

Abstract
Background
Sirt1, a class III histone deacetylase, retards aging and protects the heart from oxidative stress. We here examined whether Sirt1 is protective against myocardial ischemia/reperfusion (I/R).
Methods and Results
Protein and mRNA expression of Sirt1 is significantly reduced by I/R. Cardiac specific Sirt1 -/- mice exhibited a significant increase (44 ± 5 % vs. 15 ± 5 %, p=0.01) in the size of myocardial infarction/area at risk (MI/AAR). In transgenic mice with cardiac specific overexpression of Sirt1 (Tg-Sirt1), both MI/AAR (15 ± 4% vs. 36 ± 8%, p = 0.004) and TUNEL positive nuclei (4 ± 3 % vs. 10 ± 1%, p< 0.003) were significantly reduced compared to in non-transgenic mice. In Langendorff perfused hearts, the functional recovery during reperfusion was significantly greater in Tg-Sirt1 than in non-transgenic mice. Sirt1 positively regulates expression of pro-survival molecules, including MnSOD, thioredoxin1, and Bcl-xL, whereas it negatively regulates the pro-apoptotic molecules Bax and cleaved caspase-3. The level of oxidative stress after I/R, as evaluated by 8-OHdG staining, was negatively regulated by Sirt1. Sirt1 stimulates the transcriptional activity of FoxO1, which in turn plays an essential role in mediating Sirt1-induced upregulation of MnSOD and suppression of oxidative stress in cardiac myocytes. Sirt1 plays an important role in mediating I/R-induced increases in the nuclear localization of FoxO1 in vivo.
Conclusions
These results suggest that Sirt1 protects the heart from I/R injury through upregulation of anti-oxidants and downregulation of pro-apoptotic molecules through activation of FoxO and decreases in oxidative stress.
Keywords: Sirt1, ischemia, reperfusion, longevity factor, apoptosis, oxidative stress, FoxO
Introduction
Silent information regulator 1, or Sirt1, is a member of the Sirtuin family of class III histone deacetylases (HDACs)1. The class III HDACs are distinguished from HDACs in the other classes by their requirement of NAD+ for their enzyme activity2. Sirt1 is involved in gene silencing, differentiation, cell survival, metabolism and longevity1. Sirt1 activity extends the lifespan of lower organisms, including yeast, C. elegans, and flies3, 4. In addition, resveratrol, which stimulates Sirt1, extends the lifespan of mice fed a high fat diet, suggesting that Sirt1 may affect aging and/or lifespan in mammals5. The beneficial effects of caloric restriction may be dependent on Sirt16-8. Conversely, Sirt1 knockout mice exhibit developmental abnormalities, including septal and valvular heart defects9, 10. Sirt1 regulates the function of transcription factors and co-factors, including MyoD, Ku, p53, PGC1 and the FoxO family of transcription factors11-19, through deacetylation.
Activation of molecular mechanisms extending lifespan generally increase the ability of the organism to survive against stress, a phenomenon which is termed hormesis20. We have shown previously that upregulation of Sirt1 inhibits apoptosis, protects against oxidative stress in cardiac myocytes, and retards the progression of aging in the mouse heart21, 22. By extending this observation, one can speculate that therapeutic activation of a longevity factor, such as Sirt1, may protect the heart and the cardiac myocytes therein from pathologically relevant stress, such as ischemia and reperfusion23. However, the protective effect of Sirt1 against myocardial ischemia/reperfusion (I/R), a major cause of myocardial injury in the clinical setting, has not been clearly demonstrated. Furthermore, the molecular mechanism by which Sirt1 mediates its protective effects against I/R is unknown.
To address these issues, we used genetically altered mouse models in which expression of Sirt1 is either upregulated or downregulated in a cardiac myocyte specific manner. In particular, we asked 1) how expression of Sirt1 is affected by I/R, 2) how upregulation or downregulation of Sirt1 in cardiac myocytes affects myocardial injury and cardiac function after I/R, and 3) whether stimulation of Sirt1 activates cell protective mechanisms in the heart during I/R.
Methods
Genetically altered mouse models
Cardiac-specific Sirt1 transgenic mice (Tg-Sirt1) were generated using the α–myosin heavy chain promoter (courtesy of Dr. J. Robbins, Children's Hospital, Cincinnati, Ohio, USA) on an FVB background. The baseline cardiac phenotype of Tg-Sirt1 mice (line #40) has been described22. Cardiac specific Sirt1 knockout (Sirt1 -/-) mice were generated by crossing Sirt1flox/flox mice (Jackson Laboratory) with C57BL/6J background with α–myosin heavy chain promoter driven Cre mice (αMHC-Cre, courtesy of Dr. M. Schneider, Imperial College, London, UK). All Sirt1flox/flox (control) and Sirt 1flox/flox, αMHC-Cre (cardiac specific Sirt1 -/-) mice were backcrossed to C57BL/6J background. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Medicine and Dentistry of New Jersey.
Antibodies
The antibodies used in this study include anti-Sirt1 and anti-MnSOD antibodies (Upstate Biotechnology), anti-acetylated-p53 (Lys-382) antibody (Abcam), anti-Bcl-xL antibody (Pharmingen), anti-Bax, anti-8-OHdG and anti-FoxO1 antibodies (Santa Cruz), anti-cleaved-caspase-3, and anti-acetylated FoxO1 antibodies (Cell Signaling Technology), anti-troponin T antibody (Thermo Scientific) and anti-actinin and anti-tubulin antibodies (Sigma). The anti-thioredoxin1 (Trx1) antibody has been described previously24.
