

Abstract
The role of the SDF-1α-CXCR4 axis in response to myocardial infarction is unknown. We addressed it using the CXCR4 antagonist, AMD3100, to block SDF-1α interaction with CXCR4 after chronic coronary artery ligation. Chronic AMD3100 treatment decreased ejection fraction and fractional shortening in mice 20 days after myocardial infarction compared with vehicle-treated mice (echocardiography). Morphometric analysis showed hearts of AMD3100-treated infarcted mice to have expanded scar, to be hypertrophic (confirmed by myocyte cross-section area) and dilated, with increased LV end systolic and end diastolic dimensions, and to have decreased scar collagen content; p-AKT levels were attenuated and this was accompanied by increased apoptosis. Despite increased injury, c-kitpos cardiac progenitor cells (CPCs) were increased in the risk region of AMD3100-treated infarcted mice; CPCs were CD34neg/CD45neg with the majority undergoing symmetric cell division. c-kitpos/MHCpos CPCs also increased in the risk region of the AMD3100-treated infarcted group. In this group, GSK-3β signaling was attenuated compared to vehicle-treated, possibly accounting for increased proliferation and increased cardiac committed MHCpos CPCs. Increased proliferation following AMD3100 treatment was supported by increased levels of cyclin D1, a consequence of increased prolyl isomerase, Pin1, and decreased cyclin D1 phosphorylation. In summary, pharmacologic antagonism of CXCR4 demonstrates that SDF-1α-CXCR4 signaling plays an important role during and after myocardial infarction and that it exerts pleiotropic salubrious effects, protecting the myocardium from apoptotic cell death, facilitating scar formation, restricting CPC proliferation, and directing CPCs toward a cardiac fate.
Keywords: Cytokines, Stromal cell derived factor-1α, Receptors, CXCR4, AMD3100, myocardial infarction, progenitor cells
Introduction
The chemokine, stromal cell derived factor-1α (SDF-1α) and its receptor, CXCR4, play a significant role in hematopoiesis, HIV infection, and the regulation of hematopoietic stem and progenitor cells (HSPCs). SDF-1α levels are regulated by factors that include induction with hypoxia[1] and inactivation by extracellular proteases such as CD26/dipeptididylpeptdidase[2]. SDF-1α binding to CXCR4, a G-protein coupled receptor, regulates downstream effectors through signaling via MAPKs, AKT, STAT3, lipid rafts, and caveolin dependent mechanisms[3].
Few studies have focused on delineation of SDF-1α-CXCR4 signaling in the heart. We have recently found both SDF-1α and CXCR4 to be expressed in the heart, both in myocytes and non-myocyte cell populations, and to provide a protective effect from ischemia-reperfusion injury through the activation of ERK and AKT with a concomitant decrease in apoptosis[4]. SDF-1α has also been shown to exert a negative inotropic response via its coupling to Gi[5].
A well established action of SDF-1α-CXCR4 signaling is to maintain HSPCs in the bone marrow[6]. Maintenance of HSPCs in the bone marrow relies on the chemoattractant properties of the SDF-1α-CXCR4 signaling axis. This action of SDF-1α has been a target for therapeutic intervention after cardiac injury [7-10]. The rationale for the therapeutic use of SDF-1α is focused on the potential to increase angiogenesis as well as the potential to increase the recruitment of circulating stem cells and progenitors to the site of increased SDF-1α, or the site of injury. These early studies demonstrated that overexpression or preservation of SDF-1α was capable of improving LV function, decreasing the extent of injury and increasing vascular density as well as the number hematopoietic progenitors[7-11]. While it is clear that increased SDF-1α-CXCR4 signaling is beneficial, the physiological role of SDF-1α-CXCR4 in the heart and in the response to injury is unclear.
Clinically, SDF-1α-CXCR4 has been targeted for both the mobilization of HSPCs and the prevention of HIV infection. Mobilization of HSPCs with GCSF for transplant has led to benefits such as improved engraftment, decreased hospital stay, and improved survival. GCSF-induced mobilization relies on inactivation of SDF-1α-CXCR4 decreasing the homing signal originating in the bone marrow stroma [12]. However, these benefits come at the cost of patient comfort[13] and with variation in effect due to genetic factors [14]. In recent trials, the CXCR4 competitive antagonist, AMD3100 (Plerixafor), has been shown to be effective in mobilizing HSPCs with and without GCSF in a shorter period of time with less toxicity to the patient and cells[15]. AMD3100 has also been found to be effective in blocking HIV entry via CXCR4[16].
Although SDF-1α-CXCR4 signaling has been extensively studied in many tissues, limited information exists concerning its role in the heart. Targeted deletion of SDF-1α and CXCR4 results in embryonic lethality[6, 17]. To determine the role that SDF-1α-CXCR4 plays in response to chronic cardiac injury, we used AMD3100 to block SDF-1α-CXCR4 signaling after coronary artery ligation. We present findings that demonstrate that SDF-1α-CXCR4 signaling is important in the response to myocardial infarction to limit the extent of injury and preserve function. In addition we also find that blockade of SDF-1α-CXCR4 signaling leads to expansion and arrested maturation of committed cardiac progenitors, demonstrating a role for cardiac CXCR4 in regulating CPC proliferation and cardiac commitment.
Methods
Detailed methods are available online.
Results
CXCR4 Blockade with AMD3100 exacerbates myocardial dysfunction after coronary artery ligation
We have shown that SDF-1α-CXCR4 signaling in the heart promotes protection against acute ischemia/reperfusion injury[4], but the role of SDF-1α-CXCR4 in the myocardial response to chronic infarction is unknown. To elucidate this issue, the competitive antagonist, AMD3100, was used to block SDF-1α-CXCR4 interaction and CXCR4 activation. AMD3100 was infused via subcutaneous osmotic minipumps. In the vehicle infusion group, coronary occlusion did not affect body weight, EF, FS, or LV chamber diameter or volume compared with sham surgery (Figure 1). The preservation of LV function and morphology in the vehicle infusion group is consistent with the small infarct sizes (Figure 2). Administration of AMD3100 did, however, lead to a significant decline in LV function with decreases in both EF and FS (Figure 1A and B). The decline in LV function was accompanied by a significant increase in LV chamber diameter and volumes (Figures 1C-F and Supplemental Figures 3 and 4).
Figure 1.
