

Abstract
Proper protein function in cells, tissues and organisms depends critically on correct protein folding or interaction with partners. Over the last decade, single-molecule FRET (smFRET) has emerged as a powerful tool to probe complex distributions, dynamics, pathways and landscapes in protein folding and binding reactions, leveraging its ability to avoid averaging over an ensemble of molecules. While smFRET was practiced in a two-color form until recently, the last few years have seen the development of enhanced multicolor smFRET methods that provide additional structural information permitting us to probe more complex mechanisms. In this review, we provide a brief introduction to the smFRET technique, then follow with advanced multicolor measurements and end with ongoing methodology developments in microfluidics and protein labeling that are beginning to make these techniques more broadly applicable to answering a number of key questions about folding and binding.
Introduction
Folding and binding of proteins are critical processes that are intimately linked to the formation of a large fraction of the machinery that keeps cells and organisms in working order. The mechanisms of these complex processes have captivated the interest of chemists, physicists and biologists for several decades 1–16. Along the way many insights have been gained into how unfolded proteins find their native structures, with energy biases, pathways and interactions all playing roles. In the case of binding, mechanisms include “lock and key” and “induced fit” scenarios which distinguish whether the final bound structure exists in the free protein ensemble or is formed only following binding. Furthermore, self-association of proteins into oligomeric or amyloid structures has also been linked to several diseases and even function17–20. Figure 1 depicts a simplified version of the complex network of interactions and structural changes that are involved in the folding-binding landscapes of proteins21–24. Recent years have seen a large expansion in the application of single-molecule methods to protein folding applications25–30. These still-novel methods offer the abilities to observe and quantify a number of features of critical importance for understanding the fundamental mechanisms of biological processes. For example, dynamics, structural distributions and latent folding of denatured proteins, intrinsically disordered proteins and folded proteins and their complexes can be quantified25–38. Much of this review focuses on recent extensions of 2-color smFRET methodology from the perspective of how these can provide powerful new capabilities for protein folding and binding studies.
Figure 1.

Schematic depicting complex networks that comprise the folding and interactions of proteins. For example, the transition from unfolded to folded states can proceed directly or through intermediates, which can be partially folded or misfolded. Misfolding in proteins has been linked with aggregation-related diseases, where proteins self-assemble into toxic oligomers or fibrils. On the other hand, interactions with cellular partners can be crucial for protein activity; we depict here two possible mechanisms of coupled binding and folding, with structure formation either preceding binding or vice versa.
The remainder of this short review is structured as follows. First, we present the basics of 2-color smFRET for structural studies of biomolecules. We then discuss a simple extension of this method to three dye colors, and show how this can be used in some cases to study restructuring of protein complexes. Next, we discuss extensions of the instrumentation to additional channels for more comprehensive multicolor smFRET studies. Finally, we discuss recent advances in single molecule detection that will be useful for multicolor smFRET studies, and finish with our thoughts for the future. In this review, we focus mainly on relevant developments from our lab, and provide key references to other related work in the literature for the reader’s reference.
Two-color FRET on individual proteins
FRET is the non-radiative transfer of energy from a donor to an acceptor chromophore via a dipole-dipole coupling mechanism. The FRET rate constant and efficiency have a strong dependence on the distance between the donor and acceptor, and hence can be used as a molecular ruler on the ~30–70Å range, which is ideal for structural studies of proteins and their complexes25–30, 39. In a typical single-molecule FRET experiment, the protein of interest is labeled at two different positions using donor and acceptor dyes. These dyes are most often chemically grafted onto the proteins using cysteines residues that have been site-specifically introduced by mutagenesis. Alternately, other means of dye attachment can be used for this purpose, as briefly discussed towards the end of the review. The design of optimal dye positions is usually guided by previous knowledge of structure and activity.
Typical experimental setups have been described extensively elsewhere. We’ll focus here on experiments with a setup designed to interrogate molecules as they are diffusing or flowing in solution. An advantage of this type of experiment is that adverse interactions of molecules with surfaces are minimized (especially problematic for dynamic molecules like many folded and unfolded proteins). We note that another common type of experiment uses proteins grafted onto a surface for imaging, with the advantage that extended time-trajectories of the molecular properties may be obtained; however surface interactions must be tested for and avoided on a case-by-case basis40, 41. Briefly, FRET-labeled samples are diluted and placed in a cuvette on a confocal microscope (many such microscope setups, such as the one in our lab are home-built around a simple inverted microscope). The extremely low concentration (typically 100 pM or less) of fluorescent molecules combined with a very small detection volume (~ 1fl) together enable detection of individual molecules while reducing background and avoiding aggregation.
When a single molecule passes through the focal volume, the donor dye is excited by the illumination laser, and can transfer some of its energy to the acceptor dye. Bursts of fluorescence are collected and the contributions from the donor and acceptor dyes are separated in various detection channels. The FRET efficiency EFRET is then calculated ratiometrically for each single-molecule event as:
Necessary corrections such as for differences in fluorescence and detection quantum yields of the dyes and crosstalk between the channels are also applied25, 42, 43. The histograms of EFRET obtained allow a direct visualization of populations, which can be further identified with various conformations44 based on additional information from complementary techniques such as CD, NMR and crystallography.
