

Abstract
We recently identified Grainyhead-like 2 (GRHL2) as a novel transcription factor that binds to and regulates the activity of the human telomerase reverse transcriptase (hTERT) gene promoter. In this study, we investigated the biological functions of GRHL2 and the molecular mechanism underlying hTERT gene regulation by GRHL2. Retroviral transduction of GRHL2 in normal human keratinocytes (NHK) led to a significant extension of replicative life span, whereas GRHL2 knockdown notably repressed telomerase activity and cell proliferation. Using promoter magnetic precipitation coupled with Western blotting, we confirmed the binding of GRHL2 to the hTERT promoter and mapped the minimal binding region at −53 to −13 of the promoter. Furthermore, mutation analysis revealed the three nucleotides from −21 to −19 to be critical for GRHL2 binding. Because hTERT expression is regulated in part by DNA methylation, we determined the effects of GRHL2 on the methylation status of the hTERT promoter. Senescent NHK exhibited hypermethylation of the CpG island, which occurred with the loss of hTERT expression. On the contrary, the promoter remained hypomethylated in GRHL2-transduced NHK, irrespective of cell proliferation status. Also, knockdown of endogenous GRHL2 led to hypermethylation of the promoter. These results indicate that GRHL2 regulates the hTERT expression through an epigenetic mechanism and controls the cellular life span.
Keywords: DNA Methylation, DNA Transcription, Epithelial Cell, Telomere, Transcription Promoter, DNA Methylation, Grainyhead, Telomerase, hTERT, Keratinocytes
Introduction
Telomerase is a ribonucleoprotein complex minimally composed of the protein subunit hTERT2 and the RNA template human telomerase RNA (1). Most human tumors show a high level of telomerase activity, whereas normal somatic cells display an undetectable level (2). Telomerase activation in cancers has been implicated in the extension of cellular life span, immortalization, and carcinogenesis (3–5). Telomerase enzyme activity is closely associated with the expression of hTERT, whereas human telomerase RNA is constitutively expressed in most cells, including telomerase-negative normal cells (6). Ectopic expression of hTERT alone is sufficient for reconstitution of telomerase activity in cells, indicating that hTERT is the rate-limiting step in telomerase activation (7). Several studies have revealed transcriptional regulation of the hTERT gene by direct DNA-protein binding of the trans-regulatory factors and involvement of DNA methylation of the 5′-CpG island of the hTERT promoter (8, 9).
We recently identified novel trans-regulators of the hTERT promoter by promoter magnetic precipitation coupled with mass spectrometry (10). This is a global, high throughput screening for the nuclear proteins that physically interact with the promoter. Using this method, we identified MSH2, heterogeneous nuclear ribonucleoproteins D and K, and Grainyhead-like 2 (GRHL2) to be associated with the hTERT promoter preferentially in cells exhibiting high telomerase activity. Among them, GRHL2 was found to bind the hTERT promoter near the transcription start site and to be necessary for the hTERT mRNA expression and telomerase activity in oral squamous cell carcinoma (OSCC) cells.
Grainyhead (GRH) is a developmental transcriptional factor involved in dorsoventral patterning of the Drosophila embryo (11), epithelial morphogenesis (12), and central nervous system development (13). Notably, GRH is expressed in Drosophila epithelium and is responsible for the cuticle formation by transcriptional regulation of DOPA decarboxylase (14). DOPA decarboxylase along with tyrosine hydroxylase is responsible for the maturation of the outer cuticle, making up the impermeable protective barrier in Drosophila (15). GRH also regulates cell proliferation, as evinced in post-embryonic neuroblasts whose mitotic activity is maintained by GRH expression (16). GRH physically binds to the promoter region of proliferating cell nuclear antigen (PCNA) gene in Drosophila and exerts the regulatory effects on the promoter activity, suggesting a possible role in DNA replication (17).
Mammalian homologues of GRH include GRHL1, GRHL2, and GRHL3, which exhibit sequence and biochemical similarities with GRH (18, 19). Among them, GRHL3 is most extensively characterized. Like GRH in Drosophila, GRHL3 is necessary for the formation of epithelial impermeability barrier; mice lacking GRHL3 exhibit defective stratum corneum of the skin, compromising the epithelial barrier function (20, 21). GRHL1 plays a critical role in epithelial morphogenesis, similar to GRHL3, but by regulating the expression of desmoglein-1 (DSG1), a desmosomal cadherin necessary for intercellular adhesion in keratinocytes. A recent gene knock-out study showed defects in hair anchorage and epidermal differentiation in GRHL1-null mice resulting from the reduced expression of DSG1 (22). Although GRHL1 and GRHL3 share the physiological roles in epidermal morphogenesis and keratinocyte differentiation, GRHL2 has been implicated in carcinogenesis. In a genome-wide screening of DNA amplification in hepatocellular carcinoma samples, gain of GRHL2 in 8q22.3 was found to be associated with early recurrence of hepatocellular carcinoma, suggesting its involvement in cancer development (23). We previously showed that GRHL2 expression was markedly enhanced in OSCC cells compared with the normal counterparts (10). This finding combined with the transcriptional role of GRHL2 in hTERT regulation indicates that GRHL2 may be required for continued expression of hTERT during cellular immortalization and carcinogenesis. In this study, we investigated the biological function of GRHL2 and the mechanisms underlying its role in telomerase regulation. Our data demonstrate a novel epigenetic function of GRHL2 in the control of hTERT gene expression and cellular proliferation of human keratinocytes.
EXPERIMENTAL PROCEDURES
Cells and Cell Culture
Primary NHK were prepared from keratinized oral epithelial tissues (NHOK) or from foreskin (NHEK) according to the methods described elsewhere (24). Primary NHOK and NHEK were maintained in EpiLife medium supplemented with the growth factors (Invitrogen). These cells were serially subcultured until they reached senescence, and the replication kinetics was determined by counting the cell number and the population doubling (PD) levels as described previously (24). NHOK cultures with exogenous Bmi-1 expression (named NHOK/Bmi-1) (25) were also maintained in EpiLife. Normal human oral fibroblasts (NHOF) were obtained from gingival connective tissue explants and cultured in Dulbecco's modified Eagle's medium (DMEM) and Medium 199 (Invitrogen) supplemented with 10% fetal bovine serum (FBS) (Gemini BioProducts, West Sacramento, CA). Human OSCC cell lines (SCC4 and SCC15) and HOK-16B cells were cultured according to the methods previously described (26).
Organotypic Cultures
The organotypic epithelial tissue reconstructs were established in three-dimensional “raft” cultures using NHEK as described by Dongari-Bagtzoglou and Kashleva (27). Each culture was established with 106 cells seeded on the subepithelial equivalents consisting of type I collagen and NHOF. The raft cultures were also established with SCC15 infected with LV-EGFP or LV-GRHL2i (see below) using the same protocol. The tissue equivalents were harvested after 14 days of culture and sectioned at the sagittal direction for histological examination by hematoxylin and eosin staining.