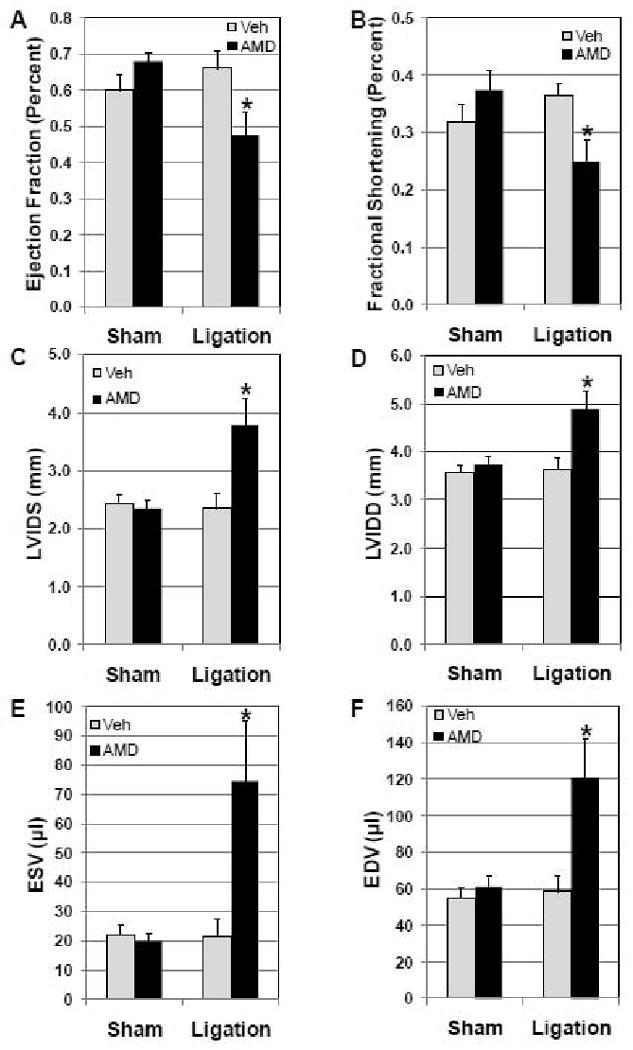
AMD3100 attenuates cardiac function and accentuates myocardial injury after coronary artery occlusion. Mice were treated chronically with AMD3100 by osmotic minipump (ALZA). Minipumps implanted 24 h prior to coronary occlusion delivered AMD3100 or vehicle for 21 days. Mice underwent coronary artery ligation or sham surgery 24 h after implantation of minipumps. LV function, ejection fraction (EF, A) and fractional shortening (FS, B), and LV morphometry, left ventricular inner dimension systolic (LVIDS, C) and diastolic (LVIDD, D),end systolic volume (ESV, E),end diastolic volume (EDV, F) were assessed 20 days after surgery in vivo by echocardiography in Sham Vehicle (n=8), Sham AMD (n=8), Ligated vehicle (n=8), and Ligated AMD (n=10) treated mice. Values are mean ± SEM *P<0.05 vs Vehicle Ligation.
Figure 2.
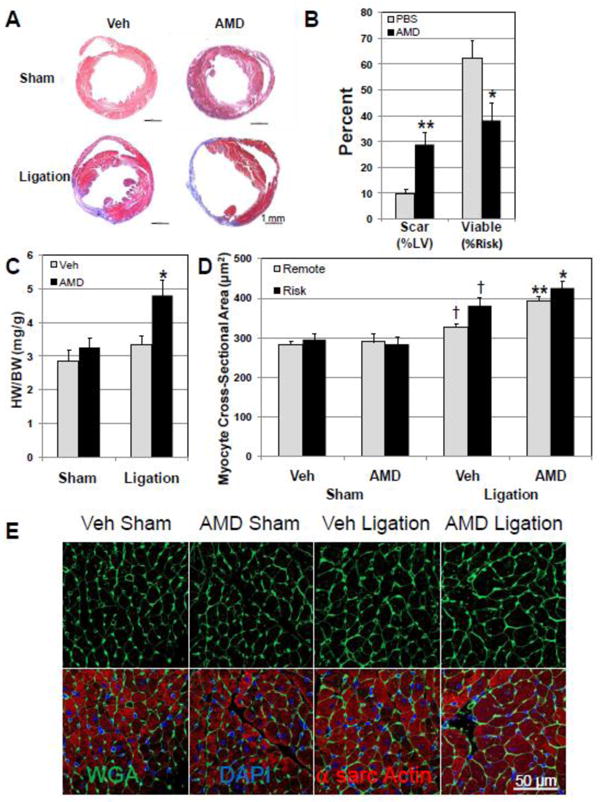
AMD3100 treatment increases myocardial injury and accentuates LV hypertrophy after infarction. Scar and viable myocardium in the risk region were determined in Masson's trichrome stained LV sections 20 days after surgery. Myocyte cross-sectional area was determined in sections stained with FITC-conjugated wheat germ agglutinin (WGA). (A) Representative Masson's trichrome stained heart short axis sections. (B) Scar as percent of LV was increased in AMD3100- treated hearts after ligation as viable myocardium in the risk region was decreased(n=8-13). (C) Heart weight (HW/BW) was significantly increased in AMD3100-treated mice after ligation (final body weights: Veh Sham, 31.6± 0.8 g; AMD Sham, 30.8± 0.6g: Veh Lig, 30.6±0.8g; AMD Lig, 31.5±0.7g.) (D) Myocyte cross-sectional area in FITC-labeled WGA stained sections was significantly increased in both risk and remote regions in AMD3100-treated hearts after ligation (n=6-8). (E) Representative FITC-labeled WGA stained cross-sections. Values are mean ± SEM. *P<0.05, **P<0.005 vs Vehicle Ligation; † P<0.05 vs Vehicle Sham.
AMD3100 increases myocardial infarction and remodeling
The effect of AMD3100 on the extent of injury was assessed in Masson's trichrome stained sections from vehicle- and AMD3100-treated hearts after sham surgery or ligation. Morphometric analysis at sacrifice 20 days after coronary ligation demonstrated that AMD3100 increased myocardial scar significantly after ligation (Figure 2A and 2B). The risk region was similar in AMD3100- and vehicle-treated groups with infarction (Supplemental Figure 3). The increase in injury was accompanied by a decrease in viable myocardium within the risk region (Figure 2B). The LV chamber was significantly dilated, confirming the increased chamber dimensions observed with echocardiography (Supplemental 4 and 5). The LV dilation, LV hypertrophy and LV dysfunction in infarcted hearts treated with AMD3100 are indicative of a progression towards heart failure, demonstrating a protective role of the SDF-1α–CXCR4 axis in response to infarction. Hearts from AMD3100-treated mice after ligation were also hypertrophic compared to vehicle-treated infarcted mice (Figure 2C and Supplemental Figures 3 and 4). Increased LV mass could be attributed to myocyte hypertrophy, because myocyte cross-sectional area was significantly increased in AMD3100-treated infarcted mice in both the risk and remote areas (Fig 2D and 2E). AMD3100 treatment had no effect on myocyte cross-sectional area in the sham group (Figure 2D).
To examine the mechanisms by which AMD3100 blockade of CXCR4 signaling could impair the response to injury leading to LV dilatation and dysfunction, capillary density, apoptosis and collagen content were assessed. Compensated hypertrophy requires an increase in capillary density to provide adequate blood supply for increased myocardial demands. Uncompensated hypertrophy and dysfunction have been attributed to failure of capillary density to increase with myocardial hypertrophy through angiogenesis[18]. Cardiac sections were stained with the endothelial marker isolectin B4 (FITC-conjugated) and capillary density determined in risk and remote regions (Figure 3A and B). In the sham surgery group, AMD3100 treatment led to a small decrease in capillary density compared to vehicle-treated. In the coronary ligation group, AMD3100 treatment resulted in a significant decline in capillary density in the risk area and a small decline in the remote area. Capillary density in the vehicle-treated ligation group was unchanged compared with vehicle-treated/sham mice. Picrosirius red staining showed that AMD-treated infarcted hearts had less collagen in the scar than Veh-treated hearts (Supplemental Figure 6). This decrease in collagen content may contribute to dilation of AMD3100-treated hearts after infarction.
Figure 3.