A key strength of smFRET for folding studies is that by directly observing and quantifying the evolution of folded/unfolded populations as a function of denaturant concentration, one can access details about the folding transitions and discriminate between various hypotheses. For example, a simple two-state transition can be observed as a transfer of molecules between two well-defined folded and unfolded states, each having distinct EFRET characteristics (See Figure 2b and the next section for an example). On the other hand, if the proteins populate an intermediate folded state, an additional FRET peak may appear in the histograms at intermediate denaturant concentrations44. The high-sensitivity of the technique may be very useful in that case, as these states may only represent a fraction of the molecules. Another interesting possibility also emerged in the folding field, as the protein may shift continuously between states, without encountering significant energy barriers 45–49. These proteins could have very fast “downhill” folding and the difficult detection of these folded states is the focus of some recent studies50.
Figure 2.
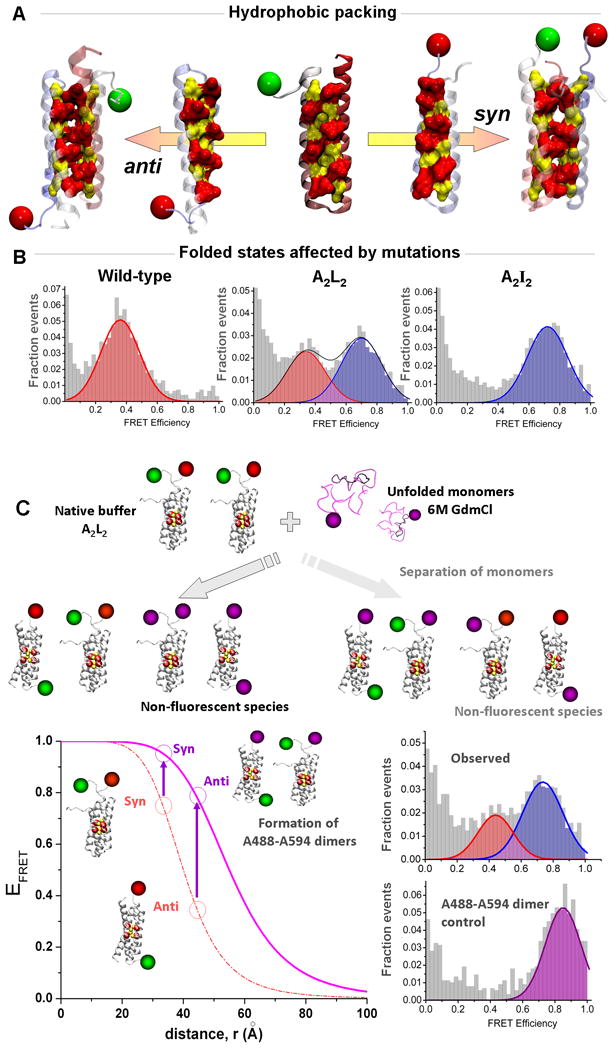
Multicolor smFRET study of the Rop mutants (adapted from reference 51: Gambin et al., Proc. Natl. Acad. Sci., USA, 2009, 106: 10153–10158)
(A) The Repressor of Primers (Rop) complexes are formed by association of two helix-turn-helix motifs. The active enzyme requires that the two monomers bind in an anti conformation to form the proper RNA interface. (B) The symmetry created by multiple mutations within the hydrophobic core of the protein pushes the two monomers to re-arrange into a stable syn conformation, as observed for the A2I2 mutant. Surprisingly, conditions were found where the A2L2 mutant can form coexisting anti and syn, as evidenced by the presence of two FRET peaks. (C) Multi-color smFRET was used to check whether monomers are separating during the interconversion between the two opposite folded states. In a dual-channel format, the experiments took advantage of the difference between two possible dye pairs to detect in a simple manner the formation of mixed species in a competition assay (see text for details). The bottom left panel shows the FRET efficiencies of the syn and anti conformations for the Rop dimers forming an A488-A647 pair (green and red dyes) or forming an A488-A594 pair (green and purple dyes). The formation of A488-A594 pairs would only occur if the original A488-A647 dimers dissociate during the structural rearrangement. When we performed this test, we observed the two FRET peaks corresponding to the syn and anti conformations of A488-A647 dimers, and no significant higher-FRET species that would indicate exchange with A594 monomers (as found in a separate control experiment, bottom right).
Next, we discuss a simple example of 3-color smFRET experiments which provide enhanced information for studying structural rearrangements within complexes.
Expanding the palette of colors–a simple case
Our recent smFRET study of the protein dimer Rop51 showcases in a simple manner the power of using different dye pairs to study mechanisms of protein association and rearrangement of protein complexes. The Rop dimer is formed by hydrophobic zipping of a four-helix bundle52, 53. In this simple system, a series of in-depth previous investigations had provided an improved understanding of the balance of hydrophobicity and steric packing on the stability and folding of such complexes54. A surprising effect was the observation that increasing the efficiency of packing by using Leucine and Alanine residues actually resulted in a destabilized mutant, with enhanced folding/unfolding kinetic properties55–57. Computational analysis of this system resulted in the prediction that certain mutations would result in enhanced symmetry58 and the ability to populate both syn and anti conformations (Figure 2a) 59, 60. We used regular 2-color smFRET experiments to probe one such mutant, and directly demonstrated that while it populated mainly one conformation under native buffer conditions, mild denaturation induces it to indeed populate the two conformations (Figure 2a–b). This interesting observation brought up the key question of how the structures interconvert, within the context of existing complexes, or following complete dissociation.
A third fluorescent label was used to distinguish between these possibilities. The experiment consisted of a “competition assay”, for which an excess of monomers labeled with Alexa594 (A594) was introduced in the solution during the switching reaction, as illustrated in Figure 2c. The idea is that if the complexes need to completely dissociate prior to rearrangement, reassociation will occur largely with the large excess of protein labeled with A594. Now, because the A488-A594 dye pair has higher FRET efficiency than the A488-A647 used to determine structure, formation of a mixed species would result in a high-EFRET peak corresponding to both conformations in a regular two-channel experiment that collects both A594 and A647 in the same acceptor channel. When the experiment was carried out, the surprising result was that a majority of molecules seemed to switch conformations without complete dissociation. Future experiments will probe in more detail the dynamic features of rearrangement of symmetrically related protein complexes.