Antibodies
We used the following antibodies for this study: GRHL2 (Abnova, Taipei City, Taiwan); hTERT (Santa Cruz Biotechnology, Santa Cruz, CA); p16INK4A (Santa Cruz Biotechnology); Sp1 (Santa Cruz Biotechnology); pRb (Santa Cruz Biotechnology), p-pRb (Cell Signaling Technology, Inc. Danvers, MA); PCNA (Calbiochem); CDK6 (Santa Cruz Biotechnology); and β-actin (Santa Cruz Biotechnology).
Retroviral Vector Construction and Infection
To transduce exogenous proteins in cultured cells, we constructed retroviral vectors expressing wild-type GRHL2. Full-length cDNA encoding GRHL2 was cloned from the cDNA library obtained from primary NHOK by PCR amplification using the primers 5′-CGTTAACAAACATGTCACAAGAGTCG-3′ (forward) and 5′-GGATCCTAGATTTCCATGAGCGTGA-3′ (reverse). GRHL2 cDNA was then subcloned into pLXSN retroviral expression vector (Clontech) at the HpaI/BamHI restriction sites (underlined sequences). Retroviral vector construction and infection were performed according to the methods described elsewhere (29).
Knockdown of Endogenous GRHL2 or hTERT by Lentiviral Vector
Endogenous GRHL2 expression was knocked down with the lentiviral vector (LV-GRHL2i) expressing short hairpin RNA (shRNA) against the GRHL2 target sequence. Construction of the LV-GRHL2i vector and the control (LV-EGFP) expressing the enhanced green fluorescence protein (EGFP) was described earlier (10). Stable knockdown of endogenous hTERT was achieved using LV-hTERTi as described in our previous work (28). All lentiviral vectors used in this study allow detection of the infected cells by GFP+ signal under the epifluorescence microscope.
For some experiments, we utilized duplex siRNA targeting GRHL2 or the control, scrambled siRNA (Santa Cruz Biotechnology), which was introduced using Lipofectin (Invitrogen). Rapidly proliferating NHEK (2 × 105) were plated in 60-mm dishes and transfected with 60 nm siRNA. The cultures were harvested after 3 days post-transfection for biochemical analyses.
PMP-WB Assay
PMP was performed using the streptavidin-coated Dynabeads® (Invitrogen) according to the methods described elsewhere (10). Biotinylated hTERT promoter DNA fragment from −544 to +5 was amplified in a PCR using the forward primer 5′-CTCCGTCCTCCCCTTCAC-3′ and the reverse primer 5′-ATCGATCAGCGCTGCCTGAAACTC-3′ with biotin conjugation on the 5′ end of the forward primer. The PCR product was then immobilized onto the beads and incubated with 200 μg of nuclear extract isolated from SCC4 cells. After a 1-h incubation at room temperature, the beads were collected by the magnetic apparatus and washed with the binding buffer containing 0.5% Nonidet P-40. Proteins that bound the hTERT promoter were co-precipitated with the beads, and were fractionated by SDS-PAGE. Subsequently, Western blotting was performed to determine the presence of GRHL2, Sp1, or RNA polymerase II in the protein samples that co-precipitated with the magnetic beads. For the regional mapping analysis of the hTERT promoter, the 145-bp hTERT promoter fragment (−97 to +48) was amplified into seven overlapping fragments using the specific primer sets. The sequence of the primers and PCR conditions will be available upon request. The PCR amplification included biotinylated forward primers to allow for immobilization of the PCR products onto Dynabeads®. Subsequently, PMP-WB was performed as described above to detect binding of GRHL2, Sp1, or polymerase II onto different PCR fragments.
To determine the specificity of binding, we incorporated specific and nonspecific competitors in the PMP-WB analyses. Specific competitor was the identical hTERT promoter sequence without the 5′-biotinylation such that it would not be immobilized onto Dynabeads®. Noncompetitor was the 55-bp unrelated DNA sequence derived from pLPCX vector (Clontech) digested with HindIII and ClaI with the sequence, 5′-TCGAATTCG-TTAGGCCATTAAGGCCTGTCGACAAGCGGCCGCCTC-GGCCAAACAT-3′.
Construction of Mutant hTERT Promoter Fragments
hTERT promoter variants were constructed by site-directed mutagenesis of the pGL3B-TRTP plasmid using the QuikChange multisite-directed mutagenesis kit (Stratagene, La Jolla, CA). We introduced three consecutive nucleotide sequence alterations by converting purines to pyrimidines and vice versa from the sequences at −53 to −13. Briefly, a 25-μl PCR was carried out with 100 ng of pGL3B-TRTP as template, 100 ng of mutagenic primer, 2.5 μl of Multireaction buffer, 0.75 μl of QuikSolution, 1 μl of dNTPs, and 1 μl of QuickChange multienzyme blend. All primers designed to introduce the site-directed mutation were labeled with phosphorylation at the 5′ end. Primers were synthesized and purified by Integrated DNA Technologies (Coralville, IA), and the sequences will be available upon request. The extension reaction was initiated by pre-heating the reaction mixture to 95 °C for 1 min, 30 cycles of 95 °C for 1 min, 55 °C for 1 min, and 65 °C for 15 min according to the length of the template. The PCR amplification products were digested with 1 μl of DpnI at 37 °C for 1 h to digest the parental double-stranded DNA. An aliquot of 1.5 μl of DpnI-treated DNA from each mutagenesis reaction was transformed into XL10-Gold ultracompetent cells. Positive colonies were selected, and their plasmids were isolated by mini-prep for DNA sequencing. The mutant hTERT promoter fragments were used directly for PMP-WB by immobilizing them onto Dynabeads® or inserted into the pGL3B-TRTP for promoter-luciferase analysis.
Western Blotting
Whole cell extracts (WCE) from cultured cells were fractionated by SDS-PAGE and transferred to Immobilon protein membrane (Millipore, Billerica, MA) and probed for the presence of various proteins using the aforementioned antibodies. Densitometric analyses were performed using the Scion Image software (Scion Corp., Frederick, MD) and normalized against β-actin.