Ischemia/reperfusion and prolonged ischemia in vivo
Mice were housed in a temperature-controlled environment with 12-hr light/dark cycles where they received food and water ad libitum. Mice were anesthetized by intraperitoneal injection of pentobarbital sodium (50 mg/kg). A rodent ventilator (model 683; Harvard Apparatus Inc.) was used with 65% oxygen. The animals were kept warm using heat lamps. Rectal temperature was monitored and maintained between 36 and 37°C. The chest was opened by a horizontal incision at the third intercostal space. I/R was achieved by ligating the anterior descending branch of the left coronary artery (LAD) using an 8-0 prolene suture, with silicon tubing (1 mm OD) placed on top of the LAD, 2 mm below the border between the left atrium and LV. Ischemia was confirmed by ECG change (ST elevation). After occlusion for 45 minutes in mice of FVB background and 20-30 minutes in mice of C57BL/6J background, the silicon tubing was removed to achieve reperfusion and the rib space and overlying muscles were closed. FVB mice were subjected to longer ischemia because they are more resistant to I/R injury. Some mice were subjected to alternating brief periods of ischemia (1.5 min, 4 times) and reperfusion (3.5 min, 4 times) before a longer period (20 min) of ischemia. When recovered from anesthesia, the mice were extubated and returned to their cages. They were housed in a climate-controlled environment. Twenty-four hours after reperfusion, the animals were reanesthetized and intubated, and the chest was opened. After arresting the heart at the diastolic phase by KCl injection, the ascending aorta was canulated and perfused with saline to wash out blood. The LAD was occluded with the same suture, which had been left at the site of the ligation. To demarcate the ischemic area at risk (AAR), Alcian blue dye (1%) was perfused into the aorta and coronary arteries. Hearts were excised, and LVs were sliced into 1-mm thick cross sections. The heart sections were then incubated with a 1% triphenyltetrazolium chloride solution at 37°C for 15 min. The infarct area (pale), the AAR (not blue), and the total LV area from both sides of each section were measured using Adobe Photoshop (Adobe Systems Inc.), and the values obtained were averaged. The percentage of area of infarction and AAR of each section were multiplied by the weight of the section and then totaled from all sections. AAR/LV and infarct area/AAR were expressed as percentages25.
Langendorff-perfused mouse heart model of global I/R
Mice were anesthetized with pentobarbital (65 mg/kg, i.p.) and treated intraperitoneally with 50 units of heparin. The heart was quickly removed and catheterized with a 22-gauge needle. The hearts were mounted on a Langendorff-type isolated heart perfusion system and subjected to retrograde coronary artery reperfusion with 37°C oxygenated Krebs-Henseleit bicarbonate buffer (NaCl 120 mmol/L, Glucose 17 mmol/L, NaHCO3 25 mmol/L, KCl 5.9 mmol/L, MgCl2 1.2 mmol/L, CaCl2 2.5 mmol/L, EDTA 0.5 mmol/L), pH 7.4, at a constant pressure of 80 mmHg. A balloon filled with water was introduced into the left ventricle (LV) through the mitral valve orifice and connected to a pressure transducer via a plastic tube primed with water. LV pressures and LV dP/dt were recorded with a strip chart recorder (Astro-Med, Inc). The LV end-diastolic pressure was set at 4-10 mmHg at the beginning of perfusion by adjusting the volume of the balloon in the LV and the volume was kept constant throughout an experiment. After a 30 min equilibration period, the heart was subjected to 30 min of global ischemia (at 37°C) followed by 60 min of reperfusion.
Evaluation of apoptosis in tissue sections
DNA fragmentation was detected in situ using TUNEL, as described25. Nuclear density was determined by manual counting of DAPI-stained nuclei in six fields for each animal using the 40x objective, and the number of TUNEL-positive nuclei was counted by examining the entire section using the same power objective.
Immunoblot analysis
For immunoblot analysis, heart samples were homogenized in lysis buffer (50 mmol/L Tris-HCL pH 7.4, 0.1% SDS, 1% Igepal CA-630, 0.15 mol/L NaCl, 0.25% Na-deoxycholate, and 1 mmol/L EDTA supplemented with protease inhibitors).
Adenovirus constructs
Adenovirus harboring Sirt1 has been described21. Adenovirus harboring sh-RNA for FoxO1 (Ad-sh-FoxO1) was generated as previously described26 using the following hairpin forming oligo 5’ – CGCCAAACTCACTACACCATTTCAAGAGAATGGTGTAGTGAGTTTGGCTTTTT A – 3’. The hairpin loop sequence is underlined.
Statistics
Data are expressed as mean ± SEM but nonparametric statistics were employed due to the small numbers of subjects. Differences in means between two groups and among more than two groups were evaluated with Mann-Whitney U test and Kruskal-Wallis test, respectively. The post-hoc comparisons were performed by Mann-Whitney U test with Bonferroni correction when the multi-group comparisons were significant. Hemodynamic data between two groups at different time of reperfusion were compared using linear mixed models. All the statistical analyses were performed using SPSS 15.0 for Windows (SPSS Inc., Chicago, IL, USA). P values of < 0.05 were considered statistically significant.
Results
Sirt1 is downregulated by ischemia/reperfusion (I/R)
C57BL/6J mice were subjected to 20 minutes of ischemia followed by 24 hours of reperfusion (I/R) (Fig. 1A). Expression of Sirt1 was significantly reduced in hearts subjected to I/R compared to sham operated hearts (66.6±1.8%, p=0.009 vs. sham operated hearts) (Fig. 1B). Downregulation of Sirt1 was normalized (113.9±12.0%) in hearts subjected to preconditioning before I/R (4 cycles of 1.5 minutes of ischemia followed by 3.5 minutes of reperfusion) (Fig. 1AB). Similar results were obtained regarding the effect of I/R and preconditioning on mRNA expression of Sirt1 (Fig. 1B).
Figure 1. Sirt1 is downregulated by I/R in vivo.
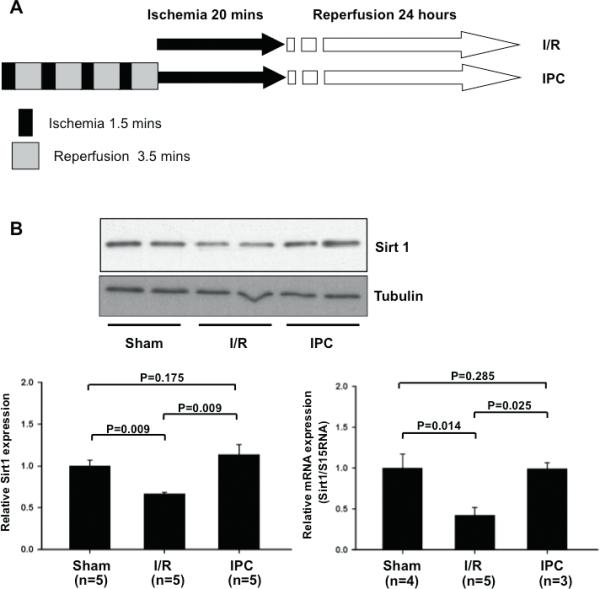
(A) The protocol used for ischemia/reperfusion (I/R) and ischemia preconditioning (IPC) for C57BL/6J mice. (B) Heart homogenates were prepared from mice subjected to sham operation, I/R and IPC. Expression of Sirt1 and tubulin was evaluated by immunoblots. mRNA expression of Sirt1 was evaluated with RT-PCR. The expression level of sham operated mice is expressed as 1.