Linking protein folding and binding with multichannel detection
More advanced versions of 3-color (or even 4-color) smFRET can provide a wealth of information on the succession of binding and folding steps occurring during the formation of protein complexes.
Let us consider for example the case of protein dimerization, beginning from the unfolded state. As depicted in Figure 3a, multiple paths can be hypothesized for the reaction, each with a different succession of events and intermediates states. In other words, a key question is whether the formation of protein structure precedes the interaction between the two monomers, or whether the proteins need to fold separately to form the binding interface. Using two-color smFRET, this question would be difficult to answer directly, since different molecules could explore different paths, and complex formation is not being directly tested for.
Figure 3.
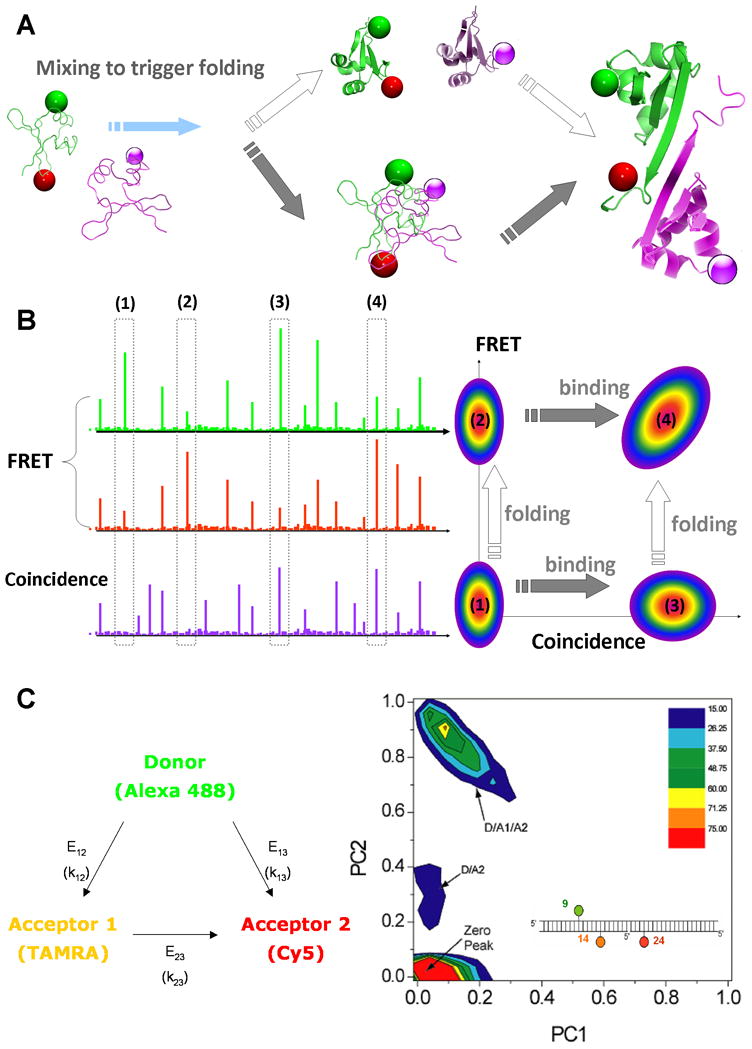
Multicolor single-molecule FRET and coincidence
(A) The structures of the Trp repressor dimer is used to illustrate the different pathways followed during protein-protein interaction and folding. Starting from two monomers kept in unfolded states (using for example denaturing conditions), one could trigger folding and observe in real time the steps of dimer formation. Two simple cases can be imagined: the monomers could fold separately then bind once the correct interface is formed, or first interact and together perform the folding steps. By using 3 fluorescent dyes, it becomes possible to distinguish between the two simple pathways, or even detect new intermediates. The first monomer would be labeled with the FRET pair in order to detect conformational changes; the second monomer would be labeled with a single dye, to detect simply its presence and the formation of a complex. (B) Schematic of single-molecule fluorescent bursts in the 3-color format. The highlighted bursts with high/low FRET and coincidence/no coincidence correspond to various folding/binding steps. In a 2D representation of FRET versus Coincidence, these different populations would be well separated and the sequence of folding/binding steps clearly identified. (C) 3-color smFRET histograms from proof-of-principle experiments with DNA (adapted from reference 62: Clamme et al., Chemphyschem, 2005, 6: 74–77).
Multi-color smFRET holds great promise to explore such multicomponent reactions, by allowing the simultaneous detection of conformational changes and interactions. As explained in Figure 3a, a donor and an acceptor pair (D1-A1) can be placed on some proteins, the EFRET measured leading to the determination of folding events within the monomer. At the same time, the remaining proteins can be labeled with a different dye, D2, with absorption/emission shifted to higher wavelengths to avoid interference with the FRET measurement. If a second laser is used to excite this particular dye, one will be able to detect whether the two species of proteins, D1-A1 and D2 are present at the same time in the focal volume, by searching for coincidence of bursts between the different detection channels. As shown in Figure 3b, by plotting the events on a 2D map of EFRET and coincidence, one could separate clearly the unfolded, dimer unfolded, folded monomers and folded dimer states.