Reverse Transcription (RT)-PCR
Total RNA was isolated from the cultured cells using the RNeasy mini kit (Qiagen, Chatsworth, CA). The RT reaction was performed with 5 μg of RNA, and the PCRs were performed for hTERT, PCNA, GRHL2, and GAPDH in a semi-quantitative manner. The PCR amplification was performed with the following primer sequences: 5′-CGAGCTGCTCAGGTCTTTCTTTTATG-3′ (forward) and 5′-CCACGACGTAGTCCATGTTCACAATC-3′ (reverse) for hTERT with the annealing temperature of 56 °C; 5′-AGGGCTCCATCCTCAAGAAG-3′ (forward) and 5′-CTCCTGGTTTGGTGCTTCAA-3′ (reverse) for PCNA at 54 °C; 5′-CTTAGTGCCCATGCCCAGTG-3′ (forward) and 5′-CGCTTGGAATTCTCCAGGTG-3′ (reverse) for GRHL2 at 54 °C; and 5′-GACCCCTTCATTGACCTCAAC-3′(forward) and 5′-CTTCTCCATGGTGGTGAAGA-3′(reverse) for GAPDH at 55 °C. The PCR products were fractionated by agarose gel electrophoresis and stained with ethidium bromide.
hTERT Promoter Luciferase Assay
A pGL3B-TRTP containing the 1670-bp fragment (−1665 to + 5) of the hTERT promoter region linked to firefly luciferase cDNA (kindly provided by Dr. J. C. Barrett, NCI, National Institutes of Health, Bethesda) was used for the hTERT promoter reporter assay. We included either wild-type hTERT promoter sequence or the mutants as mentioned above for the promoter assay. Prior to transfection, a 6-well plate with ∼5 × 104 cells per well was inoculated and cultured for 24 h. pGL3B-TRTP and the variant plasmids (1 μg per each well) were introduced into cells using Lipofectin (Invitrogen). We also included the hTERT promoter deletion from −49 to +5 using the plasmid, pGL3-TRTP(Δ−49/+5), as described in our previous report (10). Transfection efficiencies were controlled by pRL-SV40 plasmid (1 ng per each well) containing the Renilla luciferase cDNA under the control of SV40 enhancer/promoter. Cells were collected 48 h post-transfection, and the lysates were prepared using the Dual-Luciferase reporter assay system (Promega, Madison, WI). Luciferase activity was measured using a luminometer (Turner Designs, Sunnyvale, CA). The hTERT promoter activity was determined as the mean of triplicate experiments.
Telomerase Assay
Telomerase activity was detected using the TRAP-eze telomerase detection kit (Chemicon, Temecula, CA) according to the manufacturer's guidelines. This kit allows for semi-quantitative detection of telomerase enzyme activity and amplification of the internal control. WCE isolated in 1× CHAPS buffer was used with the TRAP reaction mixture in the two-step telomerase assay involving the PCR amplification (2). The PCR products were separated by 12.5% nondenaturing PAGE. The radioactive signal was detected by PhosphorImager (GE Healthcare).
Methylation-specific PCR (MSP)
Methylation of the hTERT promoter at the 5′-CpG island was studied by MSP, according to the methods described elsewhere (30). The genomic DNAs were isolated from the cultured cells using Qiagen tissue/blood kit and modified by bisulfite treatment. Promoter DNA at two different regions, R1 (from −217 to +29) and R2 (from +32 to +219), were amplified using the primer sets that can recognize only bisulfite-modified DNA sequences (U primers) or unmodified sequences (M primers). PCR products were electrophoresed in 2% agarose and visualized under UV illumination.
Real Time Quantitative MSP
Quantitative MSP analysis was performed in triplicate for each sample using LightCycler 480 SYBR Green I master kit (Roche Applied Science) according to the manufacturer's guideline. Amplification was carried out in 96-well plates using LightCycler 480, with 0.25 μm of each primer and bisulfite modified DNA in a 10-μl final reaction volume. Using the MSP primers for the hTERT promoter region 2 (R2), thermal cycling was initiated with an incubation step of 10 min at 95 °C, followed by 45 cycles (95 °C for 10 s, 55 °C for 20 s, and 72 °C for 20 s). Alu gene amplification was used to control for DNA input amount (31). 2−ΔΔCq value determination method was used to compare the fold differences in which ΔΔCq = Cq(hTERT) − Cq(Alu). Methylation index (%) was calculated as M/(M + U) × 100, where M is the quantity of methylated hTERT promoter, and U is the quantity of unmethylated hTERT promoter (32).
DNA Methyltransferase Activity Assay
NHEK were treated with or without 1 μm 5-aza-2′-deoxycytidine (5-aza-CdR, Sigma) for 5 days and then were harvested. Nuclear extracts were prepared using a commercial kit according to manufacturer's protocol (Active Motif, Carlsbad, CA). DNMT1 activity was measured using an EpiQuikTM DNA methyltransferase activity assay kit (Epigentek, Brooklyn, NY). Nuclear extracts were incubated with the methylation substrate for 1 h at 37 °C, and then exposed to the capture antibody for 60 min and the detection antibody for 30 min at room temperature. Absorbance was read using a microplate spectrophotometer at 450 nm, and the DNMT1 enzyme activity (OD/h/ml) was calculated according to the following formula: (sample OD − blank OD)/(sample volume × 1000), according to the manufacturer's guide.
RESULTS
GRHL2 Extends the Replicative Life Span of Normal Human Keratinocytes but Cannot Immortalize the Cells
We investigated the role of GRHL2 in regulation of cell proliferation by assessing the expression level of GRHL2 in cultured NHK. Endogenous GRHL2 expression was detectable in the proliferating culture of NHK and decreased during senescence (Fig. 1A). This decrease occurred with a concomitant reduction in the expression of PCNA, a cell proliferation marker (26), and a progressive increase in the expression of p16INK4A. We checked for the intracellular localization of GRHL2 by fractionating the cells at replicating and senescing phases into the cytoplasmic and nuclear fractions (Fig. 1B). GRHL2 was found mostly in the nuclear fraction of NHK and SCC4 cells, and the intranuclear protein level was reduced dramatically during senescence.
FIGURE 1.

GRHL2 expression is decreased during senescence of NHK. A, Western blotting was performed with the WCEs of primary NHOK (strain 08-2) harvested at the indicated PD levels. Intracellular levels of GRHL2, PCNA, and p16INK4A were determined. β-Actin was probed for the loading control. B, NHEK (07-2) and NHOK (07-12) cultures were fractionated into cytoplasmic (denoted as C) or nuclear (denoted as N) samples at the indicated PD levels. Western blotting was performed for GRHL2 and lamin B. Fractionated samples of SCC4 were included for comparison. C, rapidly proliferating NHEK (strain 09-1) were infected with LXSN or LXSN-GRHL2 and maintained in serial subcultures. Red arrow indicates the point of infection with LXSN-GRHL2. D, NHOK infected with LXSN or LXSN-GRHL2 were stained for senescence-associated β-galactosidase activity at the indicated PD levels, which were calculated from the point of primary culture. Original magnification, ×100.
To determine the role of GRHL2 in the control of normal cell proliferation, exogenous GRHL2 was transduced in NHK using the retroviral vectors. Primary NHEK were infected with LXSN-GRHL2 or the LXSN control vector, and maintained in serial subcultures. The parental NHEK and those infected with LXSN completed 22 PDs until senescence (Fig. 1C). GRHL2 transduction led to an extended life span for almost 50 PDs before senescence, although the cells did not become immortalized. Furthermore, GRHL2 led to a reduction in the number of cells expressing the senescence-associated β-galactosidase activity (Fig. 1D), a cytochemical marker of senescence (33).
