scholar]
- 51.Denchev C, Giraud T, Hood M. Three new species of anthericolous smut fungi on Caryophyllaceae. Mycologia Balcanica. 2009;6:79–84. [Google Scholar]
- 52.Gladieux P, Vercken E, Fontaine M, Hood ME, Jonot O, et al. Maintenance of fungal pathogen species that are specialized to different hosts: allopatric divergence and introgression through secondary contact. Mol Biol Evol. In press. 2010 doi: 10.1093/molbev/msq235. [DOI] [PubMed] [Google Scholar]
- 53.Thrall P, Jarosz A. Host-pathogen dynamics in experimental populations of Silene alba and Ustilago violacea. II. Experimental tests on theoretical models. J Ecol. 1994;82:561–570. [Google Scholar]
- 54.Thrall P, Jarosz A. Host-pathogen dynamics in experimental populations of Silene alba and Ustilago violacea. I. Ecological and genetic determinants of disease spread. J Ecol. 1994;82:549–559. [Google Scholar]
- 55.Bernasconi G, Antonovics J, Biere A, Charlesworth D, Delph L, et al. Silene as a model system in ecology and evolution. Heredity. 2009;103:5–14. doi: 10.1038/hdy.2009.34. [DOI] [PubMed] [Google Scholar]
- 56.Taylor D, Keller S. Historical range expansion determined the phylogenetic diversity introduced during contemporary species invasion. Evolution. 2007;61:334–345. doi: 10.1111/j.1558-5646.2007.00037.x. [DOI] [PubMed] [Google Scholar]
- 57.Hathaway L, Malm JU, Prentice HC. Geographically congruent large-scale patterns of plastid haplotype variation in the European herbs Silene dioica and S. latifolia (Caryophyllaceae). Bot J Linn Soc. 2009;161:153–170. [Google Scholar]
- 58.Prentice HC, Malm JU, Hathaway L. Chloroplast DNA variation in the European herb Silene dioica (red campion): postglacial migration and interspecific introgression. Plant Syst Evol. 2008;272:23–37. [Google Scholar]
- 59.Friedrich H. Caryophyllaceae. In: Rechinger KH, editor. Illustrierte Flora von Mitteleuropa. Hamburg: Parey; 1979. pp. 763–1182. [Google Scholar]
- 60.Baker HG. Melandrium album (Mill) Garcke. J Ecol. 1947;35:274–282. [Google Scholar]
- 61.Baker HG. Melandrium dioicum (L. Emend) Coss. & Germ. J Ecol. 1947;35:283–292. doi: 10.1038/168747a0. [DOI] [PubMed] [Google Scholar]
- 62.Karrenberg S, Favre A. Genetic and ecological differenciation in hybridizing campions Silene dioica and S. latifolia. Evolution. 2008;62:763–773. doi: 10.1111/j.1558-5646.2008.00330.x. [DOI] [PubMed] [Google Scholar]
- 63.Bucheli E, Gautschi B, Shykoff JA. Differences in population structure of the anther smut fungus Microbotryum violaceum on two closely related host species, Silene latifolia and S. dioica. Mol Ecol. 2001;10:285–294. doi: 10.1046/j.1365-294x.2001.01146.x. [DOI] [PubMed] [Google Scholar]
- 64.Durand E, Jay F, Gaggiotti OE, Francois O. Spatial Inference of Admixture Proportions and Secondary Contact Zones. Mol Biol Evol. 2009;26:1963–1973. doi: 10.1093/molbev/msp106. [DOI] [PubMed] [Google Scholar]
- 65.Vekemans X, Hardy OJ. New insights from fine-scale spatial genetic structure analyses in plant populations. Mol Ecol. 2004;13:921–35. doi: 10.1046/j.1365-294x.2004.02076.x. [DOI] [PubMed] [Google Scholar]