Significant progress has been made towards 3- and multicolor smFRET technologies that would permit such investigations of protein folding and binding. Early demonstrations of 3-color smFRET were reported by Hohng et al. 61 for immobilized molecules and Clamme et al. for freely diffusing molecules62, and multistep smFRET was probed by Heilemann et al. in the context of an electronic “DNA wire” 63. In the study by Clamme et al. 62, 3-color smFRET instrumentation and data analysis methods were developed and applied to the case of well-defined DNA samples. Data acquisition was via 3 channels corresponding to donor, acceptor 1 and acceptor 2. DNAs were labeled so as to span several combinations of the three possible transfer efficiencies (D→A1, D→A2 and A1→A2), and the experiments showed that the peak positions changed in keeping with expected changes in FRET efficiency. Identification and quantification of multiple subpopulations in a mixture of triply labeled DNAs was also demonstrated. Due to the ratiometric data reduction, a given 3-color peak provided only two independent values while the system had three unknowns. Hence, one of the three transfer efficiencies was determined in a separate experiment with a two-color sample. Although not exploited here, it should be noted that photobleached acceptors provide a FRET value that may also be used to solve for all three FRET values using a single experiment. The study by Hohng et al. 61 used a simplified 3-color smFRET transfer design, avoiding transfer between the two acceptors. Their work revealed the simultaneous flipping of different arms of a 4-way DNA Holliday junction. A few nice extensions and applications have also been reported. For example, rapid alternation or cycling between multiple excitations was used for enhanced studies of conformational changes and binding in DNA molecules and complexes 64–67, and DNA wires have been further probed using multicolor experiments68. There is little doubt that additional progress will soon be made on both technical improvements and also in applications in biological systems where they will have the most impact.
Next, we discuss some other considerations and adjunct technologies that are beginning to help make progress in this area.
Improving dye photophysics–reducing photobleaching and increasing brightness
The photon emission rate of a dye is a key determinant of the quality of information available from single molecule experiments. This is especially important in the case of multicolor experiments, given the additional complexity in terms of data acquisition and analysis. For examples, in FRET histograms such as those presented in Figure 2, more rapid photon flux would provide narrower peaks, increased sensitivity to structural dynamics and subpopulations, and the possibility for more rapid data collection. Factors such as the fluorescence lifetime and cycling between singlet and triplet manifolds limit the maximum available photon rates. Hence, increasing laser power is useful only below such saturation. Moreover, an increase of laser power in this region and beyond results in an increase in dye photobleaching. This in turn results in a decrease in the number of FRET capable species detected per unit time, increase in the number of photobleached signals, and a corresponding decrease in histogram quality. Photobleaching is even more problematic in the case of multicolor FRET experiments because bleaching of even one of the dyes will cause the multicolor FRET signal to be lost for the molecule. Moreover, extensions to 4 or more colors while avoiding spectral overlap and crosstalk generally will push additional dyes into the near IR; these dyes seem to be more photolabile.
Because molecular oxygen is a significant contributor to photobleaching69–72, many single-molecule experiments utilize oxygen scavengers to reduce the oxygen concentration in the solution. In particular, an enzymatic scavenger system containing glucose oxidase and catalase has been widely used. However, a key problem with such additives is that they alter the buffer conditions, and deleterious interactions with the added components are possible. Moreover, the enzyme system is not compatible with more denaturing conditions in the context of folding experiments. To improve the deoxygenation procedure for these experiments, we recently reported on-chip deoxygenation in a microfluidic device (Figure 4a) 73. The principle is simple74–77: the buffer travels in a small channel flanked by two deep channels ventilated by pure nitrogen. Because the material PDMS used is porous to gas, the oxygen present in the buffer and protein solution can be efficiently removed prior to measurement, resulting in improved quality of histograms (Figure 4c). In the context of an orthogonal smFRET pair for use in 4-color experiments, the use of the deoxygenation device allowed the first diffusion smFRET measurements of the A647-A750 dye pair (Figure 4c(ii)). This pair could be used along with existing non-overlapping smFRET pairs for 4-color smFRET experiments in the near future. Additionally, signal quality for another pair was observed to increase substantially when in-chip oxygen removal was combined with a use of a triplet quencher78, leading to brighter bursts, which should result in improved FRET resolution and faster data acquisition rates.
Figure 4.
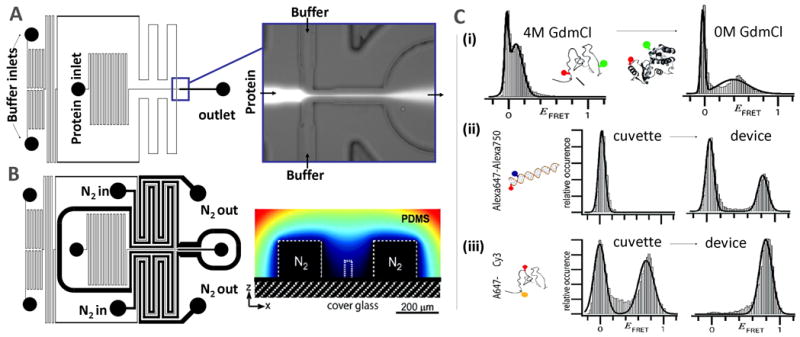
Microfluidic platform for enhanced single-molecule FRET detection (adapted from reference 73: Lemke et al., J. Amer. Chem. Soc., 2009, 131: 13610–13612).
(A) A laminar-flow mixer can remove denaturant or add binding partner to trigger folding events. (B) Large channels ventilated by nitrogen flank the channels containing buffers and proteins, efficiently removing oxygen from the solution before mixing. (C) smFRET histograms obtained with the device, showing (i) the folding of T4 lysozyme proteins upon removal of guanidinium chloride, (ii) the radical improvement of data upon deoxygenation for data from a near IR-dye (Alexa750) and (iii) for T4 lysozyme proteins labeled by using an unnatural amino-acid strategy.