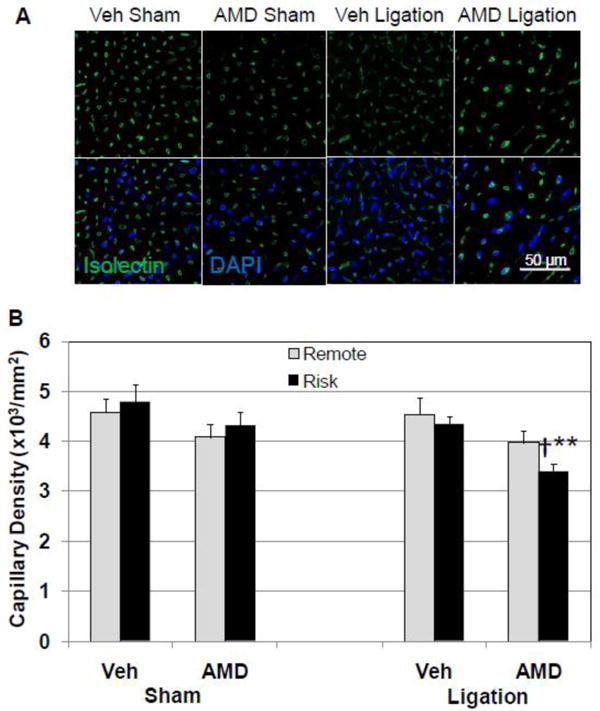
AMD3100 treatment decreases capillary density in infarcted mice. Capillaries in risk and remote regions of the LV were labeled with FITC-conjugated isolectin B4 and counted. (A) Representative fluorescent confocal images of FITC-isolectin stained cross sections from vehicle- and AMD3100-treated mice after ligation or sham surgery. All images have the same scale. (B) Quantitative analysis of capillary density demonstrating a decrease in capillary density in AMD3100-treated mice after coronary ligation. Values are mean ± SEM (n=6). **P<0.001 vs Vehicle Ligation; †P<0.01 vs AMD Sham
Heart failure is associated with an increase in apoptotic cell death that contributes to myocyte loss and decreased functional capacity[19]. AMD3100 treatment slightly increased the prevalence of apoptosis (assessed by TUNEL staining) in the sham surgery group. Although coronary ligation increased apoptosis in the LV of both vehicle- and AMD3100-treated groups, apoptosis in AMD3100-treated infarcted hearts was significantly more prevalent than in vehicle-treated infarcted hearts (Figure 4A,B, and C).
Figure 4.
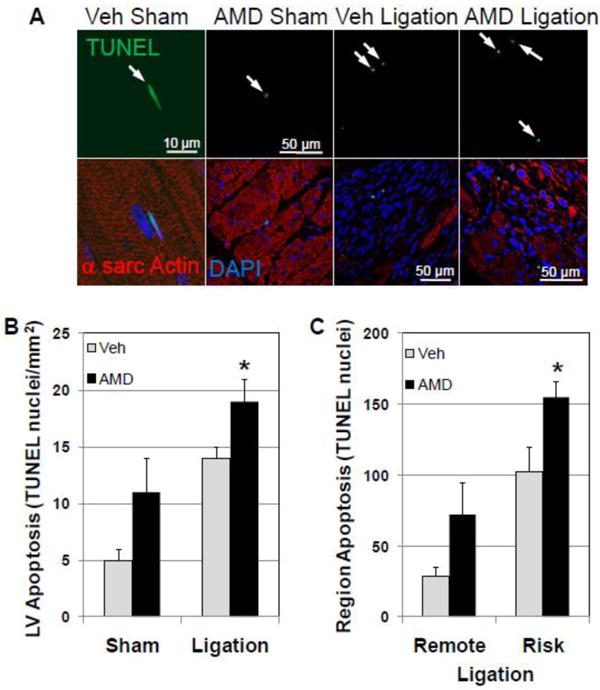
Apoptotic cell death is increased in AMD3100-treated mice after infarction. Apoptotic cell death was assessed in Vehicle- and AMD3100- treated hearts after ligation and sham surgery by TUNEL staining. (A) Representative fluorescent confocal images of TUNEL stained apoptotic nuclei from vehicle- and AMD3100-treated mice after ligation or sham surgery. (B) TUNEL staining and apoptotic cell death is increased in the LV of AMD3100-treated hearts after ligation. (C) Apoptotic cell death was predominant in the risk region of AMD treated hearts after ligation. Arrows denote apoptotic nuclei. Values are mean ± SEM (n=6-8). *P<0.05 vs Vehicle Ligation.
Activation of tissue committed c-kitpos CPCs
SDF-1α-CXCR4 signaling plays an important role in stem cell homing and maintenance in the hematopoietic system[6] and has been exploited to increase homing of stem and progenitor cells to the heart after injury[7-8]. To determine whether antagonism of the SDF-1α-CXCR4 interaction after infarction would antagonize the homing and engraftment of stem and progenitor cells to the site of injury and their contribution to repair, and thus account for the observed functional decline, the number of c-kitpos CPCs was counted in risk and remote regions. In vehicle-treated hearts, low numbers of c-kit pos CPCs were identified in the LV and this number increased significantly after coronary ligation (Figure 5A). Surprisingly, in AMD3100-treated hearts, c-kit pos CPCs in the risk region were significantly higher than in the vehicle-treated ligation group (Figure 5A). Although CPC numbers were increased in the remote area, they were not significantly different between AMD3100-treated and vehicle-treated infarcted mice.
Figure 5.
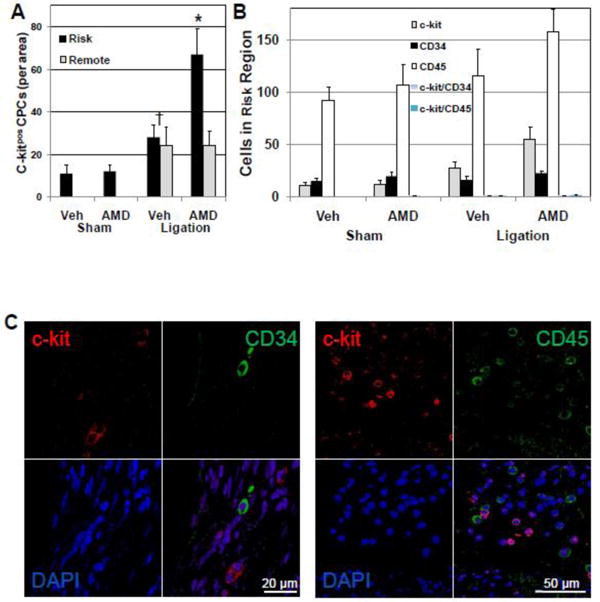
c-kitpos/CD34neg/CD45neg CPC numbers are increased in AMD3100-treated mice after ligation. CPCs were identified by staining for c-kit. Lineage of c-kitpos CPCs was determined by counterstaining and colocalization with lymphoid, endothelial, and myeloid markers CD34 or CD45. (A) AMD3100-treatment increased c-kitpos CPCs in the risk area of the LV after ligation.(B) CD34pos and CD45pos cells were present in the risk area but did not colocalize with c-kitpos CPCs. (C) Representative confocal images of c-kitpos, CD34pos and CD45pos cells in the risk area of AMD3100-treated ligated mice. Values are mean ± SEM (n=6). *P<0.05 vs Vehicle Ligation.
