

Abstract
Anhedonia is a core symptom of major depressive disorder (MDD), the neurobiological mechanisms of which remain poorly understood. Despite decades of speculation regarding the role of dopamine (DA) in anhedonic symptoms, empirical evidence has remained elusive, with frequent reports of contradictory findings. In the present review, we argue that this has resulted from an underspecified definition of anhedonia, which has failed to dissociate between consummatory and motivational aspects of reward behavior. Given substantial preclinical evidence that DA is involved primarily in motivational aspects of reward, we suggest that a refined definition of anhedonia that distinguishes between deficits in pleasure and motivation is essential for the purposes of identifying its neurobiological substrates. Moreover, bridging the gap between preclinical and clinical models of anhedonia may require moving away from the conceptualization of anhedonia as a steady-state, mood-like phenomena. Consequently, we introduce the term “decisional anhedonia” to address the influence of anhedonia on reward decision-making. These proposed modifications to the theoretical definition of anhedonia have implications for research, assessment and treatment of MDD.
Keywords: Anhedonia, Depression, Dopamine, Reward, Motivational Anhedonia, Consummatory Anhedonia, Decisional Anhedonia
1. Introduction
With a lifetime prevalence of approximately 16% (Kessler et al., 2003), Major Depressive Disorder (MDD) is predicted to become the second leading cause of death and disability in the United States by the year 2020 (Murray and Lopez, 1997). Dating back to the original Feighner criteria published in 1972, anhedonia has long been presumed as a core feature of MDD (Feighner et al., 1972). The DSM-IV-TR (American Psychiatric Association, 1994) defines anhedonia as diminished interest or pleasure in response to stimuli that were previously perceived as rewarding during a pre-morbid state (DSM-IV-TR). Along with depressed mood, anhedonia is one of two required symptoms for a diagnosis of MDD (American Psychiatric Association, 2000; World Health Organization, 1992). Recent reports estimate that approximately 37% of individuals diagnosed with MDD experience clinically significant anhedonia (Pelizza and Ferrari, 2009). Anhedonia is a particularly difficult symptom to treat, as accruing evidence suggests that current first-line pharmacotherapies (e.g., SSRIs) do not adequately address motivational and reward-processing deficits in depression (APA, 2000; Dunlop and Nemeroff, 2007; McCabe et al., 2009; Nutt et al., 2007; Price et al., 2009; Shelton and Tomarken, 2001), and the presence of anhedonic symptoms is a predictor of poor treatment response generally (Spijker et al., 2001).
In an effort to find more effective treatments for psychiatric symptoms, the National Institute of Mental Health has emphasized translational research approaches to identify neurobiological mechanisms underlying psychiatric symptoms and disorders (Insel, 2009), such as the development of the newly proposed Research Domain Criteria (RDoC) (Insel and Cuthbert, 2009; Miller). Reward-related symptoms represent an excellent opportunity for translational neuroscience, given the vast basic science literature from which to draw upon (Berridge and Robinson, 2003; Gold et al., 2008). However, application of this important preclinical work to human conditions is hampered by the enormous heterogeneity in psychiatric disorders, and the limited phenotypic characterization of clinical samples in most group studies. This is true not only for the presence or absence of specific symptoms within a disorder (diagnostic heterogeneity), but also for the presence or absence of co-morbid conditions (heterogeneity of co-morbidity), etiological pathways involved in disorders (etiological heterogeneity), and even for definitions of symptoms (symptom heterogeneity).
Attention to these multiple forms of heterogeneity is critical for elucidating the neurobiological pathways involved. For example, under the DSM-IV definition of Major Depressive Episode, which requires the presence of 5 out of 9 possible symptoms, it is possible for two individuals to both be diagnosed with major depression while only sharing a single symptom of the disorder. Such heterogeneity in how an individual meets criteria may be both practical and theoretically appropriate, but it may also mask important associations that are related to specific symptoms, rather than the whole diagnostic category. Similarly, co-morbidity may obscure disorder specific, or symptom specific associations. For instance, while multiple studies have shown that individuals with depression exhibit increased amygdala activation in response to negatively-valenced stimuli (Fu et al., 2004; Siegle et al., 2006; Siegle et al., 2002; Siegle et al., 2007), newer evidence suggests that this amygdala activity may occur primarily in individuals with MDD and co-morbid anxiety symptoms (Beesdo et al., 2009). Heterogeneity in etiological factors may also be important. In testing the role of the Hypothalamic-Pituitary-Adrenal (HPA) Axis in MDD, it has been demonstrated that individuals with depression and early life-trauma exhibit structural reductions in regions involved in HPA axis regulation, while individuals with depression but not early-life trauma do not (Treadway et al., 2009b; Vythilingam et al., 2002). Group designs in MDD research that ignore this type of etiological heterogeneity may conceal important neurobiological differences (Heim et al., 2004).