Downregulation of endogenous Sirt1 in the heart exacerbates myocardial injury caused by I/R
In order to examine the role of endogenous Sirt1 in mediating survival and death of cardiac myocytes, we used cardiac specific Sirt1 -/- mice. Expression of Sirt1 was selectively attenuated in the heart and there was no compensatory increase in Sirt3 expression in cardiac specific Sirt1 -/- mice (supplemental Fig. S1). The cardiac phenotype in cardiac specific Sirt1 -/- mice was normal at 3 months of age (supplemental Tables S1). Cardiac specific Sirt1 -/- mice or control wild type mice were subjected to 30 min of ischemia followed by 24 hours of reperfusion. The area at risk (AAR)/left ventricle in cardiac specific Sirt1 -/- mice was not significantly different from that in control mice. The size of myocardial infarction/AAR was significantly greater in cardiac specific Sirt1 -/- than in control mice (Fig. 2AB), suggesting that endogenous Sirt1 plays a protective role in the heart during I/R.
Figure 2. Myocardial injury caused by I/R is enhanced in cardiac specific Sirt1 -/- mice.
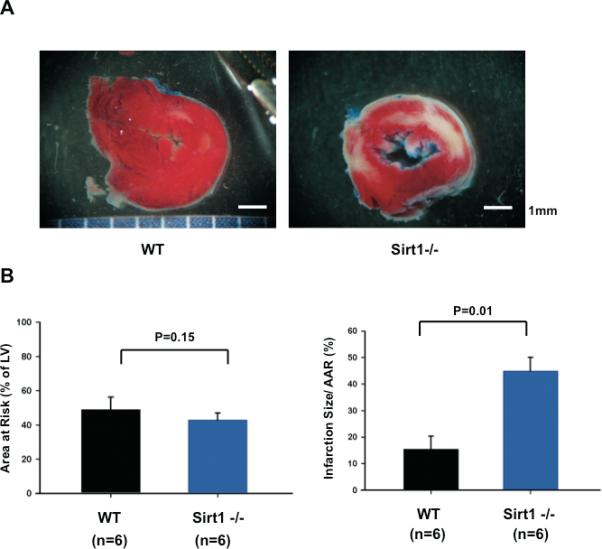
Cardiac specific Sirt1 -/- mice and wild type (+/+) mice generated on a C57BL/6J background were subjected to 30 min of ischemia and 24 hours of reperfusion. (A) Gross appearance of LV myocardial sections after Alcian blue and triphenyltetrazolium chloride (TTC) staining. (B) (left) The area at risk (AAR) (% of LV) was comparable between wild type and cardiac specific Sirt1 -/- mice. (right) The infarction area/AAR was significantly greater in cardiac specific Sirt1 -/- mice than in wild type mice.
Overexpression of Sirt1 in the heart is protective against I/R injury
Since downregulation of endogenous Sirt1 in the heart subjected to I/R appears to promote myocardial injury, we hypothesized that upregulation of Sirt1 in the heart prevents myocardial injury in response to I/R. In order to evaluate the effect of increased expression of Sirt1 upon myocardial injury caused by I/R, Tg-Sirt1 and NTg mice generated on an FVB background were subjected to 45 minutes of ischemia followed by 24 hours of reperfusion (Fig. 3A). We confirmed that Sirt1 is significantly upregulated in the myocardium of Tg-Sirt1 compared to NTg (supplemental Fig. S2AB). The level of acetylated p53 was significantly reduced in Tg-Sirt1, indicating that the activity of Sirt1 is elevated in Tg-Sirt1 (not shown). The size of AAR/LV was not significantly different between Tg-Sirt1 and NTg mice (Fig. 3B), but the size of MI/AAR after I/R was significantly smaller in Tg-Sirt1 than in NTg mice (Fig. 3B). The number of TUNEL positive cells in the ischemic border zone was also smaller in Tg-Sirt1 mice than in NTg mice (Fig. 3C).
Figure 3. I/R injury is attenuated in Tg-Sirt1 mice.
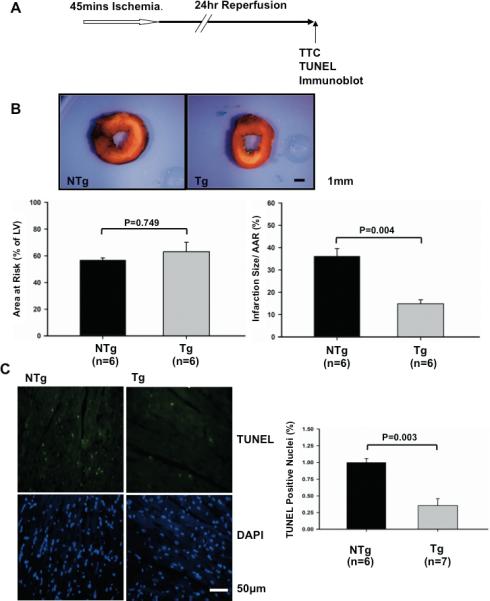
(A) The protocol used for I/R for Tg-Sirt1 and NTg. Tg-Sirt1 or NTg mice generated on an FVB background were subjected to 45 minutes of ischemia and 24 hours of reperfusion. (B) (upper) Gross appearance of LV myocardial sections after Alcian blue and triphenyltetrazolium chloride (TTC) staining. (lower left) The area at risk (AAR) (% of LV) was comparable between NTg and Tg-Sirt1. (lower right) The infarction area/AAR was significantly smaller in Tg-Sirt1 than in NTg. (C) (left) LV myocardial sections were subjected to TUNEL and DAPI staining. Representative images of the staining in the border zone. (right) The number of TUNEL-positive myocytes was expressed as a percentage of total nuclei detected by DAPI staining.