To gain insight into the mechanism underlying the enhanced cell proliferation, we performed Western blotting for the key proteins associated with keratinocyte proliferation and differentiation. NHEK infected with LXSN or LXSN-GRHL2 were harvested at the increasing PD levels, and Western blotting was performed for GRHL2, PCNA, p53, p21WAF1, involucrin, and p16INK4A (Fig. 2A). Elevated GRHL2 expression was noted in cells infected with LXSN-GRHL2, and the exogenous GRHL2 expression was maintained at this level throughout the in vitro culture. Thus, the cell division arrest beyond PD 49 was not due to the loss of exogenous GRHL2 expression. In fact, the transduced cells with higher PDs demonstrated increased level of GRHL2, suggesting selection for the cells with higher GRHL2 expression. Involucrin expression was drastically increased in the control cells during senescence but was inhibited in the cells transduced with GRHL2. Interestingly, these cells accumulated similar level of p16INK4A as the control at senescence (PD 19 and 23), yet underwent an additional 20 PDs before senescence in the presence of high p16INK4A level. Thus, extension of the replicative life span by GRHL2 did not occur through p16INK4A suppression. Rather, GRHL2 allowed the cells to gain resistance to the growth inhibitory effects of p16INK4A and replicate in the absence of keratinocyte differentiation.
FIGURE 2.

GRHL2 transduction allows NHK to proliferate in the presence of elevated p16INK4A levels while attenuating the subculture-induced keratinocyte differentiation. A, NHOK (09-1) infected with LXSN or LXSN-GRHL2 were harvested at the indicated PD levels, and Western blotting was performed for GRHL2, PCNA, p53, p21WAF1, involucrin, and p16INK4A. β-Actin was probed as loading control. B, three independent cultures of NHOK (08-3, 08-4, and 919) were infected with LXSN or LXSN-GRHL2. Western blotting was performed with WCE obtained at the indicated PD levels for GRHL2, hTERT, PCNA, pRb, and p-Rb (Ser-807/811). β-Actin and CDK6 were used for loading controls. Densitometric analysis was performed and plotted.
GRHL2 Delays the Loss of hTERT Expression and Telomerase Activity in NHK
Because GRHL2 regulates the hTERT promoter activity, we further characterized the effects of GRHL2 on hTERT expression and telomerase activity. GRHL2 transduction led to increased levels of hTERT, PCNA, and hyperphosphorylated pRb (Ser-807/811) compared with those infected with LXSN (Fig. 2B). Parental NHOK at PD 11 and those infected with LXSN or LXSN-GRHL2 demonstrated readily detectable telomerase activity (Fig. 3). Telomerase activity was then lost in the control cells during senescence at PD 23. On the contrary, the GRHL2-transduced cells maintained telomerase activity beyond PD 40, although the enzyme activity gradually decreased as the cells reduced the proliferation rate. Thus, GRHL2 significantly prolonged the time period during which telomerase activity was expressed in cells, although the level of the enzyme activity was not induced by GRHL2.
FIGURE 3.
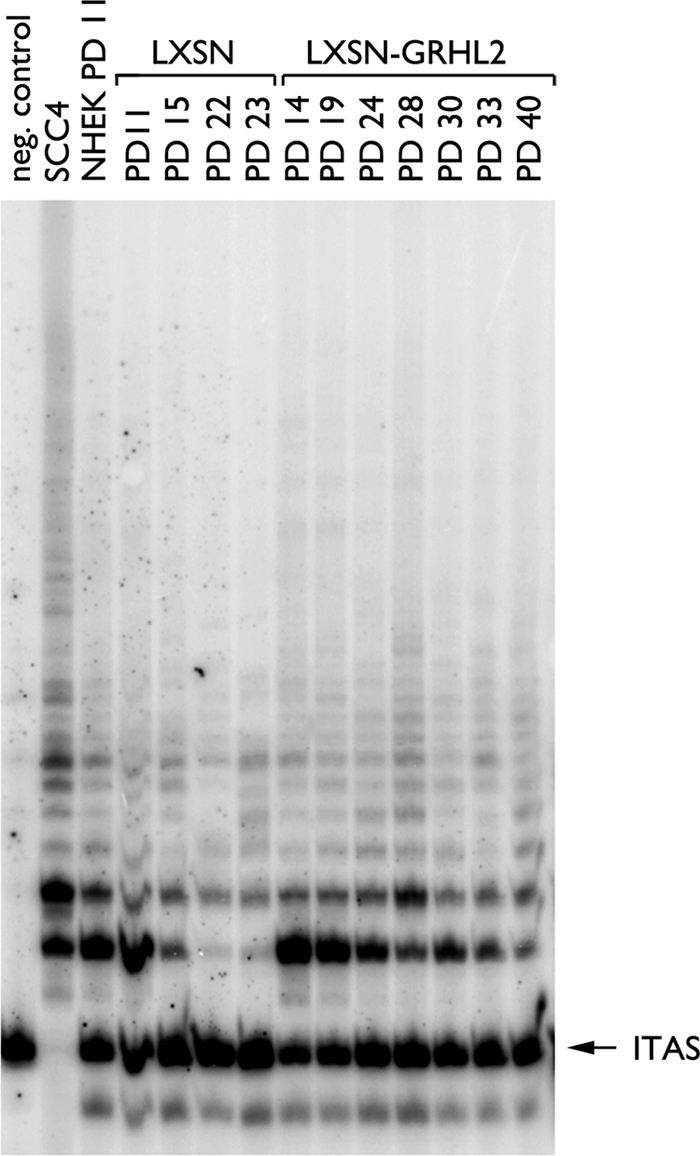
Duration of active telomerase is prolonged in NHOK by GRHL2 transduction. NHOK (09-1) infected with LXSN or LXSN-GRHL2 were harvested at the indicated PD levels and assayed for telomerase activity. WCE (1 μg) isolated in 1× CHAPS buffer was used for the PCR-based TRAP assay. The radioactively labeled PCR products were separated by 12.5% nondenaturing PAGE. ITAS, internal telomeric amplification standard.
We performed a series of GRHL2 knockdown experiments in several different cell types, including primary NHEK, SCC15, SCC4, and the NHOK/Bmi-1 cells, using the lentiviral vector targeting GRHL2 expression (LV-GRHL2i). Infection of cells with the viral vectors was highly efficient, as revealed by the EGFP labeling (Fig. 4A). SCC15 and SCC4 cells expressed higher levels of hTERT than did NHEK or the NHOK/Bmi-1 cells. Thus, after GRHL2 knockdown, the OSCC cells exhibited a greater extent of hTERT down-regulation. GRHL2 knockdown reduced the cell proliferation rate and the levels of PCNA and hTERT expression compared with the control (Fig. 4, B and C). Telomerase activity was diminished in NHEK infected with LV-GRHL2i (data not shown). To determine the effects of GRHL2 knockdown in the organotypic culture, we established the three-dimensional culture with SCC15 cells infected with LV-EGFP or with LV-GRHL2i (Fig. 4D). Compared with the three-dimensional culture prepared with NHEK, SCC15 formed an irregular epithelial sheath showing nuclear atypia and invasion into the subepithelial layer. SCC15 cells after GRHL2 knockdown showed epithelial atrophy, reduced epithelial thickness, and loss of the invasive phenotype. Thus, GRHL2 expression in cancer cells may be required for the maintenance of the cancerous phenotype.