A less commonly addressed form of heterogeneity—symptom heterogeneity—can arise from the presence of compound diagnostic criteria, or criteria that may be met in multiple ways. In the case of anhedonia, the DSM-IV-TR states that individuals meeting criteria “may report feeling less interest in hobbies, ‘not caring anymore,’ or not feeling any enjoyment in activities that were previously considered pleasurable” (American Psychiatric Association, 2000, p. 349). In other words, clinical diagnosis of anhedonia does not discriminate between a decrease in motivation and a reduction in experienced pleasure. The failure to draw such a distinction may reflect the long-held assumption that people are motivated to pursue the things they find pleasurable, and vice-versa.
In the present review, we suggest that heterogeneity at the level of symptom definition is at least as problematic as the more commonly acknowledged issues of co-morbidity or etiological variability in group samples. In making this argument, we use anhedonia in MDD as a case study, and suggest that the distinction between the motivational and hedonic aspects of anhedonia is critical, especially when attempting to elucidate neurobiological pathways underlying the expression of this symptom. Indeed, overly broad definitions may sometimes point towards spurious relationships between symptom and substrate. When Roy Wise first presented the highly influential dopamine deficiency hypothesis of anhedonia, he argued that dopamine (DA) critically mediated an organism’s experience of pleasure, or ”yumminess”, in response to rewarding stimuli (Wise, 1980). Consequently, it was posited that anhedonia in mood disorders could be explained by a reduction in DA transmission (Willner, 1983a, b, c). In the intervening quarter-century however, only half of this original hypothesis has found empirical support. Namely (and as described below), subsequent research using neuroimaging, pharmacological and genetic methods in both humans and animals has provided some support for the claim that DA function is impaired in at least a sub-population of individuals with MDD (Dunlop and Nemeroff, 2007; Yadid and Friedman, 2008). However, contrary to the original anhedonia hypothesis, the conceptualization of DA as being primarily related to pleasure has been largely abandoned (Berridge and Robinson, 2003; Salamone et al., 2007).
These two developments raise a potential problem: if alterations in DA are a significant component in the pathogenesis of MDD but are unrelated to deficits in experience of pleasure, what is their functional and clinical consequence? In the present review, we suggest that this problem may be resolved through a refined definition of anhedonia, which attends more closely to the distinction between deficits in the hedonic response to rewards (“consummatory anhedonia”) and a diminished motivation to pursue them (“motivational anhedonia”). These facets of anhedonia are intended to roughly correspond to the reward-processing components of ‘liking’ and ‘wanting” proposed in the preclinical literature (Berridge and Robinson, 1998, 2003). In addition, we suggest that bridging the gap between preclinical and clinical models of anhedonia may require moving away from the conceptualization of anhedonia as a steady-state, mood-like phenomena, and towards a more behavioral model that emphasizes the influence of anhedonic symptoms on decision-making. To address this issue, we present the concept of “decisional anhedonia”, and highlight the neurobiological networks that may underlie impaired decision-making in the context of reward. These distinctions have been largely overlooked in the extant empirical literature on MDD, which may explain why this literature is replete with inconsistent findings (Forbes, 2009). We suggest that a crucial next step in the field is to refine our assessment and diagnostic tools to better reflect the multi-faceted nature of reward deficits in MDD.
This strategy is not entirely new, and indeed echoes decades-old theoretical models and clinical observations. Neurobiological models of personality have previously emphasized dissociations between “approach” and “consumption” of rewards, with the former constituting a behavioral activation system. These models further posited that DA is primarily linked with approach emotions, and might therefore underlie individual differences in reward seeking behaviors and psychopathology (Cloninger, 1987; Depue and Collins, 1999; Depue and Iacono, 1989; Gray, 1987). Similarly, this dissociation has been noted in the clinical literature; the psychiatrist Donald Klein noted that many patients with depression and anhedonia appeared to enjoy rewards that were readily available, yet complained bitterly about feeling no desire to obtain them (Klein, 1987). While these theories and observations have existed for some time, they have yet to be integrated with the substantial animal literature on the neurobiology of reward processing into a comprehensive framework.