- 66.Sun JX, Mullikin JC, Patterson N, Reich DE. Microsatellites Are Molecular Clocks That Support Accurate Inferences about History. Mol Biol Evol. 2009;26:1017–1027. doi: 10.1093/molbev/msp025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Giraud T, Yockteng R, Lopez-Villavicencio M, Refregier G, Hood ME. Mating system of the anther smut fungus, Microbotryum violaceum: selfing under heterothallism. Eukaryotic Cell. 2008;7:765–775. doi: 10.1128/EC.00440-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Granberg A, Carlsson-Graner U, Arnqvist P, Giles BE. Variation in breeding system traits within and among populations of Microbotryum violaceum on Silene dioica. Int J Plant Sci. 2008;169:293–303. [Google Scholar]
- 69.Hood ME, Antonovics J. Intratetrad mating, heterozygosity, and the maintenance of deleterious alleles in Microbotryum violaceum ( = Ustilago violacea). Heredity. 2000;85:231–241. doi: 10.1046/j.1365-2540.2000.00748.x. [DOI] [PubMed] [Google Scholar]
- 70.Giraud T, Jonot O, Shykoff JA. Selfing propensity under choice conditions in a parasitic fungus, Microbotryum violaceum, and parameters influencing infection success in artificial inoculations. Int J Plant Sci. 2005;166:649–657. [Google Scholar]
- 71.Hood ME, Antonovics JA. Mating within the meiotic tetrad and the maintenance of genomic heterozygosity. Genetics. 2004;166:1751–1759. doi: 10.1534/genetics.166.4.1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Charlesworth D. Effects of inbreeding on the genetic diversity of populations. Phil Trans R Soc London Biol. 2003;358:1051–1070. doi: 10.1098/rstb.2003.1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pannell JR, Charlesworth B. Effects of metapopulation processes on measures of genetic diversity. Phil Trans R Soc London Biol. 2000;355:1851–1864. doi: 10.1098/rstb.2000.0740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Thrall PH, Antonovics J. Theoretical and empirical studies of metapopulations: population and genetic dynamics of the Silene - Ustilago system. Can J Bot. 1995;73:S1249–S1258. [Google Scholar]
- 75.Giles BE, Goudet J. Genetic differentiation in Silene dioica metapopulations: Estimation of spatiotemporal effects in a successional plant species. Am Nat. 1997;149:507–526. [Google Scholar]
- 76.Antonovics J. Long term study of a plant-pathogen metapopulation. In: Hanski I, Gaggiotti O, editors. Ecology, Genetics, and Evolution of Metapopulations. Amsterdam: Academic Press; 2004. pp. 471–488. [Google Scholar]
- 77.Giraud T. Patterns of within population dispersal and mating of the fungus Microbotryum violaceum parasitising the plant Silene latifolia. Heredity. 2004;93:559–65. doi: 10.1038/sj.hdy.6800554. [DOI] [PubMed] [Google Scholar]
- 78.Willis KJ, van Andel TH. Trees or no trees? The environments of central and eastern Europe during the Last Glaciation. Quat Sci Rev. 2004;23:2369–2387. [Google Scholar]
- 79.Deffontaine V, Libois R, Kotlik P, Sommer R, Nieberding C, et al. Beyond the Mediterranean peninsulas: evidence of central European glacial refugia for a temperate forest mammal species, the bank vole (Clethrionomys glareolus). Mol Ecol. 2005;14:1727–39. doi: 10.1111/j.1365-294X.2005.02506.x. [DOI] [PubMed] [Google Scholar]