The effect of Sirt1 on I/R injury was also evaluated in the Langendorff isolated perfused heart model. Hearts isolated from Tg-Sirt1 or NTg mice were subjected to 30 minutes of global ischemia followed by 60 minutes of reperfusion (Fig. 4A). The baseline LV function of Tg-Sirt1 and NTg mouse hearts was similar (supplemental Table S2). After reperfusion, recovery of LV systolic pressure (LVSP) and LV developed pressure (LVDP) was significantly greater in Tg-Sirt1 than in NTg mice (Fig. 4BC). LV end-diastolic pressure (LVDEP) did not differ significantly between Tg-Sirt1 and NTg (Fig. 4D). However, systolic and diastolic dP/dt were both significantly improved (Fig. 4EF). The size of myocardial infarction evaluated by TTC staining was significantly smaller in Tg-Sirt1 mice than in NTg mice (Fig. 4G). Taken together, these results suggest that Sirt1 plays a protective role against myocardial dysfunction and injury caused by I/R.
Figure 4. Myocardial I/R injury is attenuated in Tg-Sirt1 mouse hearts ex vivo.
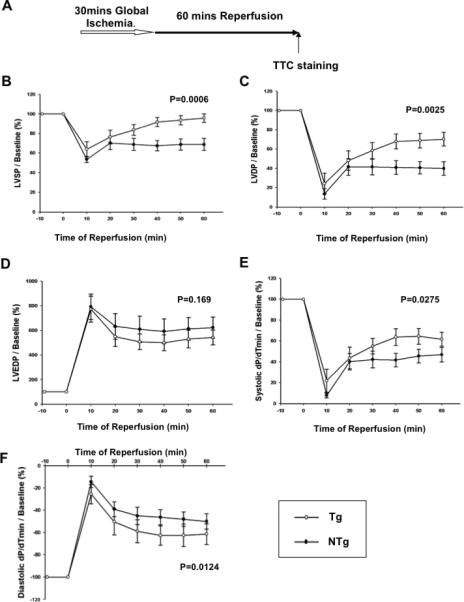
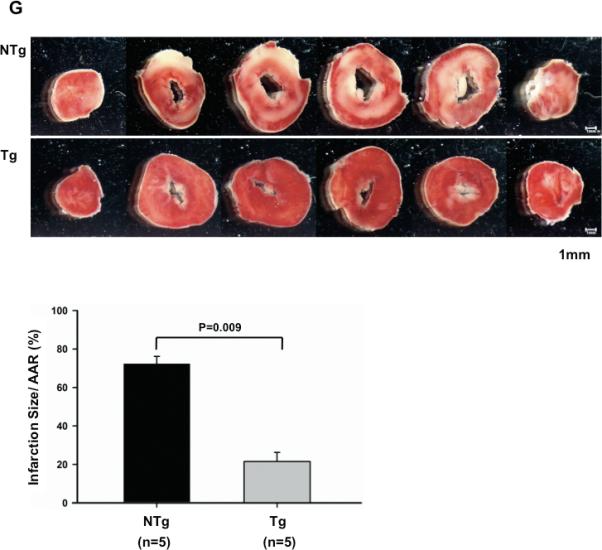
(A) The protocol used for I/R for Tg-Sirt1 and NTg control mice in the Langendorff model. The hearts of Tg-Sirt1 or NTg control mice were subjected to 30 minutes of global ischemia and 60 minutes of reperfusion. (B-F) Hemodynamics of the Langendorff-perfused isolated mouse hearts of Tg-Sirt1 and NTg control mice. (B) LVSP, left ventricular systolic pressure; (C) LVDP, left ventricular developed pressure (systolic pressure – diastolic pressure); (D) LVEDP, left ventricular end-diastolic pressure; (E,F) Systolic and diastolic dP/dt. In B-F, the level at baseline is expressed as 100%. (G) (upper) Gross appearance of LV myocardial sections after triphenyltetrazolium chloride (TTC) staining. (lower) The infarction area/AAR (where AAR= total heart) was significantly smaller in Tg-Sirt1 than in NTg.
Sirt1 upregulates anti-oxidant and anti-apoptotic molecules and downregulates pro-apoptotic molecules in the heart
We investigated the molecular mechanism by which Sirt1 protects the heart from I/R injury. Homogenates were prepared from the ischemic area in Tg-Sirt1 and NTg hearts subjected to I/R, and expression of molecules involved in survival and death of cardiac myocytes was evaluated. Expression of MnSOD (6.6 ± 1.8 fold vs. NTg) and Trx1 (2.4 ± 0.3 fold), anti-oxidants, was significantly higher in Tg-Sirt1 than in NTg (Fig. 5A-C). Expression of Bcl-xL (2.0 ± 0.3 fold), an anti-apoptotic molecule, was also increased (Fig. 5A and D). On the other hand, expression of Bax (0.2 ± 0.03 fold) and cleaved caspase-3 (0.4 ± 0.14 fold), pro-apoptotic molecules, was significantly lower in Tg-Sirt1 than in NTg (Fig. 5A, E and F). Expression of these genes was reversed in cardiac specific Sirt1 -/- mouse hearts subjected to I/R (supplemental Fig. S3).
Figure 5. Sirt1 upregulates cardioprotective molecules in the heart.