To ascertain whether the increased number of c-kit pos CPCs in the risk region of AMD3100-treated hearts were derived from the periphery as a consequence of increased mobilization and were of hematopoietic or endothelial progenitor cell (EPC) origin, c-kitpos CPCs were also stained for myeloid, lymphoid, endothelial, and hematopoietic markers, CD34 and CD45. Very few of the c-kitpos CPCs in the risk region expressed either CD34 or CD45 (Figure 5B and C). After coronary ligation and AMD3100 treatment CD34pos cells in the LV were not elevated and CD45pos cells were only slightly, nonsignificantly increased (Figure 5B and C). Levels of CD34pos or CD45pos cells were not affected in the remote region (Supplemental Figure 7). These results suggest that the increase in c-kitpos CPCs in hearts of AMD3100-treated mice after ligation originated from resident cardiac progenitors (c-kitpos CPCs).
If this is the case, increased proliferation would be a mechanism that could account for this increase in CPC number. The proliferative state of c-kitpos CPCs was assessed by immunofluorescent staining forKi67 and phosphohistone H3 as well as c-kit. c-kitpos CPCs expressing the cell cycle/mitosis marker, Ki67, were increased in the risk region of AMD3100-treated hearts after ligation compared with vehicle-treated hearts (Figure 6A and C). The increase in proliferating c-kitpos CPCs was verified with the mitosis marker, phosphohistone H3. CPCs co-expressing phosphohistone H3 and c-kitpos were also increased in AMD3100-treated hearts after ligation (Figure 6B and D). In all groups, levels of proliferating c-kitpos CPCs in remote regions were similarly low. Thus the increase in c-kitpos CPCs was due, in part, to increased proliferation of tissue resident c-kitpos CPCs in the risk region after ligation.
Figure 6.
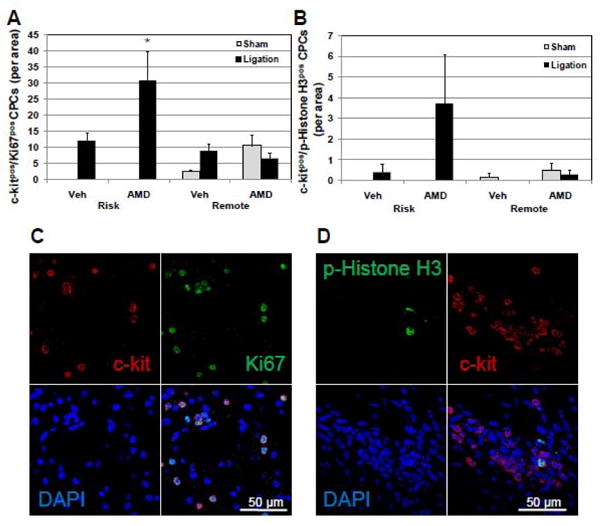
AMD3100 treatment increases proliferating c-kitpos CPCs in the risk area after ligation. To determine whether the increased number of c-kitpos CPCs in the LV of mice treated with AMD3100 after ligation was a consequence of increased proliferation, the number of c-kitpos CPCs that had entered the cell cycle 20 days after ligation was determined by immunofluorescent detection of the proliferation marker, Ki67, and the mitosis marker, phosphohistone H3. (A) c-kitpos/Ki67pos CPCs are increased in the risk area in AMD treated mice after ligation. (B) c-kitpos/phosphohistone H3pos CPCs are increased in the risk area of AMD treated mice after ligation. (C and D) Representative confocal images of proliferating c-kitpos CPCs expressing Ki67 or phosphohistone H3. Values are mean ± SEM (n=6). *P<0.05 vs Vehicle Ligation; § P<0.05 vs Vehicle Sham; †P<0.05 vs AMD Sham.
AMD3100 attenuates AKT and GSK-3β signaling after infarction
Previously we had shown that SDF-1α-CXCR4 interact to activate the pro-survival signaling pathways, AKT and ERK, in myocytes[4]. To assess this, mice were treated with AMD3100 or vehicle 24 h prior to ligation or sham surgery and hearts harvested 48 h after sham or coronary ligation surgery for analysis of activated signaling kinases. In the vehicle-treated group, ligation had little effect on AKT or GSK-3β 9Ser phosphorylation (Figure 7A, 7B and 7C). However, p-AKT levels were significantly decreased as a consequence of AMD3100 treatment (Figure 7A and B). The decline in p-AKT was a consequence of decreased AKT levels that were evident in both sham and infarcted groups with AMD3100 treatment (Supplemental Fig 8). AKT has multiple downstream targets that affect cell survival and proliferation. To validate the finding that AMD3100 decreased total and activated AKT and to determine the downstream consequences of decreased levels of p-AKT, we evaluated activation of the downstream target of AKT, GSK-3β. GSK-3 β activity is reciprocally regulated by phosphorylation at serine9 (S9, inhibited) and tyrosine216 (Y216, activated) residues[20-21]. GSK-3β S9 phosphorylation was unchanged with ligation or AMD3100 treatment although there was a slight increase in levels in sham and infarcted AMD3100-treated groups (Figure 7A and D). Conversely, GSK-3β Y216 phosphorylation levels decreased with AMD3100 treatment in sham and significantly decreased in the infarcted group (Figure 7A and C). The decrease in Y216 phosphorylation and the slight increase in S9 phosphorylation would result in a net decrease in GSK-3 β activity.
Figure 7.
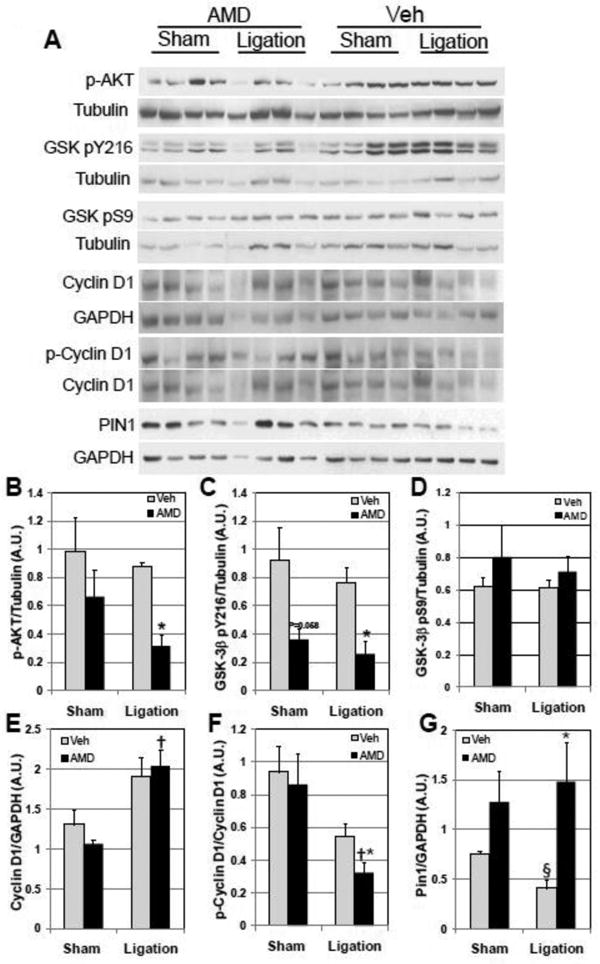
AMD3100 blockade of SDF-1α-CXCR4 signaling attenuates survival signaling while increasing proliferative signaling. (A) The effect of AMD blockade on CXCR4 signaling was assessed by Western analysis in mice treated with Vehicle or AMD with ligation or sham surgery 48 hr after surgery. Summarized data of normalized Western analysis is as follows: (B) p-AKT levels are attenuated in AMD3100-treated infarcted hearts. (C) GSK-3β pY216 is attenuated in AMD3100-treated infarcted hearts. (D) GSK-3β pS9 is insignificantly increased in AMD3100-treated hearts. (E) Cyclin D1 is significantly increased in AMD3100-treated infarcted hearts. (F) p-Cyclin D1 levels are decreased in AMD3100-treated infarcted mice. (G) Pin1 levels are increased in AMD3100-treated infarcted mice. Values are mean ± SEM (n = 4). *P<0.05 vs Vehicle Ligation; †P<0.05 vs AMD Sham.