- 80.Kotlik P, Deffontaine V, Mascheretti S, Zima J, Michaux J, et al. A northern glacial refugium for bank voles (Clethrionomys glareolus). Proc Natl Acad Sci USA. 2006;103:14860–14864. doi: 10.1073/pnas.0603237103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bhagwat SA, Willis KJ. Species persistence in northerly glacial refugia of Europe: a matter of chance or biogeographical traits? J Biogeography. 2008;35:464–482. [Google Scholar]
- 82.Provan J, Bennett K. Phylogeographic insights into cryptic glacial refugia. Trends Ecol Evol. 2008;23:564–571. doi: 10.1016/j.tree.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 83.Binney HA, Willis KJ, Edwards ME, Bhagwat SA, Anderson PM, et al. The distribution of late-Quaternary woody taxa in northern Eurasia: evidence from a new macrofossil database. Quat Sci Rev. 2009;28:2445–2464. [Google Scholar]
- 84.Stewart JR, Lister AM, Barnes I, Dalen L. Refugia revisited: individualistic responses of species in space and time. Proc R Soc B. 2010;277:661–671. doi: 10.1098/rspb.2009.1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Willis KJ, Niklas KJ. The role of Quaternary environmental change in plant macroevolution: the exception or the rule? Phil Trans R Soc London Biol. 2004;359:159–172. doi: 10.1098/rstb.2003.1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Aspock H. Postglacial formation and fluctuations of the biodiversity of Central Europe in the light of climate change. Parasitol Res. 2008;103:S7–S10. doi: 10.1007/s00436-008-1057-6. [DOI] [PubMed] [Google Scholar]
- 87.Gómez A, Lunt D. Refugia within refugia: Patterns of phylogeographic concordance in the Iberian Peninsula. In: Weiss S, Ferrand N, editors. Phylogeography of Southern European Refugia. Dordrecht, The Netherlands: Springer; 2007. pp. 155–188. [Google Scholar]
- 88.Ursenbacher S, Schweiger S, Tomovic L, Crnobrnja-Isailovic J, Fumagalli L, et al. Molecular phylogeography of the nose-horned viper (Vipera ammodytes, Linnaeus (1758)): Evidence for high genetic diversity and multiple refugia in the Balkan peninsula. Mol Phylogenet Evol. 2008;46:1116–1128. doi: 10.1016/j.ympev.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 89.Deffontaine V, Renaud S, Fontaine M, Quéré J, Libois R, et al. A relic bank vole lineage in the French Basque country highlights the biogeographical history of Pyrenean mountains in Europe. Mol Ecol. 2009;18:2489–2502. doi: 10.1111/j.1365-294X.2009.04162.x. [DOI] [PubMed] [Google Scholar]
- 90.Grill A, Amori G, Aloise G, Lisi I, Tosi G, et al. Molecular phylogeography of European Sciurus vulgaris: refuge within refugia? Mol Ecol. 2009;18:2687–2699. doi: 10.1111/j.1365-294X.2009.04215.x. [DOI] [PubMed] [Google Scholar]
- 91.Previsic A, Walton C, Kucinic M, Mitrikeski PT, Kerovec M. Pleistocene divergence of Dinaric Drusus endemics (Trichoptera, Limnephilidae) in multiple microrefugia within the Balkan Peninsula. Mol Ecol. 2009;18:634–647. doi: 10.1111/j.1365-294X.2008.04046.x. [DOI] [PubMed] [Google Scholar]
- 92.Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, et al. Glacial refugia: Hotspots but not melting pots of genetic diversity. Science. 2003;300:1563–1565. doi: 10.1126/science.1083264. [DOI] [PubMed] [Google Scholar]
- 93.Hickerson MJ, Stahl EA, Lessios HA. Test for simultaneous divergence using approximate Bayesian computation. Evolution. 2006;60:2435–53. [PubMed] [Google Scholar]