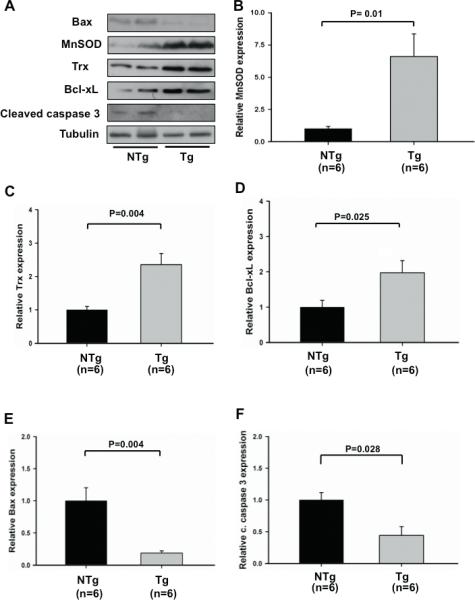
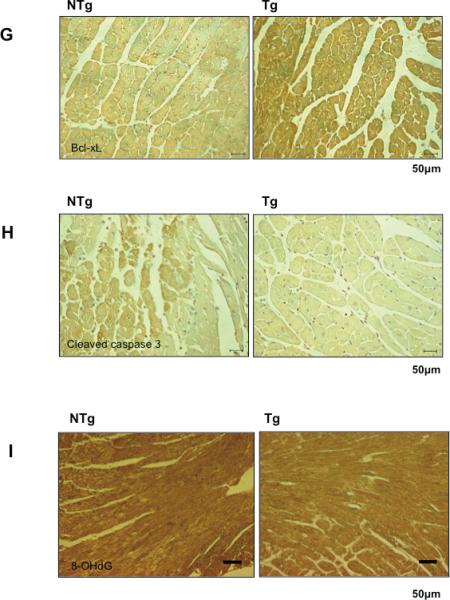
Heart homogenates were prepared from the ischemic area in Tg-Sirt1 and NTg mice subjected to I/R. Expression of Bax, MnSOD, Trx1, Bcl-xL, cleaved caspase-3 and tubulin was evaluated by immunoblots. (A) Representative immunoblots are shown. (B-F) The quantitative and statistical analyses of the immunoblots are shown. The relative expression of MnSOD (B), Trx1 (C), Bcl-xL (D), Bax (E) and cleaved caspase-3 (F) in Tg-Sirt1 versus NTg is shown. The expression level of NTg mice is expressed as 1. (G-I) Representative immunostaining of Bcl-xL (G), cleaved caspase-3 (H), and 8-OHdG (I) in the ischemic area of left ventricular myocardial sections in Tg-Sirt1 and NTg mice subjected to I/R.
In order to examine whether changes in the levels of the aforementioned molecules in Tg-Sirt1 occur in cardiac myocytes, immunostaining was conducted. We confirmed that both upregulation of Bcl-xL and downregulation of cleaved caspase-3 take place in cardiac myocytes in Tg-Sirt1 subjected to I/R (Fig. 5GH).
We have shown previously that Sirt1 reduces oxidative stress in the heart and cardiac myocytes therein under stress conditions. Since both MnSOD and Trx1 were upregulated in Tg-Sirt1 hearts subjected to I/R, we examined whether oxidative stress was attenuated in Tg-Sirt1 hearts. As expected, staining with anti-8-hydroxydeoxyguanosine (8-OHdG) antibody, a marker of oxidative DNA damage, was significantly weaker in Tg-Sirt1 than in NTg after I/R (Fig. 5I). Conversely, the level of 8-OHdG staining was significantly enhanced in cardiac specific Sirt1 -/- mice compared to wild type mice after I/R (supplemental Figure S4).
Sirt1–induced upregulation of MnSOD is partially mediated by FoxO1
Sirt1 deacetylates FoxO transcription factors, thereby stimulating FoxO-mediated transcription of anti-oxidants12. We therefore examined the role of FoxO in mediating Sirt1-mediated upregulation of MnSOD in cardiac myocytes. Overexpression of Sirt1 increases, whereas knockdown of Sirt1 decreases, total FoxO1 in cultured cardiac myocytes (Fig. 6AB). Sirt1-induced upregulation of FoxO1 was also observed at the mRNA level (supplemental Fig. S5). On the other hand, overexpression of Sirt1 decreases, whereas knockdown of Sirt1 increases, an acetylated form of FoxO1 (Fig. 6A and C). A sample prepared from heart cells treated with p300 acetyltransferase exhibited strong acetylation of FoxO1, thereby serving as a positive control for this experiment. Sirt1-mediated upregulation of MnSOD was significantly attenuated in the presence of FoxO1 knockdown in cultured cardiac myocytes (Fig. 6DE). These results suggest that FoxO1 plays an essential role in mediating Sirt1-induced upregulation of MnSOD. In order to test whether Sirt1 stimulates FoxO1-mediated transcription, we examined the effect of Sirt1 upon the activity of 3xIRS-luc, a reporter gene containing three repeats of the FoxO1 binding site in the IGFBP1 IRS27. Sirt1 significantly increased the activity of 3xIRS-luc, an effect which was abolished in the presence of sirtinol (Fig. 6F). This result suggests that Sirt1 increases FoxO1-mediated transcription. In order to test whether Sirt1-induced upregulation of anti-oxidants reduces oxidative stress, we examined the effect of Sirt1 overexpression upon increases in oxidative stress induced by chelerythrine treatment. We have shown previously that chelerythrine increases oxidative stress, thereby inducing apoptosis in cardiac myocytes. Sirt1 significantly attenuated chelerythrine-induced increases in oxidative stress, as evaluated with DFCDA staining. Importantly, inhibition of oxidative stress by Sirt1 was reversed when FoxO1 is downregulated (supplemental Fig. S6). These results suggest that Sirt1 inhibits oxidative stress in cardiac myocytes through FoxO1-dependent mechanisms.
Figure 6. Sirt1-induced upregulation of MnSOD is mediated by FoxO1.
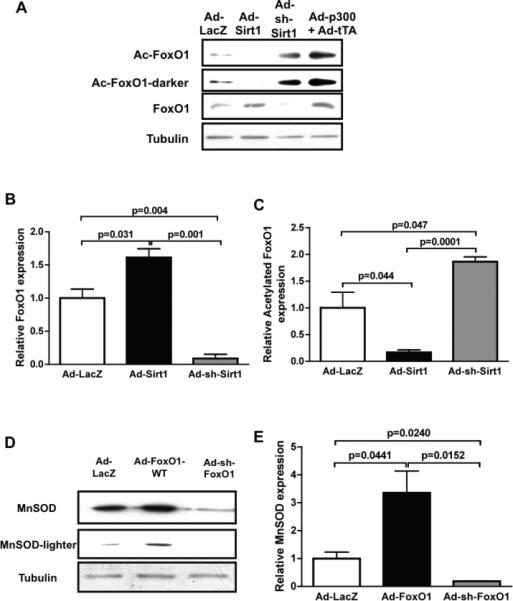
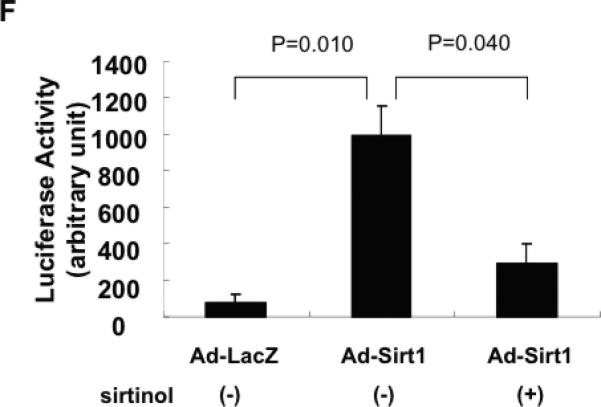
Cultured myocytes were transduced with adenovirus harboring LacZ (control, Ad-LacZ), wild type Sirt1 (Ad-Sirt1-WT), shRNA-Sirt1 (Ad-sh-Sirt1) or tet-inducible p300. Expression of p300 was induced by co-transduction with adenovirus harboring tTA without doxycycline. Forty-eight hours after transduction, cells were harvested. (A) Expression of FoxO1, acetylated FoxO1 and tubulin (internal control) was evaluated with immunoblot analyses. (B,C) The relative expression of FoxO1 and acetylated FoxO1 was evaluated by densitometric analyses. The expression level of FoxO1 and acetylated FoxO1 in Ad-LacZ transduced myocytes is expressed as 1. (D,E) Myocytes were transduced with adenovirus harboring LacZ, wild type FoxO1 (Ad-FoxO1-WT) and shRNA-FoxO1 (Ad-sh-FoxO1). (D) Expression of MnSOD and tubulin (internal control) was evaluated with immunoblot analyses. (E) The relative expression of MnSOD was evaluated by densitometric analyses. The expression level of MnSOD in Ad-LacZ transduced myocytes is expressed as 1. (F) Myocytes were transfected with 3xIRS-Luc and then transduced with Ad-LacZ or Ad-Sirt1 (5 MOI). Ad-Sirt1 transfected myocytes were treated with or without sirtinol (10 μM). Experiments were conducted three times.