Reaching out-of-equilibrium conditions
In order to directly study structural distributions under non-equilibrium conditions, e.g., during transitions between unfolded and folded states, the folding-association reaction needs to be triggered. In order to reach the first folding events that could occur on a millisecond timescale or faster, continuous-flow mixing can be used. Microfluidic mixers have been used for the fastest reaction times at the ensemble level (down to 1 μs)79–83, and adapted for the needs of single-molecule measurements 73, 84–86. In these microfluidic devices, although the flows are not turbulent, buffer exchange can still occur on very short timescales: the protein flow is squeezed laterally by the buffer, and molecules can diffuse rapidly in/out of the narrow protein layer, triggering the folding. For example, in the device shown in Figure 4a, the 2 labeled proteins can be kept unfolded in GdmCl solution, and removal of the denaturant will trigger the formation of the dimer (an example of folding on-chip is shown in Figure 4c–(i) 73). The reaction can be probed at single-molecule resolution, with observation on the ms timescale following mixing.
Site-specific labeling strategies
Site-specific labeling of proteins is often a non-trivial and time-consuming task. Dual-cysteine labeling of proteins has worked for many smFRET experiments because the observed FRET efficiency for a given distance is often very similar for the Donor-Acceptor and Acceptor-Donor labeling isomers obtained in a random labeling scheme (i.e., the photophysical properties of the dyes are similar at the two labeling positions). However, random cysteine labeling of a protein with 3 dyes will result in a mixture of 6 different 3-color labeling isomers with up to 6 different combinations of distances being measured for the dye pairs. In other words, this would result in an extremely complex labeling mixture that would needlessly (and perhaps impossibly) complicate an already difficult experiment. The way to overcome such a problem is to attach dyes in a site-specific manner. A number of possible strategies have been reported that may be tailored for this purpose, including total synthesis, selective cysteine labeling 87, ligation of labeled fragments of the protein88, and use of a site-specifically introduced unnatural amino acid to achieve orthogonal labelling chemistry89. These strategies, each with its own advantages and potential disadvantages, can be combined and adapted for the more complex demands of triple labelling in multi-color smFRET experiments.
Concluding remarks
This short review presented how technical advances recently made in multicolor smFRET detection are beginning to create new opportunities for studying detailed mechanistic issues in biological folding and binding which are ubiquitous and critically important in biology. It can be seen that while some preliminary advances can be made by relatively simple extensions of prior single-molecule instrumentation, full use of the methodologies require improvements in many dimensions including in instrumentation, better dyes and photophysics, as well as attachment chemistries. In addition, the richer and more complex data from these experiments will benefit from better analysis algorithms, and also allow better comparisons with theory. One area of current and expanding interest11, 90–98 is in the class of proteins which are intrinsically disordered, which exhibit dynamic and complex structural and binding-folding behaviour, and with some also forming amyloids. Multicolor smFRET experiments will very likely reveal novel information about the structural landscapes of these complex proteins, with strong biological and health implications. Finally, while many of these technical advances are being made in vitro, there is little doubt that in vivo experiments will also benefit from multicolor smFRET experiments to directly understand the structural properties of multicomponent complexes in live cells and organisms.
Acknowledgments
We thank personnel and collaborators who contributed greatly to the corresponding papers from the Deniz lab discussed here. We also gratefully acknowledge the NSF and NIH for support for work from the Deniz Lab reviewed in this article.
Contributor Information
Yann Gambin, Email: gambin@scripps.edu.
Ashok A. Deniz, Email: deniz@scripps.edu.
References
- 1.Frauenfelder H, Sligar SG, Wolynes PG. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 2.Leopold PE, Montal M, Onuchic JN. Proc Natl Acad Sci USA. 1992;89:8721–8725. doi: 10.1073/pnas.89.18.8721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bryngelson JD, Onuchic JN, Socci ND, Wolynes PG. Proteins-Structure Function and Genetics. 1995;21:167–195. doi: 10.1002/prot.340210302. [DOI] [PubMed] [Google Scholar]
- 4.Onuchic JN, Luthey-Schulten Z, Wolynes PG. Annu Rev Phys Chem. 1997;48:545–600. doi: 10.1146/annurev.physchem.48.1.545. [DOI] [PubMed] [Google Scholar]