It is the goal of this review to provide such an integrative framework. Central to our approach is the claim that symptom definitions should be grounded in a neurobiological context. In the sections that follow, we begin by summarizing the available empirical literature regarding reward-processing deficits in depression. We argue that the distinction between motivation and pleasure has critical implications for both interpreting the existing literature and for the design of future studies of MDD. Following this review, we summarize the preclinical literature regarding the neural mechanisms of motivational, hedonic and decisional aspects of reward, and compare these mechanisms to neurobiological studies of human MDD. Based on existing preclinical and clinical data of reward processing we suggest that multiple nodes involved in reward circuits are impacted in MDD, and that this dysfunction may underlie anhedonic symptoms.
2. Diagnostic and Behavioral Studies of Anhedonia in Depression
In his original definition, Theodule Ribot described anhedonia as the inability to experience pleasure (Ribot, 1896). However, across multiple domains of the social and neural sciences, it has become clear that embedded within the idea of pleasure are multiple constructs, including reinforcement, desire, predicted utility, subjective pleasure, experienced utility, remembered utility, and so forth, each of which describes a unique aspect of reward processing. Attention to these distinctions is important when evaluating self-report and laboratory studies of anhedonia and related constructs (e.g., positive affect) in depression. In this section, we summarize the different ways in which anhedonia has been measured in MDD. Notable, we show that empirical work has focused heavily on exploring hedonic experience in depression, while studies of motivation are relatively absent.
2.1 Anhedonia and the diagnosis of Depression
As already noted, the DSM-IV treats anhedonia as one of two symptoms required for a diagnosis of MDD. However, the psychometric properties of anhedonia and other symptoms of depression have only recently been subjected to rigorous empirical analysis. In 2006, the Rhode Island Methods to Improve Diagnostic Assessment and Services (MIDAS) project published a series of papers exploring the psychometric aspects of DSM criteria as assessed using a structured interview in a sample of 1523 subjects (Zimmerman et al., 2006). As part of this study, they also made head-to-head comparisons between current DSM-IV symptom criteria and theoretical criteria, such as helplessness/hopelessness, lack of emotional reactivity, and diminished drive (a construct similar to motivational anhedonia). This additional diminished drive criterion is distinct from the typical Structural Clinical Interview for DSM Axis-I Disorders (SCID) assessment of anhedonia, which does not dissociate between motivational or consummatory aspects of reward; in keeping with DSM-IV criteria, the SCID simply asks patients whether they “have lost interest or pleasure in things that they usually enjoy”. Strikingly, diminished drive criterion had the second highest odds-ratio for predicting a diagnosis of depression (50.1), ranking only below sad mood (61.2) and significantly greater than anhedonia as assessed by the SCID (29.7) (McGlinchey et al., 2006). This finding is all the more impressive when considering the fact that the criterion of diminished drive is handicapped in comparison to the DSM anhedonia criteria, as only the latter bears directly on diagnostic outcome.
2.2 Clinical Assessment of Anhedonia in Depression
Dimensional assessment of anhedonic symptom severity has primarily been achieved through self-report instruments. A content review of items used in the most common anhedonia measures reveals that they unanimously emphasize the experience of pleasure in response to positive stimuli, with little or no attention to diminished drive or motivation. This includes the Chapman Anhedonia Scale (Chapman et al., 1976), the Scale of Negative Symptoms (SANS; (Andreasen, 1982), the Fawcett-Clark Pleasure Scale, (FCPS; (Fawcett et al., 1983) and the Snaith-Hamilton Pleasure Scale (SHAPS; (Snaith et al., 1995). It is also worth noting that several of these scales were developed with a primary focus on schizophrenia (Chapman, SANS) rather than depression. Symptom severity instruments specific to depression often assess anhedonia with a small number of items; a single question in the case of the 17-item Hamilton Depression Rating Scale (Hamilton, 1960), four items on the 21-item Beck Depression Inventory (BDI anhedonia scale; (Beck et al., 1996)) and four on the 30-item Inventory of Depressive Symptoms. Importantly, none of these scales have made an explicit attempt to dissociate between pleasure and motivational aspects of anhedonia. More recently, the Temporal Experience of Pleasure Scale (TEPS; (Gard, 2006) was developed to assess anticipatory and consummatory pleasure. This scale is a promising advance, though it is unclear whether the experience of pleasure when anticipating rewards is an identical construct to reward motivation, and its application in clinical populations will be necessary to determine its utility for parsing clinical anhedonia.