FIGURE 4.

GRHL2 knockdown reduces cell proliferation, hTERT mRNA expression, and telomerase activity. A, rapidly proliferating NHEK and SCC15 cells were infected with LV-EGFP or LV-GRHL2i. Shown in the panel are the phase contrast and fluorescence images of the cells at 72 h after infection to demonstrate the efficiency of infection. Original magnification, ×100. B, NHEK, SCC15, and NHOK/Bmi-1 cells were infected with LV-EGFP or LV-GRHL2i and harvested after 72 h post-infection. Semi-quantitative RT-PCR was performed to determine the level of GRHL2, PCNA, and hTERT mRNA with or without GRHL2 knockdown. GRHL2 knockdown in SCC4 cells was accomplished in our previous report (10). GAPDH was amplified as a loading control. C, effects of GRHL2 knockdown on cell proliferation were determined in NHEK, NHOK/Bmi-1, and SCC15 cells by counting the cells after 72 h post-infection. D, organotypic raft cultures were established with NHEK and SCC15 infected with LV-EGFP or LV-GRHL2i. After 14 days of culture, the reconstructs were harvested for histological examination in sagittal orientation.
In the next experiment, we determined whether GRHL2 enhances cell proliferation through an hTERT-dependent manner. Endogenous hTERT expression was knocked down in the GRHL2-transduced cells. NHEK infected with LXSN or LXSN-GRHL2 were superinfected with LV-hTERTi or LV-EGFP (empty vector). The cells infected with the lentiviral vectors were identified by the GFP+ signal (Fig. 5A). As expected, GRHL2 transduction led to enhanced cell proliferation compared with the control cells, but the cell proliferation was notably inhibited after hTERT knockdown in the GFP+ cells (Fig. 5, A and B). Notably, NHEK infected with LXSN-GRHL2 exhibited nonproliferative, flattened morphology after hTERT knockdown. PCNA expression was strongly enhanced in cells transduced with GRHL2, but hTERT knockdown inhibited such an effect (Fig. 5C). By comparing the cell counts of GFP+ and GFP− cells, it appears that the GFP+ cells were outgrown by the GFP− cells after hTERT knockdown, and this was more evident in the GRHL2-transduced culture than the control. hTERT knockdown was efficient (∼ 80%) as shown in the RT-quantitative PCR data in the LXSN-infected NHEK (Fig. 5C), but GRHL2-transduced NHEK showed moderate level of hTERT knockdown. This may be due to enhanced hTERT expression resulting from GRHL2 transduction and/or overgrowth of GFP− cells in the same culture. These data suggest that GRHL2 promotes NHEK cell proliferation in part through the hTERT-dependent mechanism.
FIGURE 5.

Knockdown of endogenous hTERT counteracts the enhanced proliferation by GRHL2 transduction. A, NHEK were infected with LXSN or LXSN-GRHL2. After selection with 200 μg/ml G418, the cells were infected with LV-EGFP (empty vector) or LV-hTERTi. Subsequently, the cells were maintained in culture for 8 days and photographed for phase contrast view or under epifluorescence. Original magnification, ×100. B, all test groups were started with 1.0 × 105 cells per 100-mm dish. After 8 days post-infection with the lentiviral vectors, cell numbers were counted under phase contrast (total cells) or under epifluorescence (GFP+). Shown in the bar graph are the cell counts in each group relative to that of NHEK/LXSN infected with LV-EGFP. C, relative expression of PCNA and hTERT mRNA was determined in cells by RT-quantitative PCR. The graph shows the mean of triplicates and the standard deviation.
Characterization of the Interaction between GRHL2 and the hTERT Promoter
To delineate the role of GRHL2 in hTERT regulation, we characterized the protein-DNA interactions between GRHL2 and the hTERT promoter region by PMP-WB. For these series of experiments, we compared the binding of GRHL2 to the hTERT promoter with that of Sp1, which is a well characterized transcription factor known to bind the hTERT promoter (34). The specificity of the interaction was confirmed using the nonbiotinylated DNA competitor, which reduced the binding of GRHL2 and Sp1 to the immobilized hTERT DNA (Fig. 6A). PMP-WB using the cells with varying levels of GRHL2 revealed congruent levels of GRHL2 binding to the promoter DNA, although the binding of Sp1 was not affected (Fig. 6B). These data ruled out the mutual dependence of binding between GRHL2 and Sp1 to the promoter DNA.
FIGURE 6.

GRHL2 binds in the proximal regions of the hTERT promoter. A, PMP was performed with 200 μg of nuclear extracts of SCC4 cells in the presence of the 62-mer DNA containing the hTERT promoter sequences from −52 to +9. The competitor was made of the identical 62-mer DNA but without conjugation to Dynabeads® due to the absence of 5′-biotinylation. The nonspecific competitor DNA was composed of unrelated vector DNA sequences. The presence of GRHL2 and Sp1 in the magnetic precipitates was determined by Western blotting. B, PMP-WB was performed as described in A but using the nuclear extracts from different cells, such as NHOK/Bmi-1, parental SCC4, and SCC4 infected with LV-EGFP or LV-GRHL2i. C, regional map is shown for the seven overlapping fragments of the hTERT promoter amplified by PCR. D, PMP was performed with the seven hTERT fragments using nuclear extract of SCC4 cells. Western blotting detected GRHL2, Sp1, and RNA polymerase II (Pol II) in the magnetic precipitates associated with different fragments.
We generated seven overlapping fragments of the hTERT promoter DNA from −97 to +48 by PCR amplification (Fig. 6C). These fragments were independently tested for their ability to bind GRHL2. The strongest interaction was noted with the fragment 4, and GRHL2 weakly interacted with the fragments 2 and 3. Hence, the minimal sequences required for GRHL2 binding spans from −53 to −13. We then made mutations in three consecutive nucleotides from −53 to −13 and assessed the GRHL2 binding (Fig. 7A). The mutations at −21 to −19 notably reduced the binding, although the others did not (Fig. 7B). Sp1 binding was abolished by the mutations at −33 to −31 and −27 to −25; these mutations coincide precisely with the Sp1 binding region reported previously (34). The mutations that abolished the binding of Sp1 and GRHL2 led to marked loss of hTERT promoter luciferase activity (Fig. 7C). These data indicate that the 40-bp fragment from −53 to −13 is sufficient for GRHL2 and Sp1 binding and that the GRHL2-binding site would require the three nucleotides from −21 to −19 of the hTERT promoter.