AMD3100 increases cyclin D1 levels
The finding that c-kitpos CPCs have increased mitotic activity with AMD3100 treatment after ligation suggests that AMD3100 antagonism of CXCR4 would affect regulation of cell cycle. Cyclin D1 is a key regulator of G1/S phase transition and is a downstream target of GSK-3β[22-23]. Cyclin D1 phosphorylation increases association with CRM1 resulting in export of the complex from the nucleus, effectively attenuating cell division[23]. Because decreased levels of GSK-3β Y216 and increased levels of GSK-3β S9 in the infarcted AMD3100-treated group would be expected to decrease overall GSK-3β activity, phospho-cyclin D1 levels would be expected to be decreased; thus, we assessed cyclin D1 phosphorylation at Thr286. Coronary ligation increased levels of cyclin D1, with levels in AMD3100-treated hearts increased significantly compared with sham AMD3100-treated hearts (Figure 7A and E). In contrast, levels of p-cyclin D1 were significantly decreased in AMD3100-treated hearts after ligation compared with vehicle-treated infarcted hearts (Figure 7A and F). This is consistent with decreased GSK-3β activity. Increased cyclin D1 and decreased cyclin D1 phosphorylation contribute to increasing cell proliferation by advancing cells through the G1/S transition. Regulation of cyclin D1 may also occur via increased stabilization by the prolyl isomerase, Pin1[24]. Pin1 levels were significantly attenuated in infarcted vehicle-treated mice (Figure 7A and G). In contrast, AMD3100 treatment increased Pin1 expression in both sham and infarcted hearts. These observations provide a basis for the increase in cyclin D1 dependent proliferation of c-kitpos/CD34neg/CD45neg CPCs in the risk region of AMD3100-treated hearts after ligation and demonstrate that CXCR4 exerts its antiproliferative effect on cell cycle via multiple mechanisms.
AMD3100 increases cardiac committed c-kitpos CPCs
The increase in c-kitpos CPCs in AMD3100-treated hearts after ligation in the context of increased cardiac dysfunction and injury suggests that these progenitors are unable, or have limited capacity, to contribute any beneficial response to injury. Stem and progenitor cells have been shown to contribute to the myocardial response to injury,[25-26] specifically regenerating new myocytes in areas bordering myocardial infarction. Recent studies describing the role of GSK-3β in cardiomyocyte differentiation in the heart and in mesenchymal stem cells would suggest that attenuated GSK-3β signaling during AMD3100 treatment may also lead to an attenuated capacity of CPCs to commit to relevant cardiovascular lineages that contribute to repair of the injured myocardium[27-28]. In response to injury the CPC population would be expected to rapidly expand to maintain myocardial function. Such expansion would be expected to occur via asymmetric division, generating one committed daughter cell while the other maintains the capacity for self-renewal[29-30], as opposed to symmetric division, where both progeny would maintain the capacity for self renewal. To assess whether the proportion of symmetric vs asymmetric division played a role in this expansion, we determined the expression of the segregating determinants Numb and α-adaptin and their localization in c-kitpos CPCs (Figure 8). Although a relative paucity of these cells was observed, two observations were clear. First, the number of c-kitpos CPCs expressing either Numb or α-adaptin in the risk region of AMD3100-treated hearts was significantly increased compared with vehicle-treated hearts after ligation (Figure 8A-D), corresponding with increased proliferation. c-kitpos/Numbpos and c-kitpos/α-adaptin pos CPCs were not increased in remote areas in vehicle- or AMD3100-treated groups after ligation. Second, assessment of symmetric and asymmetric localization of Numb and α-adaptin in c-kitpos CPCs demonstrated that an increased number of c-kitpos cells in the risk region in AMD3100-treated mice with ligation underwent predominantly symmetric division (66% c-kitpos/Numbpos, 82% c-kitpos/α-adaptin pos). The fact that the preponderance of c-kitpos CPCs undergo symmetric division suggests that these CPCs maintain their capacity to self-renew, thereby limiting the production of cells committed to lineages that will participate in repair of injured myocardium.
Figure 8.

c-kitpos CPCs divide predominantly by symmetric mitotic division in AMD3100-treated hearts after ligation. The balance between self-renewal and commitment during cell division of the c-kitpos CPC pool was assessed by determining whether cell division was symmetric, indicative of self-renewal, or non-symmetric, indicative of the generation of a committed cell based on the polarized expression of Numb and α-adaptin. Myocardial commitment was assessed in c-kitpos CPCs by counterstaining for sarcomeric MHC and CPCs expressing both counted in risk and remote areas. Symmetric localization of Numb (A) and α-adaptin (B) in c-kitpos CPCs in AMD3100-treated hearts after ligation is increased in the risk area. (C and D) Representative confocal images of c-kitpos CPCs displaying symmetric (arrows) and asymmetric (arrow head) localization of Numb and α-adaptin. (E) Myocardial committed CPCs, c-kitpos/MHCpos are significantly elevated in AMD3100-treated infarcted hearts. (F) Representative confocal images of c-kitpos/MHCpos CPC (arrow). Values are mean ± SEM (n =6). *P<0.05 vs Vehicle; †P<0.05 vs Asymmetric.
To investigate whether c-kitpos CPCs maintained pluripotency or committed to a myogenic lineage, these cells were counterstained for MHC, and c-kitpos/MHCpos CPCs counted (Figure 8E and 8F). Low levels of c-kitpos/MHCpos CPCs were identified in the remote areas of vehicle- and AMD3100-treated hearts with sham surgery. In the risk region of AMD3100-treated hearts after infarction, c-kitpos/MHCpos CPCs were strikingly increased compared with vehicle-treated hearts (Figure 8E). CPCs expressing MHC appeared phenotypically similar to the progenitors that did not. When expressed as a fraction of total c-kitpos CPCs, CPCs expressing MHC were much higher in AMD3100-treated infarcted hearts (34% vs. 7% in vehicle-treated infarcted). Within the AMD3100-treated group, the uncommitted c-kitpos/MHCneg fraction was predominant (66%) and is consistent with the number of uncommitted CPCs predicted by Numb and α-adaptin symmetric localization (66-82%).