In seeking to assess the relevance of these commonly used anhedonia assessment inventories, one recent study used a 10-indicator, 3-factor confirmatory factor analysis model to assess multiple measures of depression and anhedonia in a sample of controls and individuals with MDD. Anhedonia questionnaires included the Chapman, FCPS, and SHAPS, as well as clinical symptom inventories (BDI and Beck Anxiety Inventory; BAI). Using this approach, they identified three latent variables reflecting hedonic capacity, depressive symptoms and anxiety symptoms, and found that the hedonic capacity and depression variables were only moderately associated (factor loading = −.20) (Leventhal et al., 2006).
Finally, the Mood-Anxiety Symptoms Questionnaire (MASQ) developed by Watson and Clark (Watson et al., 1995a; Watson et al., 1995b), includes a number of items related to lowered positive affect and interest, some of which appear related to aspects of anhedonia. However, these items are generally not treated separately from the larger scales that contain them, which remain relatively heterogeneous. Therefore, collapsing across these different forms of reward deficits may obfuscate the results, and may contribute to weaknesses in fitting a three-factor model across samples (Buckby et al., 2008; Burns and Eidelson, 1998; Kiernan et al., 2001).
2.3 Laboratory Studies of Anhedonia in Depression
In laboratory settings, a number of studies have examined affective responses to positively-valenced stimuli as a means of exploring the nature of anhedonic symptoms. These studies have suggested that individuals with depression generally rate positively-valenced stimuli as being less positive, less arousing, or less able to affect their mood as compared to controls (Berenbaum, 1992b, a; Dunn et al., 2004; Rottenberg et al., 2005; Rottenberg et al., 2002; Sigmon and Nelson-Gray, 1992; Sloan et al., 1997; Sloan et al., 2001; Wexler et al., 1994) although a larger number of studies have reported no group differences in these ratings (Allen et al., 1999; Dichter et al., 2004; Forbes and Dahl, 2005; Gehricke and Shapiro, 2000; Kaviani et al., 2004; Keedwell et al., 2005a, b; Mitterschiffthaler et al., 2003; Renneberg et al., 2005; Surguladze et al., 2005; Tremeau et al., 2005; Tsai et al., 2003).
A potential caveat to this approach is whether reductions in affective responsiveness to positively-valenced stimuli are specific to experienced pleasure. One alternative explanation is that individuals with depression simply show a global flattening that encompasses both positive and negative emotions. Supporting the affective-flattening hypothesis, a recent meta-analysis of studies that measured physiologic or subjective affective responses found that depression was associated with blunted reactivity to both positively- and negatively-valenced stimuli (Bylsma et al., 2008). Although it is notable that in the Bylsma analysis the effect size for positive stimuli is roughly double that for negative stimuli, their results suggest that at least part of the decline in hedonic responses may be due to a generalized affective blunting, rather than a specific deficit in experienced pleasure.
The “sweet taste test” provides another approach to assessing hedonic capacity. As part of the sweet taste test, participants rate the pleasantness of different sucrose concentrations. An advantage of this test is that it closely mirrors animal measures of hedonic experience. As such, it is notable that across four separate studies using the sweet taste test, individuals with depression and matched controls have shown no differences in reported hedonic impact (Amsterdam et al., 1987; Berlin, 1998; Dichter et al.; Kazes et al., 1994). On the surface this suggests that there is no deficit in hedonic capacity to experience a natural reinforcer in MDD. A concern may be raised, however, as there are substantial individual differences in taste sensitivity (Duffy and Bartoshuk, 2000) that may make such measures insensitive to state changes in hedonic perceptions. In summary, the literature suggests reductions in hedonic capacity in MDD, although the generalizability of such deficits remains unclear.
Additional laboratory studies have used reinforcement paradigms to explore anhedonia in depression. One well-replicated finding has been that individuals with depression fail to develop a response bias towards rewarded stimuli (Henriques et al., 1994; Pizzagalli et al., 2008; Pizzagalli et al., 2005). These paradigms use discrimination tasks in which subjects must categorize a briefly presented stimulus as belonging to category A or B. Importantly, these paradigms use a pay-off matrix so that subjects are more rewarded for correctly guessing category A, as opposed to category B, with no punishment associated with incorrect guesses. Healthy control subjects typically develop a response bias toward the more rewarding option, whereas MDD patients do not. These elegant studies provide strong evidence for an insensitivity to reward-relevant information in MDD. One limitation, however, is whether these reinforcement deficits are driven by reduced hedonic capacity, diminished motivation, or both.