FIGURE 7.

Mutational analysis of the hTERT sequences associated with GRHL2. A, hTERT promoter sequences from −53 to −13 were mutated in three consecutive nucleotide sequences, yielding seven different mutant sequences as indicated in red. Underlined sequences are the Sp1-binding sites reported elsewhere (34). B, PMP was performed with the seven mutant and the wild-type hTERT promoter sequences. Western blotting revealed GRHL2 and Sp1 binding with the DNA. C, SCC4 cells were transfected with pGL3-Luc, pGL3-TRTP, or the pGL3-TRTP containing the mutant sequences (either mutant 5 or mutant 6), and pGL3-TRTPΔ(−49 to +5) containing the deletion of the sequences from −49 to +5. Firefly and Renilla luciferase activities were determined at 48 h post-transfection and plotted as normalized values. Error bars represent standard errors.
GRHL2 Inhibits the DNA Methylation at the 5′-CpG Island of the hTERT Promoter
We previously showed that the hTERT promoter is inactivated in senescent NHOK and NHOF by DNA hypermethylation at the 5′-CpG island (9). Because GRHL2 maintained the hTERT mRNA expression during the extended life span, we tested for the possible inhibitory effects of GRHL2 on DNA methylation. MSP was performed with the bisulfite-modified genomic DNA at two different regions designated R1 and R2. Rapidly proliferating NHEK/LXSN cells showed hypomethylation in both regions at PD12, and methylation was increased at PD 23 when the cells underwent senescence (Fig. 8A). On the contrary, R1 and R2 remained primarily hypomethylated in the NHEK/GRHL2 cells throughout the in vitro culture beyond PD 38. Our previous study showed that exogenous Bmi-1 transduction in NHOK led to enhanced cellular proliferation and extension of the replicative life span without prolonging the hTERT mRNA expression (25). Thus, we determined the methylation status of the hTERT promoter in the NHOK/Bmi-1 cells to test whether the promoter hypomethylation in the NHEK/GRHL2 cells is a by-product of enhanced proliferation or linked with GRHL2 overexpression. Rapidly proliferating NHOK/B0 (control cells) and NHOK/Bmi-1 showed a hypomethylated promoter at PDs 15–16, and the promoter became hypermethylated in both cultures with the PD levels 22–25 at which time the NHOK/Bmi-1 cells maintained active cell proliferation (Fig. 8B). Therefore, the reduced DNA methylation in the NHEK/GRHL2 cells appears to be linked with GRHL2 transduction and not a by-product of enhanced cell proliferation. Furthermore, knockdown of endogenous GRHL2 in rapidly proliferating NHEK (PD 19) led to hypermethylation at the R2 region of the hTERT promoter, although the scrambled siRNA had no effect (supplemental Fig. S1). This finding further supports the involvement of GRHL2 in hTERT regulation in part via modulation of DNA methylation.
FIGURE 8.

GRHL2 inhibits DNA methylation of the hTERT promoter. A, MSP was performed with the bisulfite-modified genomic DNA isolated from the cells at the indicated PD levels. The NHEK/LXSN cells (09-1) were rapidly proliferating at PD 12 and senesced at PD 25, and the NHEK/GRHL2 cells proliferated exponentially and entered the senescing phase near PD 38 (Fig. 1). R1 and R2 indicate the two different regions of the hTERT promoter amplified by MSP (see “Experimental Procedures”). U and M indicate amplification with the primer sets recognizing the hypo- and hypermethylated DNA, respectively. B, MSP was also performed with the NHK/B0 cells harvested at PD 15 (replicating) and PD 22 (senescing) and with actively replicating NHK/Bmi-1 cells harvested at PDs 16 and 25. C, MSP was performed with quantitative PCR for the R2 region, and the methylation index was calculated by taking the % ratio of the M amplification signal to the combined M and U signals. D, DNMT1 enzyme activity was performed with the nuclear extracts of NHEK (PD 15) and the cells infected with LXSN (PDs 21 and 27) and LXSN-GRHL2 (PDs 20 and 34). The parent NHEK (PD 15) treated with 5-aza-CdR (1 μm) for 5 days was included as a control (denoted as PD15-z).
Using quantitative MSP, we quantitated the level of the promoter methylation at the varying stages and determined the methylation index, which is the % ratio of the methylation amplification signal to that of the combined signals (32). As shown in Fig. 8C, the methylation index was drastically increased in the control cells nearing senescence, whereas it was notably suppressed in the GRHL2-transduced cells even at the higher PD level. Furthermore, the enzyme activity of DNMT1, which is responsible for the CpG methylation (35), was strongly induced in the NHEK control cells at high PDs nearing senescence and correlated well with the hTERT promoter methylation status (Fig. 8D). Treatment of the replicating cells at PD 15 with 1 μm 5-aza-CdR for 5 days inhibited the DNMT1 activity. GRHL2 transduction notably inhibited the DNMT1 enzyme activity compared with the control cells, even at the higher PDs. These data support the notion that GRHL2 maintains the hTERT expression in NHK by inhibiting DNA methylation, possibly by interfering with the DNMT enzyme activity.
DISCUSSION
We showed that GRHL2 is a novel transcription factor of hTERT that maintains the gene expression and the telomerase activity in NHK and modulates the replicative life span of cells. This study was performed by transduction of exogenous full-length GRHL2, which enhanced cell proliferation, prolonged expression of hTERT and telomerase activity, and reduced keratinocyte differentiation. GRHL2 transduction also led to inactivation of pRb by phosphorylation at Ser-807/811 and increased expression of PCNA, although its effect on p16INK4A expression was not noted. This is interesting because p16INK4A is a major determinant of senescence in NHK (36), yet GRHL2 allowed the cells to continue to replicate in the presence of high p16INK4A levels. GRHL2 knockdown led to reduction of hTERT expression, loss of telomerase activity, and reduced cell proliferation without affecting the cell viability. Endogenous GRHL2 was found most predominantly in the nuclei of both primary NHK and OSCC cells, and the expression level was heavily dependent on cellular replication status. Senescent NHK demonstrated almost complete loss of GRHL2 expression. We previously reported notable telomerase activity in rapidly proliferating NHK and complete loss of the enzyme activity during senescence (26, 37). Because GRHL2 is necessary for hTERT mRNA expression and telomerase activity, the reduced GRHL2 expression may be responsible for the loss of telomerase activity during senescence in NHK. Also, our data suggest that the positive effect of GRHL2 on cell proliferation is dependent on hTERT expression because hTERT knockdown suppressed the proliferation of the GRHL2-transduced cells.