Sirt1 plays an important role in mediating nuclear localization of FoxO1 in the mouse heart
In order to clarify the role of Sirt1 in regulating FoxO1-mediated transcription in the heart in vivo, we examined localization of FoxO1 in the heart with immunostaining. In sham operated NTg or wild type hearts, nuclear staining of FoxO1 was not prominent. However, I/R significantly increased nuclear staining of FoxO1 in control hearts. I/R-induced increases in the nuclear staining of FoxO1 were significantly enhanced in Tg-Sirt1 hearts, whereas they were significantly attenuated in cardiac specific Sirt1 -/-hearts (Fig. 7). These results suggest that Sirt1 positively mediates nuclear localization of FoxO1 in the mouse heart in vivo.
Figure 7. Sirt1 enhances nuclear localization of FoxO1 in response to I/R.
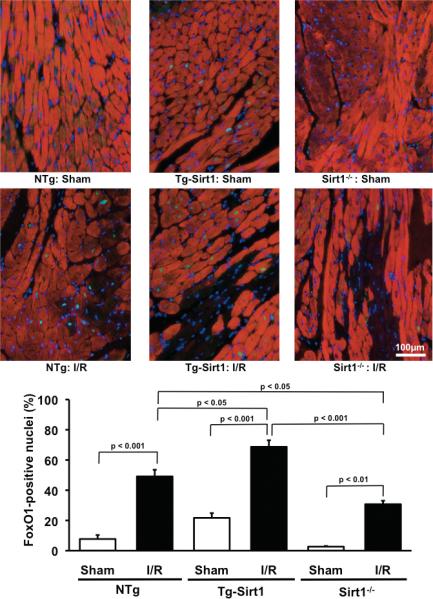
NTg, Tg-Sirt1 and cardiac specific Sirt1 -/- mice were subjected to I/R or sham operation. Twenty-four hours after reperfusion, the heart was subjected to immunostaining with anti-troponin T antibody (red), anti-FoxO1 antibody (green) and DAPI (blue). FoxO1 positive nuclei/total nuclei was evaluated and expressed as %.
Discussion
Our results suggest that Sirt1 plays a protective role against myocardial I/R in the heart in vivo. Together with the cardio-protective actions of Sirt1 against aging and oxidative stress22, our results suggest that stimulation of Sirt1 could represent a novel modality to protect the heart from ischemic heart disease.
Our results suggest that the level of Sirt1 is downregulated by I/R. We have shown previously that expression of Sirt1 is upregulated by certain stresses in the heart, including aging, oxidative stress and heart failure21, 22, thereby protecting the heart as a compensatory mechanism. Thus, the regulation of Sirt1 expression appears to be stimulus-specific and the compensatory mechanism of Sirt1 against stress does not operate in response to I/R. Since ischemic preconditioning induced by repetitive brief periods of ischemia prevented I/R-mediated downregulation of Sirt1, it is possible that expression of Sirt1 is also regulated by the extent of ischemia. At present, the molecular mechanism mediating downregulation of Sirt1 during I/R remains to be elucidated.
Both loss and gain of function experiments suggest that Sirt1 plays a protective role against I/R injury in the heart. Since the reversal of Sirt1 downregulation through overexpression of Sirt1 in transgenic mice reduced I/R-induced myocardial injury, downregulation of endogenous Sirt1 during I/R may contribute to myocardial injury. We have shown previously that overexpression of Nampt, an enzyme critically regulating NAD+ synthesis in cardiac myocytes, in transgenic mice also reduced I/R-induced myocardial injury28. Therefore, interventions to increase either protein expression or activity of Sirt1 during I/R appear to be protective.
Resveratrol protects the heart from I/R injury in experimental animals29. Although resveratrol has the ability to stimulate Sirt13, whether stimulation of Sirt1 is mediated directly through interaction between the two molecules or secondarily through intermediates remains to be elucidated30. Furthermore, the cardioprotective effect of resveratrol could be mediated by Sirt1-independent mechanisms, such as the anti-oxidant effect of resveratrol31. Our results suggest that stimulation of Sirt1 can reduce I/R injury. It would be interesting to compare the molecular mechanisms through which specific stimulation of Sirt1 and resveratrol inhibit I/R injury, using the animal models described here.