- 5.Dill KA, Chan HS. Nature Structural Biology. 1997;4:10–19. doi: 10.1038/nsb0197-10. [DOI] [PubMed] [Google Scholar]
- 6.Snow CD, Nguyen N, Pande VS, Gruebele M. Nature. 2002;420:102–106. doi: 10.1038/nature01160. [DOI] [PubMed] [Google Scholar]
- 7.Fersht AR. Nature reviews. 2008;9:650–654. doi: 10.1038/nrm2446. [DOI] [PubMed] [Google Scholar]
- 8.Lindorff-Larsen K, Rogen P, Paci E, Vendruscolo M, Dobson CM. Trends in biochemical sciences. 2005;30:13–19. doi: 10.1016/j.tibs.2004.11.008. [DOI] [PubMed] [Google Scholar]
- 9.Dill KA, Ozkan SB, Shell MS, Weikl TR. Annual review of biophysics. 2008;37:289–316. doi: 10.1146/annurev.biophys.37.092707.153558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wright PE, Dyson HJ. Journal of molecular biology. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 11.Wright PE, Dyson HJ. Current Opinion in Structural Biology. 2009;19:31–38. doi: 10.1016/j.sbi.2008.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thirumalai D, O’Brien EP, Morrison G, Hyeon C. Annual review of biophysics. 2010 doi: 10.1146/annurev-biophys-051309-103835. [DOI] [PubMed] [Google Scholar]
- 13.Horwich AL, Fenton WA. Quarterly reviews of biophysics. 2009;42:83–116. doi: 10.1017/S0033583509004764. [DOI] [PubMed] [Google Scholar]
- 14.Hartl FU, Hayer-Hartl M. Nature structural & molecular biology. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 15.Cabrita LD, Dobson CM, Christodoulou J. Current opinion in structural biology. 2010;20:33–45. doi: 10.1016/j.sbi.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 16.Powers ET, Morimoto RI, Dillin A, Kelly JW, Balch WE. Annual review of biochemistry. 2009;78:959–991. doi: 10.1146/annurev.biochem.052308.114844. [DOI] [PubMed] [Google Scholar]
- 17.Lashuel HA, Hartley D, Petre BM, Walz T, Lansbury PT., Jr Nature. 2002;418:291. doi: 10.1038/418291a. [DOI] [PubMed] [Google Scholar]
- 18.Lashuel HA, Lansbury PT., Jr Q Rev Biophys. 2006;39:167–201. doi: 10.1017/S0033583506004422. [DOI] [PubMed] [Google Scholar]
- 19.Fowler DM, Koulov AV, Balch WE, Kelly JW. Trends in biochemical sciences. 2007;32:217–224. doi: 10.1016/j.tibs.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 20.Luheshi LM, Crowther DC, Dobson CM. Current opinion in chemical biology. 2008;12:25–31. doi: 10.1016/j.cbpa.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 21.Dobson CM. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 22.Bennett MJ, Choe S, Eisenberg D. Proc Natl Acad Sci USA. 1994;91:3127–3131. doi: 10.1073/pnas.91.8.3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang SC, Cho SS, Levy Y, Cheung MS, Levine H, Wolynes PG, Onuchic JN. Proc Natl Acad Sci USA. 2004;101:13786–13791. doi: 10.1073/pnas.0403724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolynes PG, Onuchic JN, Thirumalai D. Science. 1995;267:1619–1620. doi: 10.1126/science.7886447. [DOI] [PubMed] [Google Scholar]
- 25.Deniz AA, Laurence TA, Beligere GS, Dahan M, Martin AB, Chemla DS, Dawson PE, Schultz PG, Weiss S. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5179–5184. doi: 10.1073/pnas.090104997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Michalet X, Weiss S, Jager M. Chemical reviews. 2006;106:1785–1813. doi: 10.1021/cr0404343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muller DJ, Sapra KT, Scheuring S, Kedrov A, Frederix PL, Fotiadis D, Engel A. Current opinion in structural biology. 2006;16:489–495. doi: 10.1016/j.sbi.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 28.Borgia A, Williams PM, Clarke J. Annual review of biochemistry. 2008;77:101–125. doi: 10.1146/annurev.biochem.77.060706.093102. [DOI] [PubMed] [Google Scholar]
- 29.Schuler B, Eaton WA. Current opinion in structural biology. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deniz AA, Mukhopadhyay S, Lemke EA. Journal of the Royal Society Interface. 2008;5:15–45. doi: 10.1098/rsif.2007.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Torres T, Levitus M. The journal of physical chemistry. 2007;111:7392–7400. doi: 10.1021/jp070659s. [DOI] [PubMed] [Google Scholar]
- 32.Aleman EA, Lamichhane R, Rueda D. CurrOpin Chem Biol. 2008;12:647–654. doi: 10.1016/j.cbpa.2008.09.010. [DOI] [PubMed] [Google Scholar]
- 33.Dudko OK, Hummer G, Szabo A. Proc Natl Acad Sci USA. 2008;105:15755–15760. doi: 10.1073/pnas.0806085105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Joo C, Balci H, Ishitsuka Y, Buranachai C, Ha T. Annu Rev Biochem. 2008;77:51–76. doi: 10.1146/annurev.biochem.77.070606.101543. [DOI] [PubMed] [Google Scholar]
- 35.Karymov MA, Bogdanov A, Lyubchenko YL. Biophys J. 2008;95:1239–1247. doi: 10.1529/biophysj.107.127522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mickler M, Dima RI, Dietz H, Hyeon C, Thirumalai D, Rief M. Proc Natl Acad Sci USA. 2007;104:20268–20273. doi: 10.1073/pnas.0705458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steiner M, Karunatilaka KS, Sigel RKO, Rueda D. Proc Natl Acad Sci USA. 2008;105:18071–18071. doi: 10.1073/pnas.0804034105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cornish PV, Ha T. Acs Chemical Biology. 2007;2:53–61. doi: 10.1021/cb600342a. [DOI] [PubMed] [Google Scholar]