Discussion
SDF-1α-CXCR4 signaling is important for survival and protection from acute ischemia-reperfusion injury in the myocardium[4] and for stem and progenitor cell homing and survival[31].However, the role of SDF-1α-CXCR4 signaling in the response to chronic myocardial infarction is unknown. Using the CXCR4 antagonist, AMD3100, to block SDF-1α binding and activation of CXCR4 before and after coronary artery ligation, we found that myocardial injury was exacerbated, leading to LV dysfunction and dilatation, implying progression to heart failure. Dysfunction and dilatation were accompanied by LV hypertrophy, increased myocyte cross-sectional area, decreased collagen content, decreased capillary density, increased apoptosis and attenuated AKT and GSK-3β signaling, all of which indicate that SDF-1α-CXCR4 signaling is important in the myocardial response to infarction. However, contrary to our initial hypothesis that blockade of SDF-1α-CXCR4 signaling would prevent homing of peripheral or resident stem/progenitor cells to the injured myocardium and limit stem/progenitor based regeneration after injury, we found, surprisingly, increased numbers of c-kitpos CPCs in the risk region. This c-kitpos CPC population was CD34neg/CD45neg and likely derived from resident progenitors. The expansion of this c-kitpos CPC population can be attributed to increased proliferation, as suggested by increased cyclin D1 levels. Increased numbers of c-kitpos/MHCpos CPCs may suggest that myocardial commitment or differentiation is impaired. Taken together, these observations indicate that SDF-1α-CXCR4 signaling is important for the myocardial response to infarction and that it participates in this response by increasing survival and commitment or differentiation of CPCs.
We and others have previously reported that SDF-1α interaction with and activation of CXCR4 is important for survival signaling of the myocardium to prevent acute injury during ischemia reperfusion[4, 32]. The results of this investigation support these previous studies and extend them to a chronic infarction model. The attenuated pro-survival response to injury is supported by increased apoptosis, attenuated AKT and GSK-3β levels and activation, decreased capillary density, and increased hypertrophy in the AMD3100-treated mice 20 days after ligation. These changes in response to injury in the presence of AMD3100 are hallmarks of cardiac dysfunction and are indicative of progression to heart failure. Although attenuated GSK-3β activity is typically associated with myocardial protection in acute injury[33], attenuation of GSK-3β activity has been associated with LV dysfunction and increased apoptosis in response to chronic stress with pressure overload[34]. Further, the relevance of GSK-3β in cardioprotection may be limited to rodents as recent studies have demonstrated a role for GSK-3β regulation is absent in large animals[35]. Thus chronic attenuation of GSK-3β is consistent with dysfunction and increased apoptosis observed with AMD3100 treatment after infarction. The decrease in cardiac function associated with greater hypertrophy, LV chamber dilation and decreased wall thickness further support the progression of AMD3100-treated infarcted hearts towards failure.
The worsened function and remodeling observed here with chronic AMD3100 infusion prior to and during the injury and recovery phase contrast with the beneficial effect of daily AMD3100 administration initiated after MI. Daily administration of AMD3100 after MI in the study by Proulx et al. resulted in decreased infarct size and improved function [36] Although data are lacking to define the mechanism of such action, one might speculate the intermittent dosing facilitates increased mobilization of progenitors from the periphery. Mobilization is then followed by a permissive phase as levels of AMD31100 decline. This would include increased homing and engraftment of mobilized cells to the site of injury and the benefits of survival signaling.
Although mobilization protocols that use GCSF, Flt-3 ligand, and SCF to increase levels of circulating progenitors have been found to increase viable myocardium and vascular density and decrease remodeling while improving function[37], our results suggest that intrinsic cardiac SDF-1α-CXCR4 signaling is necessary for CPC function to repair myocardium in addition to limiting myocardial apoptotic cell death and remodeling. As AMD3100 was able to mobilize from the periphery, demonstrated by increased peripheral WBCs 20 days after ligation, the increase in c-kitpos CPCs that were CD34neg and CD45neg was somewhat surprising in view of the role that SDF-1α has been shown to play in homing. The increase in c-kitpos CPCs in the AMD3100-treated hearts after ligation thus appears to be a consequence of an increase in proliferation of CPCs, a decrease in their commitment or differentiation, or both. Others have also found that c-kitpos CPCs are predominantly negative for CD34 and CD45 [10, 25, 38]. Targeted deletion of CXCR4 in adult primitive hematopoietic cells resulted in increased proliferation and expansion of bone marrow hematopoietic progenitors[39]. CXCR4 has also been shown to drive expression of cardiac–specific genes in adherent bone marrow derived cells[40]. This may be a direct consequence of CXCR4 dependent GSK-3β modulation, since GSK-3β directly regulates cardiac commitment via a Wnt/β-catenin dependent pathway[27]. Another possibility that cannot be excluded is that circulating c-kitpos progenitors may lose CD34 and CD45 expression after engraftment within the 20 day period post-ligation.
Our data demonstrating the predominance of symmetric division of Numbpos and α-adaptin pos c-kitpos CPCs suggest that the proliferation of c-kitpos CPCs favors the maintenance of the self-renewal and uncommitted properties of these cells. This preference towards self-renewal occurs at the expense of limiting the number of CPCs that commit and contribute to regeneration/repair of the injured myocardium, and thus effectively contributes to the observed increase in the numbers of progenitors. This finding resembles the action of attenuated GSK-3β, which is known to bias cell fate toward self renewal in the heart[28] and in MSCs[27]. The predominance of c-kitpos/MHCneg CPCs supports the observed predominance of symmetric division. The increased numbers of c-kitpos CPCs in the AMD3100-treated group after ligation supports a paradigm in which CXCR4 plays a dual role in the regulation of CPCs in response to injury, restricting proliferation and facilitating commitment.
The changes in signaling that occur with AMD3100 blockade of CXCR4 signaling during and after myocardial infarction converge to establish an environment that favors proliferation as demonstrated by the increased numbers of c-kitpos CPCs that were still proliferating 20 days after ligation (Ki67pos and phosphohistone H3pos). The attenuation in AKT combined with that of GSK-3β Y216 initiates events that establish convergent actions on cell cycle. AKT is known to regulate GSK-3β and other regulators of cell cycle such as p27Kip[41], Myt1[42], Wee1[43] and p21Cip[44]. The decrease in GSK-3β Y216 phosphorylation with slightly elevated S9 phosphorylation would result in decreased GSK-3β activity. With cyclin D1 being a downstream target, the decrease in GSK-3β activity results in decreased phospho-cyclin D1 (Figure 7D). Cyclin D1 phosphorylation triggers its export from the nucleus, ubiquitinization, and proteosomal degradation[22-23]. Thus decreased GSK-3β-dependent phosphorylation of cyclin D1 will facilitate increased nuclear cyclin D1 levels and cell cycle progression which was observed in the AMD3100 treated infarcted hearts. Increased levels of Pin1 also supports increased levels of cyclin D1 through its stabilization[24]. Although cyclin D1 was markedly increased in vehicle-treated infarcted mice, the actions of decreased GSK-3β activity and increased Pin1 in AMD3100-treated mice would serve to facilitate increased proliferation by increasing nuclear cyclin D1.