Our results suggest that Sirt1 upregulates cardioprotective molecules, including MnSOD, Trx1 and Bcl-xL, and downregulates pro-apoptotic molecules, including Bax. We have shown previously that Sirt1 deacetylates p53, thereby inhibiting apoptosis of cardiac myocytes in response to serum starvation in vitro21. Since Bax is positively regulated by p5332, the protective effect of Sirt1 may be mediated in part through deacetylation and suppression of p53. In fact, the level of acetylated p53 is significantly lower in Tg-Sirt1 than in NTg mice22. In addition, Sirt1 deacetylates FoxO, thereby stimulating expression of genes mediating cell protective effects12. We show that Sirt1 stimulates transcription through FoxO1 and enhances I/R-induced nuclear localization of FoxO1 in the heart. Further, Sirt1 stimulates expression of MnSOD in cultured cardiac myocytes and FoxO1 plays an essential role in mediating Sirt1-induced upregulation of MnSOD. Importantly, Sirt1 inhibits oxidative stress in cardiac myocytes through FoxO1-dependent mechanisms (supplemental Fig. S6). We propose that coordinated regulation of these cell protective and cell death promoting molecules mediates the protective effect of Sirt1 under I/R. Consequently, upregulation of Sirt1 significantly attenuates oxidative stress and activation of caspase-3 in the heart during I/R. Many of the molecular changes by either upregulation or downregulation of Sirt1 were observed only in the context of I/R but not at basal. Thus, additional I/R-dependent mechanisms regulating expression of anti-/pro-apoptotic molecules and/or nuclear localization of FoxO1 appear to exist. How Sirt1 protects the heart during I/R in vivo and the role of FoxO1 in mediating the protective effect of Sirt1 require further investigation.
We have shown recently that Nampt, an enzyme stimulating NAD+ synthesis, protects the heart from prolonged ischemia28. Since Nampt increases the activity of Sirt128, Sirt1 may protect the heart from I/R not only by reducing reperfusion injury but also by protecting the heart from prolonged ischemia. We have found recently that Sirt1 and FoxOs coordinately activate autophagy in response to glucose starvation in cardiac myocytes in vitro (manuscript in preparation). Although the molecular mechanism through which Sirt1 protects against myocardial injury caused by prolonged ischemia remains to be elucidated in vivo, Sirt1 might stimulate autophagy, thereby inhibiting myocardial cell death during prolonged ischemia. In any case, stimulation of Sirt1 is an attractive modality to reduce myocardial injury which can be caused by both ischemia and reperfusion in patients suffering from acute myocardial infarction.
Sirt1 is involved in lifespan extension in lower organisms under conditions of calorie restriction33. Stimulation of Sirt1 by a small molecule compound reduces the mortality of obese mice on a high calorie diet34. Although the molecular mechanism mediating the lifespan extension remains to be elucidated in mammals, currently well-accepted hypotheses include the hormesis hypothesis in which accumulation of stress resistance conferred by non-lethal stress leads to lifespan extension20. Stimulation of Sirt1 may confer resistance to I/R injury to the heart, which could be a novel mechanism of cardioprotection against cardiac stress23. Endogenous mechanisms of lifespan extension are stimulated by low grade stress, such as calorie restriction. The fact that downregulation of Sirt1 under I/R is attenuated in the presence of preconditioning suggests that stimulation of Sirt1 by a low grade of repetitive stress may partly mediate the beneficial effect of preconditioning.
We have shown previously that the anti-aging effect of Sirt1 is dose-dependent and that a very high dose of Sirt1 actually induces heart failure22. It has also been shown that the cardioprotective effect of resveratrol is dose-dependent35. Thus, the extent of Sirt1 stimulation should be carefully addressed when stimulation of Sirt1 is considered for treatment of I/R injury in the future.
In summary, Sirt1 has a cardioprotective effect against ischemia and reperfusion. Sirt1 upregulates cardioprotective molecules and downregulates pro-apoptotic molecules during I/R, thereby attenuating oxidative stress and inhibiting apoptosis. Thus, activation of Sirt1 could be a novel method of cardioprotection against cardiac I/R.
CLINICAL PERSPECTIVE
Reperfusion injury is a significant health problem in western countries but there is no medical treatment to effectively reduce it. We have shown previously that Sirt1, a class III histone deacetylase and a member of the sirtuin family, inhibits cell death in cardiomyocytes in response to stress and retards aging in the heart. The sirtuin family proteins play an important role in mediating lifespan extension induced by caloric restriction in lower organisms and stimulation of sirtuins appears to increase lifespan in vertebrates in certain conditions. Since many mechanisms inducing lifespan extension make organisms resistant to stress, we hypothesized that stimulation of sirtuin may also make the heart more resistant to ischemic injury. In this study, we investigated the role of Sirt1 in mediating cardioprotection in a mouse model of ischemia/reperfusion. Although downregulation of endogenous Sirt1 exacerbated myocardial injury, upregulation of Sirt1 attenuated it, in response to ischemia/reperfusion. Sirt1 enhanced nuclear accumulation of FoxO1, a transcription factor regulating anti-oxidants and cell death/survival mechanisms, which in turn attenuated oxidative stress in the heart. Our results suggest that enhancing the function of Sirt1 is a promising modality to reduce myocardial injury in patients with acute myocardial infarction.
Supplementary Material
Acknowledgement
The authors thank Daniela Zablocki for critical reading of the manuscript.
Funding Sources: This work was supported in part by U.S. Public Health Service Grants HL59139, HL67724, HL69020, HL91469, and AG27211. This work was supported by the Foundation of Leducq Transatlantic Network of Excellence.