- 39.Schuler B. Chemphyschem. 2005;6:1206–1220. doi: 10.1002/cphc.200400609. [DOI] [PubMed] [Google Scholar]
- 40.Rasnik I, Mckinney SA, Ha T. Acc Chem Res. 2005;38:542–548. doi: 10.1021/ar040138c. [DOI] [PubMed] [Google Scholar]
- 41.Friedel M, Baumketner A, Shea JE. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8396–8401. doi: 10.1073/pnas.0601210103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Deniz AA, Dahan M, Grunwell JR, Ha TJ, Faulhaber AE, Chemla DS, Weiss S, Schultz PG. Proc Natl Acad Sci USA. 1999;96:3670–3675. doi: 10.1073/pnas.96.7.3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Deniz AA, Laurence TA, Dahan M, Chemla DS, Schultz PG, Weiss S. Annu Rev Phys Chem. 2001;52:233–253. doi: 10.1146/annurev.physchem.52.1.233. [DOI] [PubMed] [Google Scholar]
- 44.Ferreon ACM, Gambin Y, Lemke EA, Deniz AA. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eaton WA. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:5897–5899. doi: 10.1073/pnas.96.11.5897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferguson N, Schartau PJ, Sharpe TD, Sato S, Fersht AR. Journal of molecular biology. 2004;344:295–301. doi: 10.1016/j.jmb.2004.09.069. [DOI] [PubMed] [Google Scholar]
- 47.Dyer RB. Current opinion in structural biology. 2007;17:38–47. doi: 10.1016/j.sbi.2007.01.001. [DOI] [PubMed] [Google Scholar]
- 48.Gruebele M. Comptes rendus biologies. 2005;328:701–712. doi: 10.1016/j.crvi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 49.Munoz V. Annual review of biophysics and biomolecular structure. 2007;36:395–412. doi: 10.1146/annurev.biophys.36.040306.132608. [DOI] [PubMed] [Google Scholar]
- 50.Huang F, Sato S, Sharpe TD, Ying L, Fersht AR. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:123–127. doi: 10.1073/pnas.0609717104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gambin Y, Schug A, Lemke EA, Lavinder JJ, Ferreon ACM, Magliery TJ, Onuchic JN, Deniz AA. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:10153–10158. doi: 10.1073/pnas.0904461106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Banner DW, Cesareni G, Tsernoglou D. Journal of molecular biology. 1983;170:1059–1060. doi: 10.1016/s0022-2836(83)80207-1. [DOI] [PubMed] [Google Scholar]
- 53.Banner DW, Kokkinidis M, Tsernoglou D. Journal of molecular biology. 1987;196:657–675. doi: 10.1016/0022-2836(87)90039-8. [DOI] [PubMed] [Google Scholar]
- 54.Magliery TJ, Regan L. Protein Eng Des Sel. 2004;17:77–83. doi: 10.1093/protein/gzh010. [DOI] [PubMed] [Google Scholar]
- 55.Munson M, Anderson KS, Regan L. Fold Des. 1997;2:77–87. doi: 10.1016/S1359-0278(97)00008-4. [DOI] [PubMed] [Google Scholar]
- 56.Munson M, Balasubramanian S, Fleming KG, Nagi AD, Obrien R, Sturtevant JM, Regan L. Protein Sci. 1996;5:1584–1593. doi: 10.1002/pro.5560050813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Munson M, Obrien R, Sturtevant JM, Regan L. Protein Sci. 1994;3:2015–2022. doi: 10.1002/pro.5560031114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wolynes PG. Proc Natl Acad Sci USA. 1996:14249–14255. doi: 10.1073/pnas.93.25.14249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levy Cho SS, Shen T, Onuchic JN, Wolynes PG. Proc Natl Acad Sci USA. 2005;102:2373–2378. doi: 10.1073/pnas.0409572102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schug A, Whitford PC, Levy Y, Onuchic JN. Proc Natl Acad Sci USA. 2007;104:17674–17679. doi: 10.1073/pnas.0706077104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hohng S, Joo C, Ha T. Biophysical journal. 2004;87:1328–1337. doi: 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clamme JP, Deniz AA. Chemphyschem. 2005;6:74–77. doi: 10.1002/cphc.200400261. [DOI] [PubMed] [Google Scholar]
- 63.Heilemann M, Tinnefeld P, Sanchez Mosteiro G, Garcia Parajo M, Van Hulst NF, Sauer M. Journal of the American Chemical Society. 2004;126:6514–6515. doi: 10.1021/ja049351u. [DOI] [PubMed] [Google Scholar]
- 64.Lee NK, Kapanidis AN, Koh HR, Korlann Y, Ho SO, Kim Y, Gassman N, Kim SK, Weiss S. Biophysical journal. 2007;92:303–312. doi: 10.1529/biophysj.106.093211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ross J, Buschkamp P, Fetting D, Donnermeyer A, Roth CM, Tinnefeld P. The journal of physical chemistry. 2007;111:321–326. doi: 10.1021/jp066082g. [DOI] [PubMed] [Google Scholar]
- 66.Lee NK, Koh HR, Han KY, Kim SK. Journal of the American Chemical Society. 2007;129:15526–15534. doi: 10.1021/ja0725145. [DOI] [PubMed] [Google Scholar]
- 67.Person B, Stein IH, Steinhauer C, Vogelsang J, Tinnefeld P. Chemphyschem. 2009;10:1455–1460. doi: 10.1002/cphc.200900109. [DOI] [PubMed] [Google Scholar]