- 94.Giraud T, Refregier G, de Vienne DM, Le Gac MA, Hood ME. Speciation in fungi. Fungal Genet Biol. 2008;45:791–802. doi: 10.1016/j.fgb.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 95.Tatarenkov A, Gao H, Mackiewicz M, Taylor DS, Turner BJ, et al. Strong population structure despite evidence of recent migration in a selfing hermaphroditic vertebrate, the mangrove killifish (Kryptolebias marmoratus). Mol Ecol. 2007;16:2701–2711. doi: 10.1111/j.1365-294X.2007.03349.x. [DOI] [PubMed] [Google Scholar]
- 96.Boileau MG, Hebert PDN, Schwartz SS. Non-equilibrium gene frequency divergence: persistent founder effects in natural populations. J Evol Biol. 1992;5:25–39. [Google Scholar]
- 97.Antonovics J, Hood ME, Thrall PH, Abrams JY, Duthie GM. Herbarium studies on the distribution of anther-smut fungus (Microbotryum violaceum) and Silene species (Caryophyllaceae) in the eastern United States. Am J Bot. 2003;90:1522–1531. doi: 10.3732/ajb.90.10.1522. [DOI] [PubMed] [Google Scholar]
- 98.Hood ME, Antonovics J, Heishman H. Karyotypic similarity identifies multiple host-shifts of a pathogenic fungus in natural populations. Infect Genet Evol. 2003;2:167–172. doi: 10.1016/s1567-1348(02)00154-5. [DOI] [PubMed] [Google Scholar]
- 99.Slippers B, Stenlid J, Wingfield MJ. Emerging pathogens: fungal host jumps following anthropogenic introduction. Trends Ecol Evol. 2005;20:420–421. doi: 10.1016/j.tree.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 100.Tellier A, de VIenne DM, Giraud T, Hood ME, Refrégier G. 37-77. NY: Nova Science Publishers; 2010. Theory and examples of reciprocal influence between hosts and pathogens, from short-term to long term interactions: coevolution, cospeciation and pathogen speciation following host shifts. In: Host-Pathogen Interactions: Genetics, Immunology and Physiology. [Google Scholar]
- 101.Lopez-Villavicencio M, Jonot O, Coantic A, Hood ME, Enjalbert J, et al. Multiple infections by the anther smut pathogen are frequent and involve related strains. PLoS Pathogens. 2007;3:e176. doi: 10.1371/journal.ppat.0030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bucheli E, Gautschi B, Shykoff JA. Isolation and characterization of microsatellite loci in the anther smut fungus Microbotryum violaceum. Mol Ecol. 1998;7:665–666. [PubMed] [Google Scholar]
- 103.Giraud T, Yockteng R, Marthey S, Chiapello H, Jonot O, et al. Isolation of 60 polymorphic microsatellite loci in EST libraries of four sibling species of the phytopathogenic fungal complex Microbotryum. Mol Ecol Res. 2008;8:387–392. doi: 10.1111/j.1471-8286.2007.01967.x. [DOI] [PubMed] [Google Scholar]
- 104.Refrégier G, Hood ME, Giraud T. No evidence of reproductive character displacement between two sister fungal species causing anther smut disease in Silene. Int J Plant Sci. 2010;171:847–859. [Google Scholar]
- 105.van Putten WF, Biere A, van Damme JM. Host-related genetic differentiation in the anther smut fungus Microbotryum violaceum in sympatric, parapatric and allopatric populations of two host species Silene latifolia and S. dioica. J Evol Biol. 2005;18:203–12. doi: 10.1111/j.1420-9101.2004.00771.x. [DOI] [PubMed] [Google Scholar]
- 106.Goudet J. FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). 2001. Available from http://www2.unil.ch/popgen/softwares/fstat.htm.