In this study, PMP-WB enabled detailed molecular analyses of the interaction between GRHL2 and the hTERT promoter in a highly quantitative manner. This is indeed a major advantage of PMP-WB over other approaches, such as electrophoretic mobility shift assay (EMSA). EMSA relies on the degree of the antibody-associated altered mobility to reveal the specific protein-DNA interaction (38). PMP-WB, on the other hand, allows for direct and quantitative detection of the proteins bound to DNA. Using this approach, we found that GRHL2 binds to the hTERT promoter in a sequence-specific manner within the region from −53 to −13, possibly interacting with the three nucleotides from −21 to −19. Also, we mapped the proximal binding site of Sp1, a transcription regulator of hTERT (39), located within the region from −33 to −25 of the hTERT promoter (Fig. 7B). These nucleotide sequences precisely match the known Sp1-binding site reported earlier (34), further adding confidence to the PMP-WB approach. Mutations of the GRHL2- and Sp1-binding sites abolished the hTERT promoter activity. Other mutations within the proximal regions from −53 to −13 also reduced the promoter activity (data no shown), suggesting occupancy of other unknown essential transcription factors in this region.
This study showed that GRHL2 inhibits DNA methylation of the hTERT promoter. Because hypermethylation of the CpG island is associated with hTERT promoter inactivation in senescent NHK (9), GRHL2 may utilize this epigenetic mechanism to prolong the hTERT expression during the extended replicative life span. We previously showed that chemical inhibition of DNMT1 with 5-aza-CdR in senescent NHOK and NHOF led to re-expression of the hTERT mRNA (9). Thus, the hTERT CpG methylation is likely due to the enzyme activity of the DNMT. The finding that GRHL2 inhibits the DNMT1 activity further supports the involvement of GRHL2 in this epigenetic regulation of the hTERT promoter.
It is noteworthy that GRHL2 transduction did not enhance the level of telomerase activity, although it maintained the level of the enzyme activity during replication. On the contrary, human papillomavirus type 16 E6 protein markedly increased telomerase activity beyond that of rapidly proliferating NHOK and led to cellular immortalization (25). This may explain the fact that GRHL2 alone could not immortalize the cells in this study. Hence, GRHL2 appears to be required for the maintenance hTERT expression and telomerase activity, yet not sufficient to further enhance them.
We previously showed that Bmi-1 overexpression in NHOK markedly increased cell proliferation without enhancing or prolonging the hTERT expression (25). Interestingly, Bmi-1 transduction had no effect on the hTERT promoter methylation despite the robust proliferation of the transduced cells. Thus, the promoter hypomethylation in the GRHL2-transduced cells is unlikely a by-product of enhanced cell proliferation. Our study revealed the hTERT gene as the first known transcriptional target of GRHL2. In fact, we identified numerous other target genes of GRHL2 by microarray and chromatin immunoprecipitation analyses.3 It is possible that this epigenetic mechanism involving inhibition of DNA methylation is commonly used by GRHL2 for its target gene regulation. Also, the phenotypic effects of GRHL2, i.e. enhanced cell proliferation, may result from altered expression of other GRHL2 target genes in addition to hTERT.
GRHL2 is also known as Brother-of-Mammalian Grainyhead identified based on the sequence similarity to Drosophila GRH, especially within the DNA binding and dimerization domains (18). GRH transcriptionally regulates the target gene expression by physical binding to the promoters. GRH binds the enhancer element of the DOPA decarboxylase promoter in Drosophila, and this binding is required for the wound-responsive promoter activation of DOPA decarboxylase (40). GRH also binds the upstream regulatory element of the PCNA promoter and enhances the gene expression (17). The sequence of the GRH-binding site in the Drosophila PCNA promoter is known as AAACCAGTTGGCA and that of Drosophila DOPA decarboxylase as ATAACCGGTTTCC (17, 41). Perhaps due to the amino acid sequence homology, human GRHL1 can also bind these promoters, indicating functional conservation between Drosophila GRH and the mammalian GRHL proteins (18). A recent study revealed the consensus binding sequence of GRHL1 as AACCGGTT (22). The binding site for GRHL2 is not known; it may likely be different from those of GRHL1 or GRH due to the sequence dissimilarity between the above mentioned binding sequences and the hTERT promoter sequence revealed in this study.
When GHRL2 was knocked down in SCC15 cells, the three-dimensional culture demonstrated atrophic epithelium that had lost the invasive phenotype (Fig. 4D). Furthermore, a recent study reported the association between the gain of GRHL2 and recurrence of hepatocellular carcinoma (23). These findings suggest possible involvement of GRHL2 in carcinogenesis, differently from GRHL1 or GRHL3, both of which are involved in epithelial morphogenesis, differentiation, and barrier formation (15). Our current data showed that GRHL2 inhibits keratinocyte differentiation that occurs during serial subcultures (Fig. 2B). We also performed microarray experiments to profile differential gene expression induced by GRHL2 transduction in NHEK. GRHL2 overexpression led to marked reduction of GRHL1 and GRHL3 mRNA expression and numerous other genes involved in keratinocyte differentiation, such as involucrin, loricrin, and the proteins required for cornified envelope formation.3 Therefore, GRHL2 may negatively target the expression of these genes to impede keratinocyte differentiation and epithelial morphogenesis while enhancing keratinocytes proliferation and possibly promoting carcinogenesis.
Supplementary Material
Acknowledgment
We thank Dr. J. C. Barrett for the hTERT promoter construct (pGL3B-TRTP).
This work was supported, in whole or in part, by National Institutes of Health Grants R01DE18295 and K02DE18959 from NIDCR.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
W. Chen, Q. Dong, K.-H. Shin, R. H. Kim, J.-E. Oh, N.-H. Park, and M. K. Kang, unpublished data.
- hTERT
- human telomerase reverse transcriptase
- OSCC
- oral squamous cell carcinomas
- GRH
- grainyhead
- PCNA
- proliferating cell nuclear antigen
- NHK
- normal human keratinocytes
- NHOK
- normal human oral keratinocytes
- NHEK
- normal human epidermal keratinocytes
- WCE
- whole cell extract
- PD
- population doublings
- MSP
- methylation-specific PCR
- 5-aza-CdR
- 5-aza-2′-deoxycytidine
- LV
- lentiviral vector
- WB
- Western blot
- EGFP
- enhanced GFP.