- 68.Heilemann M, Kasper R, Tinnefeld P, Sauer M. Journal of the American Chemical Society. 2006;128:16864–16875. doi: 10.1021/ja065585x. [DOI] [PubMed] [Google Scholar]
- 69.Donnert G, Eggeling C, Hell SW. Nat Methods. 2007;4:81–86. doi: 10.1038/nmeth986. [DOI] [PubMed] [Google Scholar]
- 70.Eggeling C, Widengren J, Brand L, Schaffer J, Felekyan S, Seidel CAM. J Phys Chem A. 2006;110:2979–2995. doi: 10.1021/jp054581w. [DOI] [PubMed] [Google Scholar]
- 71.Kong XX, Nir E, Hamadani K, Weiss S. J Am Chem Soc. 2007;129:4643–4654. doi: 10.1021/ja068002s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rasnik I, McKinney SA, Ha T. Nat Methods. 2006;3:891–893. doi: 10.1038/nmeth934. [DOI] [PubMed] [Google Scholar]
- 73.Lemke EA, Gambin Y, Vandelinder V, Brustad EM, Liu HW, Schultz PG, Groisman A, Deniz AA. Journal of the American Chemical Society. 2009;131:13610–13612. doi: 10.1021/ja9027023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mehta G, Mehta K, Sud D, Song JW, Bersano-Begey T, Futai N, Heo YS, Mycek MA, Linderman JJ, Takayama S. Biomed Microdevices. 2007;9:123–134. doi: 10.1007/s10544-006-9005-7. [DOI] [PubMed] [Google Scholar]
- 75.Merkel TC, Bondar VI, Nagai K, Freeman BD, Pinnau I. J Polymer Sci Part B. 2000;38:415–434. [Google Scholar]
- 76.Sud D, Mehta G, Mehta K, Linderman J, Takayama S, Mycek MA. J Biomed Opt. 2006:11. doi: 10.1117/1.2355665. [DOI] [PubMed] [Google Scholar]
- 77.Vollmer AP, Probstein RF, Gilbert R, Thorsen T. Lab On A Chip. 2005;5:1059–1066. doi: 10.1039/b508097e. [DOI] [PubMed] [Google Scholar]
- 78.Vogelsang J, Kasper R, Steinhauer C, Person B, Heilemann M, Sauer M, Tinnefeld P. Angew Chem-Int Ed. 2008;47:5465–5469. doi: 10.1002/anie.200801518. [DOI] [PubMed] [Google Scholar]
- 79.Knight JB, Vishwanath A, Brody JP, Austin RH. Physical Review Letters. 1998;80:3863–3866. [Google Scholar]
- 80.Hertzog DE, Michalet X, Jager M, Kong XX, Santiago JG, Weiss S, Bakajin O. Analytical chemistry. 2004;76:7169–7178. doi: 10.1021/ac048661s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hertzog DE, Ivorra B, Mohammadi B, Bakajin O, Santiago JG. Analytical chemistry. 2006;78:4299–4306. doi: 10.1021/ac051903j. [DOI] [PubMed] [Google Scholar]
- 82.Park HY, Qiu XY, Rhoades E, Korlach J, Kwok LW, Zipfel WR, Webb WW, Pollack L. Analytical chemistry. 2006;78:4465–4473. doi: 10.1021/ac060572n. [DOI] [PubMed] [Google Scholar]
- 83.Gambin Y, Simonnet C, VanDelinder V, Deniz AA, Groisman A. Lab on a Chip. 2010 doi: 10.1039/b914174j. [DOI] [PubMed] [Google Scholar]
- 84.Lipman EA, Schuler B, Bakajin O, Eaton WA. Science (New York, NY. 2003;301:1233–1235. doi: 10.1126/science.1085399. [DOI] [PubMed] [Google Scholar]
- 85.Hamadani KM, Weiss S. Biophysical journal. 2008;95:352–365. doi: 10.1529/biophysj.107.127431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pfeil SH, Wickersham CE, Hoffmann A, Lipman EA. The Review of scientific instruments. 2009;80:055105. doi: 10.1063/1.3125643. [DOI] [PubMed] [Google Scholar]
- 87.Jager M, Michalet X, Weiss S. Protein Sci. 2005;14:2059–2068. doi: 10.1110/ps.051384705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Deniz AA, Laurence TA, Beligere GS, Dahan M, Martin AB, Chemla DS, Dawson PE, Schultz PG, Weiss S. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5179–5184. doi: 10.1073/pnas.090104997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Brustad EM, Lemke EA, Schultz PG, Deniz AA. J Am Chem Soc. 2008;130:17664–17665. doi: 10.1021/ja807430h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Stein A, Pache RA, Bernado P, Pons M, Aloy P. The FEBS journal. 2009;276:5390–5405. doi: 10.1111/j.1742-4658.2009.07251.x. [DOI] [PubMed] [Google Scholar]
- 91.Dunker AK, Oldfield CJ, Meng J, Romero P, Yang JY, Chen JW, Vacic V, Obradovic Z, Uversky VN. BMC genomics. 2008;9(Suppl 2):S1. doi: 10.1186/1471-2164-9-S2-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Uversky VN, Dunker AK. Biochimica et biophysica acta [Google Scholar]
- 93.Mukhopadhyay S, Krishnan R, Lemke EA, Lindquist S, Deniz AA. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:2649–2654. doi: 10.1073/pnas.0611503104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yu J, Malkova S, Lyubchenko YL. J Mol Biol. 2008;384:992–1001. doi: 10.1016/j.jmb.2008.10.006. [DOI] [PubMed] [Google Scholar]
- 95.Brucale M, Sandal M, Di Maio S, Rampioni A, Tessari I, Tosatto L, Bisaglia M, Bubacco L, Samori B. Chembiochem. 2009;10:176–183. doi: 10.1002/cbic.200800581. [DOI] [PubMed] [Google Scholar]
- 96.Ferreon AC, Gambin Y, Lemke EA, Deniz AA. Proc Natl Acad Sci U S A. 2009;106:5645–5650. doi: 10.1073/pnas.0809232106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Trexler AJ, Rhoades E. Biochemistry. 2009;48:2304–2306. doi: 10.1021/bi900114z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Veldhuis G, Segers-Nolten I, Ferlemann E, Subramaniam V. Chembiochem. 2009;10:436–439. doi: 10.1002/cbic.200800644. [DOI] [PubMed] [Google Scholar]