Conclusions
These studies present novel findings that demonstrate SDF-1α-CXCR4 signaling is a necessary component of the natural myocardial response to injury; we show that blockade of this axis leads to a marked increase in extent of myocardial damage after coronary occlusion. The role that CXCR4 plays in response to injury can be attributed to pleiotropic actions, which include effects on survival, remodeling, proliferation and differentiation/commitment. These results suggest that SDF-1α-CXCR4 elicits effects that serve to preserve existing myocardium and facilitate replacement of lost myocardium through cardiac commitment of resident c-kitpos CPCs. These findings provide a strong rationale for further studies aimed at delineating the mechanisms that underlie the role of SDF-1α-CXCR4 in myocardial injury.
Supplementary Material
Acknowledgments
Funding Sources: This study was supported by NIH grants RO1-HL91202, R01-HL-68088, R01-HL-70897, R01-HL-76794, R01-HL-78825, R01-HL-55757, and U24HL094373.
Footnotes
Disclosures: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME, et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004 Aug;10(8):858–64. doi: 10.1038/nm1075. [DOI] [PubMed] [Google Scholar]
- 2.Christopherson KW, 2nd, Hangoc G, Broxmeyer HE. Cell surface peptidase CD26/dipeptidylpeptidase IV regulates CXCL12/stromal cell-derived factor-1 alpha-mediated chemotaxis of human cord blood CD34 + progenitor cells. J Immunol. 2002 Dec 15;169(12):7000–8. doi: 10.4049/jimmunol.169.12.7000. [DOI] [PubMed] [Google Scholar]
- 3.Kucia M, Jankowski K, Reca R, Wysoczynski M, Bandura L, Allendorf DJ, et al. CXCR4-SDF-1 signalling, locomotion, chemotaxis and adhesion. J Mol Histol. 2004 Mar;35(3):233–45. doi: 10.1023/b:hijo.0000032355.66152.b8. [DOI] [PubMed] [Google Scholar]
- 4.Hu X, Dai S, Wu WJ, Tan W, Zhu X, Mu J, et al. Stromal cell derived factor-1 alpha confers protection against myocardial ischemia/reperfusion injury: role of the cardiac stromal cell derived factor-1 alpha CXCR4 axis. Circulation. 2007 Aug 7;116(6):654–63. doi: 10.1161/CIRCULATIONAHA.106.672451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pyo RT, Sui J, Dhume A, Palomeque J, Blaxall BC, Diaz G, et al. CXCR4 modulates contractility in adult cardiac myocytes. J Mol Cell Cardiol. 2006 Nov;41(5):834–44. doi: 10.1016/j.yjmcc.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zou YR, Kottmann AH, Kuroda M, Taniuchi I, Littman DR. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature. 1998 Jun 11;393(6685):595–9. doi: 10.1038/31269. [DOI] [PubMed] [Google Scholar]
- 7.Abbott JD, Huang Y, Liu D, Hickey R, Krause DS, Giordano FJ. Stromal cell-derived factor-1alpha plays a critical role in stem cell recruitment to the heart after myocardial infarction but is not sufficient to induce homing in the absence of injury. Circulation. 2004 Nov 23;110(21):3300–5. doi: 10.1161/01.CIR.0000147780.30124.CF. [DOI] [PubMed] [Google Scholar]
- 8.Askari AT, Unzek S, Popovic ZB, Goldman CK, Forudi F, Kiedrowski M, et al. Effect of stromal-cell-derived factor 1 on stem-cell homing and tissue regeneration in ischaemic cardiomyopathy. Lancet. 2003 Aug 30;362(9385):697–703. doi: 10.1016/S0140-6736(03)14232-8. [DOI] [PubMed] [Google Scholar]
- 9.Segers VF, Tokunou T, Higgins LJ, MacGillivray C, Gannon J, Lee RT. Local delivery of protease-resistant stromal cell derived factor-1 for stem cell recruitment after myocardial infarction. Circulation. 2007 Oct 9;116(15):1683–92. doi: 10.1161/CIRCULATIONAHA.107.718718. [DOI] [PubMed] [Google Scholar]
- 10.Zaruba MM, Theiss HD, Vallaster M, Mehl U, Brunner S, David R, et al. Synergy between CD26/DPP-IV inhibition and G-CSF improves cardiac function after acute myocardial infarction. Cell Stem Cell. 2009 Apr 3;4(4):313–23. doi: 10.1016/j.stem.2009.02.013. [DOI] [PubMed] [Google Scholar]
- 11.Ara T, Tokoyoda K, Sugiyama T, Egawa T, Kawabata K, Nagasawa T. Long-term hematopoietic stem cells require stromal cell-derived factor-1 for colonizing bone marrow during ontogeny. Immunity. 2003 Aug;19(2):257–67. doi: 10.1016/s1074-7613(03)00201-2. [DOI] [PubMed] [Google Scholar]
- 12.Christopherson KW, 2nd, Cooper S, Broxmeyer HE. Cell surface peptidase CD26/DPPIV mediates G-CSF mobilization of mouse progenitor cells. Blood. 2003 Jun 15;101(12):4680–6. doi: 10.1182/blood-2002-12-3893. [DOI] [PubMed] [Google Scholar]
- 13.Anderlini P, Przepiorka D, Seong D, Miller P, Sundberg J, Lichtiger B, et al. Clinical toxicity and laboratory effects of granulocyte-colony-stimulating factor (filgrastim) mobilization and blood stem cell apheresis from normal donors, and analysis of charges for the procedures. Transfusion. 1996 Jul;36(7):590–5. doi: 10.1046/j.1537-2995.1996.36796323057.x. [DOI] [PubMed] [Google Scholar]
- 14.Broxmeyer HE, Orschell CM, Clapp DW, Hangoc G, Cooper S, Plett PA, et al. Rapid mobilization of murine and human hematopoietic stem and progenitor cells with AMD3100, a CXCR4 antagonist. J Exp Med. 2005 Apr 18;201(8):1307–18. doi: 10.1084/jem.20041385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liles WC, Rodger E, Broxmeyer HE, Dehner C, Badel K, Calandra G, et al. Augmented mobilization and collection of CD34 + hematopoietic cells from normal human volunteers stimulated with granulocyte-colony-stimulating factor by single-dose administration of AMD3100, a CXCR4 antagonist. Transfusion. 2005 Mar;45(3):295–300. doi: 10.1111/j.1537-2995.2005.04222.x. [DOI] [PubMed] [Google Scholar]
- 16.Donzella GA, Schols D, Lin SW, Este JA, Nagashima KA, Maddon PJ, et al. AMD3100, a small molecule inhibitor of HIV-1 entry via the CXCR4 co-receptor. Nat Med. 1998 Jan;4(1):72–7. doi: 10.1038/nm0198-072. [DOI] [PubMed] [Google Scholar]
- 17.Nagasawa T, Hirota S, Tachibana K, Takakura N, Nishikawa S, Kitamura Y, et al. Defects of B-cell lymphopoiesis and bone-marrow myelopoiesis in mice lacking the CXC chemokine PBSF/SDF-1. Nature. 1996 Aug 15;382(6592):635–8. doi: 10.1038/382635a0. [DOI] [PubMed] [Google Scholar]
- 18.Shiojima I, Sato K, Izumiya Y, Schiekofer S, Ito M, Liao R, et al. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J Clin Invest. 2005 Aug;115(8):2108–18. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olivetti G, Abbi R, Quaini F, Kajstura J, Cheng W, Nitahara JA, et al. Apoptosis in the failing human heart. N Engl J Med. 1997 Apr 17;336(16):1131–41. doi: 10.1056/NEJM199704173361603. [DOI] [PubMed] [Google Scholar]