REFERENCES
- 1.Counter C. M., Meyerson M., Eaton E. N., Ellisen L. W., Caddle S. D., Haber D. A., Weinberg R. A. (1998) Oncogene 16, 1217–1222 [DOI] [PubMed] [Google Scholar]
- 2.Kim N. W., Piatyszek M. A., Prowse K. R., Harley C. B., West M. D., Ho P. L., Coviello G. M., Wright W. E., Weinrich S. L., Shay J. W. (1994) Science 266, 2011–2015 [DOI] [PubMed] [Google Scholar]
- 3.Counter C. M., Avilion A. A., LeFeuvre C. E., Stewart N. G., Greider C. W., Harley C. B., Bacchetti S. (1992) EMBO J. 11, 1921–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hahn W. C., Counter C. M., Lundberg A. S., Beijersbergen R. L., Brooks M. W., Weinberg R. A. (1999) Nature 400, 464–468 [DOI] [PubMed] [Google Scholar]
- 5.Boldrini L., Pistolesi S., Gisfredi S., Ursino S., Ali G., Pieracci N., Basolo F., Parenti G., Fontanini G. (2006) Int. J. Oncol. 28, 1555–1560 [DOI] [PubMed] [Google Scholar]
- 6.Kyo S., Kanaya T., Takakura M., Tanaka M., Inoue M. (1999) Int. J. Cancer 80, 60–63 [DOI] [PubMed] [Google Scholar]
- 7.Dickson M. A., Hahn W. C., Ino Y., Ronfard V., Wu J. Y., Weinberg R. A., Louis D. N., Li F. P., Rheinwald J. G. (2000) Mol. Cell. Biol. 20, 1436–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horikawa I., Michishita E., Barrett J. C. (2004) Cytotechnology 45, 23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shin K. H., Kang M. K., Dicterow E., Park N. H. (2003) Br. J. Cancer 89, 1473–1478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang X., Chen W., Kim R. H., Kang M. K., Park N. H. (2009) Oncogene 28, 565–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang J. D., Dubnicoff T., Liaw G. J., Bai Y., Valentine S. A., Shirokawa J. M., Lengyel J. A., Courey A. J. (1995) Genes Dev. 9, 3177–3189 [DOI] [PubMed] [Google Scholar]
- 12.Narasimha M., Uv A., Krejci A., Brown N. H., Bray S. J. (2008) J. Cell Sci. 121, 747–752 [DOI] [PubMed] [Google Scholar]
- 13.Almeida M. S., Bray S. J. (2005) Mech. Dev. 122, 1282–1293 [DOI] [PubMed] [Google Scholar]
- 14.Bray S. J., Kafatos F. C. (1991) Genes Dev. 5, 1672–1683 [DOI] [PubMed] [Google Scholar]
- 15.Stramer B., Martin P. (2005) Curr. Biol. 15, R425-R427 [DOI] [PubMed] [Google Scholar]
- 16.Cenci C., Gould A. P. (2005) Development 132, 3835–3845 [DOI] [PubMed] [Google Scholar]
- 17.Hayashi Y., Yamagishi M., Nishimoto Y., Taguchi O., Matsukage A., Yamaguchi M. (1999) J. Biol. Chem. 274, 35080–35088 [DOI] [PubMed] [Google Scholar]
- 18.Wilanowski T., Tuckfield A., Cerruti L., O'Connell S., Saint R., Parekh V., Tao J., Cunningham J. M., Jane S. M. (2002) Mech. Dev. 114, 37–50 [DOI] [PubMed] [Google Scholar]
- 19.Ting S. B., Wilanowski T., Cerruti L., Zhao L. L., Cunningham J. M., Jane S. M. (2003) Biochem. J. 370, 953–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ting S. B., Caddy J., Hislop N., Wilanowski T., Auden A., Zhao L. L., Ellis S., Kaur P., Uchida Y., Holleran W. M., Elias P. M., Cunningham J. M., Jane S. M. (2005) Science 308, 411–413 [DOI] [PubMed] [Google Scholar]
- 21.Yu Z., Lin K. K., Bhandari A., Spencer J. A., Xu X., Wang N., Lu Z., Gill G. N., Roop D. R., Wertz P., Andersen B. (2006) Dev. Biol. 299, 122–136 [DOI] [PubMed] [Google Scholar]
- 22.Wilanowski T., Caddy J., Ting S. B., Hislop N. R., Cerruti L., Auden A., Zhao L. L., Asquith S., Ellis S., Sinclair R., Cunningham J. M., Jane S. M. (2008) EMBO J. 27, 886–897 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanaka Y., Kanai F., Tada M., Tateishi R., Sanada M., Nannya Y., Ohta M., Asaoka Y., Seto M., Shiina S., Yoshida H., Kawabe T., Yokosuka O., Ogawa S., Omata M. (2008) J. Hepatol. 49, 746–757 [DOI] [PubMed] [Google Scholar]
- 24.Kang M. K., Bibb C., Baluda M. A., Rey O., Park N. H. (2000) Exp. Cell Res. 258, 288–297 [DOI] [PubMed] [Google Scholar]
- 25.Kim R. H., Kang M. K., Shin K. H., Oo Z. M., Han T., Baluda M. A., Park N. H. (2007) Exp. Cell Res. 313, 462–472 [DOI] [PubMed] [Google Scholar]
- 26.Kang M. K., Guo W., Park N. H. (1998) Cell Growth & Differ. 9, 85–95 [PubMed] [Google Scholar]
- 27.Dongari-Bagtzoglou A., Kashleva H. (2006) Nat. Protoc. 1, 2012–2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang M. K., Kim R. H., Kim S. J., Yip F. K., Shin K. H., Dimri G. P., Christensen R., Han T., Park N. H. (2007) Br. J. Cancer 96, 126–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kang M. K., Park N. H. (2007) Methods Mol. Biol. 371, 151–165 [DOI] [PubMed] [Google Scholar]
- 30.Dessain S. K., Yu H., Reddel R. R., Beijersbergen R. L., Weinberg R. A. (2000) Cancer Res. 60, 537–541 [PubMed] [Google Scholar]
- 31.Kristensen L. S., Wojdacz T. K., Thestrup B. B., Wiuf C., Hager H., Hansen L. L. (2009) BMC Cancer 9, 453–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lo Y. M., Wong I. H., Zhang J., Tein M. S., Ng M. H., Hjelm N. M. (1999) Cancer Res. 59, 3899–3903 [PubMed] [Google Scholar]
- 33.Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., Pereira-Smith O. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Horikawa I., Cable P. L., Afshari C., Barrett J. C. (1999) Cancer Res. 59, 826–830 [PubMed] [Google Scholar]
- 35.Svedruziæ Z. M. (2008) Curr. Med. Chem. 15, 92–106 [DOI] [PubMed] [Google Scholar]
- 36.Rheinwald J. G., Hahn W. C., Ramsey M. R., Wu J. Y., Guo Z., Tsao H., De Luca M., Catricalà C., O'Toole K. M. (2002) Mol. Cell. Biol. 22, 5157–5172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang M. K., Kameta A., Shin K. H., Baluda M. A., Park N. H. (2004) J. Cell. Physiol. 199, 364–370 [DOI] [PubMed] [Google Scholar]
- 38.Cann J. R. (1998) Electrophoresis 19, 127–141 [DOI] [PubMed] [Google Scholar]
- 39.Wooten L. G., Ogretmen B. (2005) J. Biol. Chem. 280, 28867–28876 [DOI] [PubMed] [Google Scholar]
- 40.Mace K. A., Pearson J. C., McGinnis W. (2005) Science 308, 381–385 [DOI] [PubMed] [Google Scholar]
- 41.Uv A. E., Thompson C. R., Bray S. J. (1994) Mol. Cell. Biol. 14, 4020–4031 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.