- 107.Raymond M, Rousset F. GENEPOP (Version 1.2): Population genetics software for exact tests and eucumenism. J Hered. 1995;86:248–249. [Google Scholar]
- 108.Rousset F. genepop'007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Res. 2008;8:103–106. doi: 10.1111/j.1471-8286.2007.01931.x. [DOI] [PubMed] [Google Scholar]
- 109.Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F. Genetix 4.02, Logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, population, interaction, CNRS UMR 5000, Université de Montpellier II (France) 2001 [Google Scholar]
- 110.Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Falush D, Stephens M, Pritchard J. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003;164:1567–1587. doi: 10.1093/genetics/164.4.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Gao H, Williamson S, Bustamante CD. A Markov Chain Monte Carlo Approach for Joint Inference of Population Structure and Inbreeding Rates From Multilocus Genotype Data. Genetics. 2007;176:1635–1651. doi: 10.1534/genetics.107.072371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Francois O, Ancelet S, Guillot G. Bayesian clustering using hidden Markov random fields in spatial population genetics. Genetics. 2006;174:805–816. doi: 10.1534/genetics.106.059923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Chen C, Durand E, Forbes F, Francois O. Bayesian clustering algorithms ascertaining spatial population structure: a new computer program and a comparison study. Mol Ecol Notes. 2007;7:747–756. [Google Scholar]
- 115.Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–6. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- 116.Rosenberg N. DISTRUCT: a program for graphical display for population structure. Mol Ecol Notes. 2004;4:137–138. [Google Scholar]
- 117.Pritchard JK, Wen W. Documentation for Structure software: Version. 2004;2 [Google Scholar]
- 118.Spiegelhalter DJ, Best NG, Carlin BP, Linde AVD. Bayesian measures of model complexity and fit. J Roy Stat Soc B. 2002;64:583–639. [Google Scholar]
- 119.Jombart T. Package ‘adegenet’. a R package for the multivariate analysis of genetic markers. 2010. http://cran.r-project.org/web/packages/adegenet/index.html. [DOI] [PubMed]
- 120.Chessel D, Dufour A, Dray S. Package ‘ade4’. Analysis of Ecological Data: Exploratory and Euclivian methods in Environmental sciences. 2009. http://cran.r-project.org/web/packages/ade4/index.html.
- 121.R Development Core Team. R: A Language and Environment for Statistical Computing 2010 [Google Scholar]
- 122.Nei M, Tajima F, Tateno Y. Accuracy of estimated phylogenetic trees from molecular data. J Mol Evol. 1983;19:153–170. doi: 10.1007/BF02300753. [DOI] [PubMed] [Google Scholar]
- 123.Chakraborty R, Jin L. Determination of relatedness between individuals using DNA fingerprinting. Hum Biol. 1993;65:875–895. [PubMed] [Google Scholar]
- 124.Bowcock AM, Ruiz-linares A, Tomfohrde J, Minch E, Kidd JR, et al. High-resolution of human evolutionary trees with polymorphic microsatellites. Nature. 1994;368:455–457. doi: 10.1038/368455a0. [DOI] [PubMed] [Google Scholar]
- 125.Cavalli-Sforza L, Edwards A. Phylogenetic analysis: Models and estimation procedures. Am J Hum Genet. 1967;19:233–257. [PMC free article] [PubMed] [Google Scholar]
- 126.Goldstein DB, Ruiz Linares A, Cavalli-Sforza LL, Feldman MW. Genetic absolute dating based on microsatellites and the origin of modern humans. Proc Natl Acad Sci USA. 1995;92:6723–6727. doi: 10.1073/pnas.92.15.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu K, Muse SV. PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics. 2005;21:2128–2129. doi: 10.1093/bioinformatics/bti282. [DOI] [PubMed] [Google Scholar]
- 128.Tamura K, Dudley J, Nei M, Kumar S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol Biol Evol. 2007;24:1596–1599. doi: 10.1093/molbev/msm092. [DOI] [PubMed] [Google Scholar]
- 129.Takezaki N, Nei M. Genetic distances and reconstruction of phylogenetic trees from microsatellite DNA. Genetics. 1996;144:389–399. doi: 10.1093/genetics/144.1.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Szpiech ZA, Jakobsson M, Rosenberg NA. ADZE: A rarefaction approach for counting alleles private to combinations of populations. Bioinformatics. 2008;24:2498–2504. doi: 10.1093/bioinformatics/btn478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hardy OJ, Vekemans X. SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes. 2002;2:618–620. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Map of sampled localities for Microbotryum lychnidis-dioicae (MvSl, n = 701) and M. silenes-dioicae (MvSd, n = 342).