- 20.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995 Dec 21-28;378(6559):785–9. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 21.Wang QM, Fiol CJ, DePaoli-Roach AA, Roach PJ. Glycogen synthase kinase-3 beta is a dual specificity kinase differentially regulated by tyrosine and serine/threonine phosphorylation. J Biol Chem. 1994 May 20;269(20):14566–74. [PubMed] [Google Scholar]
- 22.Diehl JA, Cheng M, Roussel MF, Sherr CJ. Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev. 1998 Nov 15;12(22):3499–511. doi: 10.1101/gad.12.22.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alt JR, Cleveland JL, Hannink M, Diehl JA. Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000 Dec 15;14(24):3102–14. doi: 10.1101/gad.854900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liao Y, Wei Y, Zhou X, Yang JY, Dai C, Chen YJ, et al. Peptidyl-prolyl cis/trans isomerase Pin1 is critical for the regulation of PKB/Akt stability and activation phosphorylation. Oncogene. 2009 Jul 2;28(26):2436–45. doi: 10.1038/onc.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Beltrami AP, Barlucchi L, Torella D, Baker M, Limana F, Chimenti S, et al. Adult cardiac stem cells are multipotent and support myocardial regeneration. Cell. 2003 Sep 19;114(6):763–76. doi: 10.1016/s0092-8674(03)00687-1. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh PC, Segers VF, Davis ME, MacGillivray C, Gannon J, Molkentin JD, et al. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007 Aug;13(8):970–4. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cho J, Rameshwar P, Sadoshima J. DISTINCT ROLES OF GSK-3{alpha} AND GSK-3{beta} IN MEDIATING CARDIOMYOCYTE DIFFERENTIATION IN MURINE BONE MARROW DERIVED MESENCHYMAL STEM CELLS. J Biol Chem. 2009 Oct 26; doi: 10.1074/jbc.M109.019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kerkela R, Kockeritz L, Macaulay K, Zhou J, Doble BW, Beahm C, et al. Deletion of GSK-3beta in mice leads to hypertrophic cardiomyopathy secondary to cardiomyoblast hyperproliferation. J Clin Invest. 2008 Nov;118(11):3609–18. doi: 10.1172/JCI36245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Luo D, Renault VM, Rando TA. The regulation of Notch signaling in muscle stem cell activation and postnatal myogenesis. Semin Cell Dev Biol. 2005 Aug-Oct;16(4-5):612–22. doi: 10.1016/j.semcdb.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 30.Berdnik D, Torok T, Gonzalez-Gaitan M, Knoblich JA. The endocytic protein alpha-Adaptin is required for numb-mediated asymmetric cell division in Drosophila. Dev Cell. 2002 Aug;3(2):221–31. doi: 10.1016/s1534-5807(02)00215-0. [DOI] [PubMed] [Google Scholar]
- 31.Guo Y, Hangoc G, Bian H, Pelus LM, Broxmeyer HE. SDF-1/CXCL12 enhances survival and chemotaxis of murine embryonic stem cells and production of primitive and definitive hematopoietic progenitor cells. Stem Cells. 2005 Oct;23(9):1324–32. doi: 10.1634/stemcells.2005-0085. [DOI] [PubMed] [Google Scholar]
- 32.Saxena A, Fish JE, White MD, Yu S, Smyth JW, Shaw RM, et al. Stromal cell-derived factor-1alpha is cardioprotective after myocardial infarction. Circulation. 2008 Apr 29;117(17):2224–31. doi: 10.1161/CIRCULATIONAHA.107.694992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest. 2004 Jun;113(11):1535–49. doi: 10.1172/JCI19906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuda T, Zhai P, Maejima Y, Hong C, Gao S, Tian B, et al. Distinct roles of GSK-3alpha and GSK-3beta phosphorylation in the heart under pressure overload. Proc Natl Acad Sci U S A. 2008 Dec 30;105(52):20900–5. doi: 10.1073/pnas.0808315106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Skyschally A, van Caster P, Boengler K, Gres P, Musiolik J, Schilawa D, et al. Ischemic postconditioning in pigs: no causal role for RISK activation. Circ Res. 2009 Jan 2;104(1):15–8. doi: 10.1161/CIRCRESAHA.108.186429. [DOI] [PubMed] [Google Scholar]
- 36.Proulx C, El-Helou V, Gosselin H, Clement R, Gillis MA, Villeneuve L, et al. Antagonism of stromal cell-derived factor-1alpha reduces infarct size and improves ventricular function after myocardial infarction. Pflugers Arch. 2007 Nov;455(2):241–50. doi: 10.1007/s00424-007-0284-5. [DOI] [PubMed] [Google Scholar]
- 37.Dawn B, Guo Y, Rezazadeh A, Huang Y, Stein AB, Hunt G, et al. Postinfarct cytokine therapy regenerates cardiac tissue and improves left ventricular function. Circ Res. 2006 Apr 28;98(8):1098–105. doi: 10.1161/01.RES.0000218454.76784.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altarche-Xifro W, Curato C, Kaschina E, Grzesiak A, Slavic S, Dong J, et al. Cardiac c-kit+AT2+ cell population is increased in response to ischemic injury and supports cardiomyocyte performance. Stem Cells. 2009 Oct;27(10):2488–97. doi: 10.1002/stem.171. [DOI] [PubMed] [Google Scholar]
- 39.Nie Y, Han YC, Zou YR. CXCR4 is required for the quiescence of primitive hematopoietic cells. J Exp Med. 2008 Apr 14;205(4):777–83. doi: 10.1084/jem.20072513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen M, Xie HQ, Deng L, Li XQ, Wang Y, Zhi W, et al. Stromal cell-derived factor-1 promotes bone marrow-derived cells differentiation to cardiomyocyte phenotypes in vitro. Cell Prolif. 2008 Apr;41(2):336–47. doi: 10.1111/j.1365-2184.2008.00519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Larrea MD, Liang J, Da Silva T, Hong F, Shao SH, Han K, et al. Phosphorylation of p27Kip1 regulates assembly and activation of cyclin D1-Cdk4. Mol Cell Biol. 2008 Oct;28(20):6462–72. doi: 10.1128/MCB.02300-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Okumura E, Fukuhara T, Yoshida H, Hanada Si S, Kozutsumi R, Mori M, et al. Akt inhibits Myt1 in the signalling pathway that leads to meiotic G2/M-phase transition. Nat Cell Biol. 2002 Feb;4(2):111–6. doi: 10.1038/ncb741. [DOI] [PubMed] [Google Scholar]
- 43.Katayama K, Fujita N, Tsuruo T. Akt/protein kinase B-dependent phosphorylation and inactivation of WEE1Hu promote cell cycle progression at G2/M transition. Mol Cell Biol. 2005 Jul;25(13):5725–37. doi: 10.1128/MCB.25.13.5725-5737.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Y, Dowbenko D, Lasky LA. AKT/PKB phosphorylation of p21Cip/WAF1 enhances protein stability of p21Cip/WAF1 and promotes cell survival. J Biol Chem. 2002 Mar 29;277(13):11352–61. doi: 10.1074/jbc.M109062200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.