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol. 5:253–295. doi: 10.1146/annurev.pathol.4.110807.092250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 3.Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, Zipkin RE, Chung P, Kisielewski A, Zhang LL, Scherer B, Sinclair DA. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature. 2003;425:191–196. doi: 10.1038/nature01960. [DOI] [PubMed] [Google Scholar]
- 4.Tissenbaum HA, Guarente L. Increased dosage of a sir-2 gene extends lifespan in Caenorhabditis elegans. Nature. 2001;410:227–230. doi: 10.1038/35065638. [DOI] [PubMed] [Google Scholar]
- 5.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen HY, Miller C, Bitterman KJ, Wall NR, Hekking B, Kessler B, Howitz KT, Gorospe M, de Cabo R, Sinclair DA. Calorie restriction promotes mammalian cell survival by inducing the SIRT1 deacetylase. Science. 2004;305:390–392. doi: 10.1126/science.1099196. [DOI] [PubMed] [Google Scholar]
- 7.Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. 2000;289:2126–2128. doi: 10.1126/science.289.5487.2126. [DOI] [PubMed] [Google Scholar]
- 8.Rogina B, Helfand SL. Sir2 mediates longevity in the fly through a pathway related to calorie restriction. Proc Natl Acad Sci U S A. 2004;101:15998–16003. doi: 10.1073/pnas.0404184101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McBurney MW, Yang X, Jardine K, Hixon M, Boekelheide K, Webb JR, Lansdorp PM, Lemieux M. The mammalian SIR2alpha protein has a role in embryogenesis and gametogenesis. Mol Cell Biol. 2003;23:38–54. doi: 10.1128/MCB.23.1.38-54.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng HL, Mostoslavsky R, Saito S, Manis JP, Gu Y, Patel P, Bronson R, Appella E, Alt FW, Chua KF. Developmental defects and p53 hyperacetylation in Sir2 homolog (SIRT1)-deficient mice. Proc Natl Acad Sci U S A. 2003;100:10794–10799. doi: 10.1073/pnas.1934713100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y, Hoffman E, Veech RL, Sartorelli V. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Mol Cell. 2003;12:51–62. doi: 10.1016/s1097-2765(03)00226-0. [DOI] [PubMed] [Google Scholar]
- 12.Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW, Greenberg ME. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science. 2004;303:2011–2015. doi: 10.1126/science.1094637. [DOI] [PubMed] [Google Scholar]
- 13.Daitoku H, Hatta M, Matsuzaki H, Aratani S, Ohshima T, Miyagishi M, Nakajima T, Fukamizu A. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004;101:10042–10047. doi: 10.1073/pnas.0400593101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Motta MC, Divecha N, Lemieux M, Kamel C, Chen D, Gu W, Bultsma Y, McBurney M, Guarente L. Mammalian SIRT1 represses forkhead transcription factors. Cell. 2004;116:551–563. doi: 10.1016/s0092-8674(04)00126-6. [DOI] [PubMed] [Google Scholar]
- 15.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 16.Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 17.Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R, Alt FW, Wu Z, Puigserver P. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. Embo J. 2007;26:1913–1923. doi: 10.1038/sj.emboj.7601633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nemoto S, Fergusson MM, Finkel T. SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1{alpha}. J Biol Chem. 2005;280:16456–16460. doi: 10.1074/jbc.M501485200. [DOI] [PubMed] [Google Scholar]
- 19.Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, Leid M, McBurney MW, Guarente L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771–776. doi: 10.1038/nature02583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gems D, Partridge L. Stress-response hormesis and aging: “that which does not kill us makes us stronger”. Cell Metab. 2008;7:200–203. doi: 10.1016/j.cmet.2008.01.001. [DOI] [PubMed] [Google Scholar]
- 21.Alcendor RR, Kirshenbaum LA, Imai S, Sadoshima J. Sir2α, a longevity factor and a class III histone decetylase, is an essential endogenous inhibitor of apoptosis in cardiac myocytes. Circ Res. 2004;95:971–980. doi: 10.1161/01.RES.0000147557.75257.ff. [DOI] [PubMed] [Google Scholar]
- 22.Alcendor RR, Gao S, Zhai P, Zablocki D, Holle E, Yu X, Tian B, Wagner T, Vatner SF, Sadoshima J. Sirt1 regulates aging and resistance to oxidative stress in the heart. Circ Res. 2007;100:1512–1521. doi: 10.1161/01.RES.0000267723.65696.4a. [DOI] [PubMed] [Google Scholar]
- 23.Hsu CP, Odewale I, Alcendor RR, Sadoshima J. Sirt1 protects the heart from aging and stress. Biol Chem. 2008;389:221–231. doi: 10.1515/BC.2008.032. [DOI] [PubMed] [Google Scholar]
- 24.Yamamoto M, Yang G, Hong C, Liu J, Holle E, Yu X, Wagner T, Vatner SF, Sadoshima J. Inhibition of thioredoxin in the heart increases oxidative stress and cardiac hypertrophy. J. Clin. Invest. 2003;112:1395–1406. doi: 10.1172/JCI17700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto S, Yang G, Zablocki D, Liu J, Hong C, Kim SJ, Soler S, Odashima M, Thaisz J, Yehia G, Molina CA, Yatani A, Vatner DE, Vatner SF, Sadoshima J. Activation of Mst1 causes dilated cardiomyopathy by stimulating apoptosis without compensatory ventricular myocyte hypertrophy. J Clin Invest. 2003;111:1463–1474. doi: 10.1172/JCI17459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–922. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]
- 27.Tang ED, Nunez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741–16746. doi: 10.1074/jbc.274.24.16741. [DOI] [PubMed] [Google Scholar]
- 28.Hsu CP, Oka S, Shao D, Hariharan N, Sadoshima J. Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ Res. 2009;105:481–491. doi: 10.1161/CIRCRESAHA.109.203703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ray PS, Maulik G, Cordis GA, Bertelli AA, Bertelli A, Das DK. The red wine antioxidant resveratrol protects isolated rat hearts from ischemia reperfusion injury. Free Radic Biol Med. 1999;27:160–169. doi: 10.1016/s0891-5849(99)00063-5. [DOI] [PubMed] [Google Scholar]
- 30.Pacholec M, Bleasdale JE, Chrunyk B, Cunningham D, Flynn D, Garofalo RS, Griffith D, Griffor M, Loulakis P, Pabst B, Qiu X, Stockman B, Thanabal V, Varghese A, Ward J, Withka J, Ahn K. SRT1720, SRT2183, SRT1460, and Resveratrol Are Not Direct Activators of SIRT1. J Biol Chem. 285:8340–8351. doi: 10.1074/jbc.M109.088682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das S, Khan N, Mukherjee S, Bagchi D, Gurusamy N, Swartz H, Das DK. Redox regulation of resveratrol-mediated switching of death signal into survival signal. Free Radic Biol Med. 2008;44:82–90. doi: 10.1016/j.freeradbiomed.2007.09.008. [DOI] [PubMed] [Google Scholar]
- 32.Miyashita T, Reed JC. Tumor suppressor p53 is a direct transcriptional activator of the human bax gene. Cell. 1995;80:293–299. doi: 10.1016/0092-8674(95)90412-3. [DOI] [PubMed] [Google Scholar]
- 33.Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 34.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dudley J, Das S, Mukherjee S, Das DK. Resveratrol, a unique phytoalexin present in red wine, delivers either survival signal or death signal to the ischemic myocardium depending on dose. J Nutr Biochem. 2009;20:443–452. doi: 10.1016/j.jnutbio.2008.05.003. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.