(0.68 MB TIF)
Estimated number of populations in Microbotryum lychnidis-dioicae (MvSl) from STRUCTURE (a and b), InStruct (c) and TESS (d) analyses. STRUCTURE analyses: (a) mean (± SD) probabilities of the data [LnPr(X|K)] over 10 Structure replicated runs plotted as a function of the putative number of clusters (K). (b) Mean variations of probabilities of the data (Δ(LnPr(X|K)) between successive K considered in STRUCTURE analyses. Deviation Index Criterion (DIC) for InStruct (c) and TESS (d) analyses.
(0.16 MB TIF)
Estimated population structure from the STRUCTURE, InStruct and TESS analyses assuming 5 clusters. Each individual is represented by a thin horizontal line divided into K coloured segments that represent the individual's estimated membership fractions in K clusters. Black lines separate individuals from different geographic areas labelled on the right. Each plot is based on the dominant clustering solution identified at that value of K.
(0.46 MB TIF)
Population structure inferred from InStruct analyses assuming 2 to 8 clusters. Each individual is represented by a thin vertical line divided into K coloured segments that represent the individual's estimated membership fractions in K clusters. Black lines separate individuals from different geographic areas. Several barplots are shown for each K and represent the distinct modal solutions observed.
(5.37 MB TIF)
Principal component analysis on microsatellite allelic frequencies of Microbotryum lychnidis-dioicae (MvSl). Scatter plots for the first four principal components are shown using a colour labelling of genotypes defined according to the membership probability to belong to the 5 identified clusters using Bayesian clustering analyses. Each genotype that received a probability above 0.7 was coloured according to the colour pattern used in Bayesian clustering, otherwise it was considered as admixed and coloured in orange.
(0.33 MB TIF)
Principal component analysis on microsatellite allelic frequencies of Microbotryum lychnidis-dioicae (MvSl). Maps for the first four principal components scores (PC1 to 4) are shown.
(0.97 MB TIF)
Principal component analysis on microsatellite allelic frequencies of Microbotryum silenes-dioicae (MvSd) for K = 2, 3 and 5. Scatter plots for the first four principal components are shown using a colour labelling of genotypes defined according to the membership probability to belong to the K identified clusters using Bayesian clustering analyses. Each genotype that received a probability above 0.7 was coloured according to the colour pattern used in Bayesian clustering, otherwise it was considered as admixed and coloured in orange.
(0.21 MB TIF)
Level of inter-sites differentiation, expressed a FST value, as a fonction of their geographic distance in Microbotryum silenes-dioicae (MvSd).
(0.05 MB TIF)
Mapped values overall loci for several descriptive statistics in Microbotryum lychnidis-dioicae (MvSl) population where the sample size was at least of 10 teliospores. The statistics shown include the sample size per locations (N), the mean allelic richness (Ar) per population, the mean observed and expected heterozygosity (Ho and He), the average number of fixed loci per populations (Nb. fixed loci), the overall loci inbreeding coefficient (FIS) per population, and the linkage disequilibrium across loci estimated using the correlation coefficient (r2) averaged across loci. We used the function quilt.plot in the R “Fields” package to plot the values on a map according to a grid system of 50×33 pixels. Locations that fall into the same grid box will have their values averaged.
(0.75 MB TIF)
Same as Figure S11, but for Microbotryum silenes-dioicae (MvSd). The locus SL19 was excluded in the calculation of Ho, He, FIS, to avoid bias related to its almost complete fixation in a heterozygous state (see the text).
(0.58 MB TIF)
Description of genetic polymorphism in Microbotryum lychnidis-dioicae (MvSl) and M. silenes-dioicae (MvSd).
(0.07 MB PDF)
Level of pairwise differentiation, estimated as FST value, among samples of M. lychnidis-dioicae where n was higher than 10 strains. All pairwise values were significantly different at p<0.0001.
(0.05 MB PDF)
Same as Table S1 but for Microbotryum silenes-dioicae (MvSd) populations. All pairwise values but one (*) were significantly different at p<0.0001.
(0.05 MB PDF)