Finally, one recent study compared ratings of experienced emotion in individuals with current depression, remitted depression and never depressed controls across conditions that involved anticipating and experiencing rewards and punishments (McFarland and Klein, 2009). Participants rated their emotions across ten dimensions in response to four experimental conditions: anticipating monetary rewards, anticipating an unpleasant sensory stimulus (cold press), no change, and avoiding an unpleasant sensory stimulus. No differences between the three groups were reported for anticipating unpleasant stimuli, no change, or the avoidance of an unpleasant stimulus. In contrast, during reward anticipation, individuals with current MDD showed significantly reduced ratings of positive emotions as compared to controls, and slightly reduced ratings compared to individuals with prior depression. Although this study did not test motivation per se, these data provide novel evidence of a deficit in experienced emotion during reward anticipation in MDD.
Taken together, the reviewed evidence suggests that while diagnosis of anhedonia assesses both motivation and experience of pleasure, current questionnaire and laboratory measures of anhedonia have largely emphasized the latter; the authors are aware of no laboratory studies that have directly assessed motivation in MDD. In laboratory settings, a number of studies have found evidence for diminished responsiveness to positively-valenced stimuli, but the work of Bylsma et al., suggests that this may reflect a general affective flattening. Moreover, it remains unclear how closely related measures of affective responses to positively-valenced stimuli are to the construct of hedonic capacity. Importantly, the lack of group differences on the sweet-taste test raise potential doubts as to whether or not depression is associated with a specific deficit in the capacity to feel pleasure, at least at the level of basic sensory experience.
3. Neurobiological Bases of Motivational and Consummatory Anhedonia
In a striking contrast to the behavioral literature, which has largely focused on deficits in hedonic capacity (‘liking’), preclinical neurobiological studies of anhedonia have primarily targeted neural substrates involved in motivation and reinforcement (‘wanting’). Across a variety of studies, ‘liking’ and ‘wanting’ have been linked to a variety of brain regions, neural circuits and neurotransmitters. These include the neurotransmitter dopamine and opioid neuropeptides, sub-cortical structures such as the basal ganglia and striatum (particularly the nucleus accumbens (NAcc), ventral pallidum (VP), ventral tegmental area (VTA), substantia nigra (SN), amygdala and hippocampus, as well as cortical regions such as the ventromedial prefrontal cortex (vmPFC), encompassing aspects of orbital frontal cortex (OFC), anterior cingulate cortex (ACC) and medial prefrontal cortex (mPFC). In this section we review the neural circuitry involved in both ‘liking’ and ‘wanting’ as well as the evidence to support its role in consummatory and motivational aspects of anhedonia in MDD.
3.1 Reward ‘Wanting’: Dopamine and Striatal Circuitry
Located within the pars compacta of the substantia nigra (SNpc) and VTA, DA neurons give rise to three ascending pathways: the nigrostriatal, mesolimbic and mesocortical pathways, as depicted in figure 1. The nigrostriatal pathway terminating in the dorsal caudate and putamen is heavily implicated in motor control, and habit learning. The mesolimbic pathway terminates in the ventral striatum (including the NAcc), the amygdala and hippocampus, and is most closely associated with associative learning, reward motivation and reinforcement. The mesocortical pathway projects to cortical regions, including dense innervation of the ACC, with additional terminals in orbital frontal cortex, medial prefrontal cortex and the insula. This third pathway is strongly associated with working memory, attention, and inhibitory control.
Figure 1.

Schematic illustration of dopamine projection pathways and circuitry regulating DA release in the human brain. DA firing rates are maintained at tonic levels in part due to steady-state inhibitory firing from the ventral pallidum. Excitatory projections from prefrontal cortex project, amygdala and hippocampus synapse on striatal targets, including the nucleus accumbens. The nucleus accumbens sends GABAergic projections to the ventral pallidum, suppressing VP inhibition of VTA, thereby facilitating phasic burst-firing of VTA DA neurons. Note: Placement of structure labels is approximate. Amyg = amygdala; Caud = Caudate; DA = Dopamine; GABA = GABAergic projections; Glu = glutamatergic projections; Hipp = hippocampus; NAcc = nucleus accumbens; Put = Putamen; SN = substantia nigra; VP = ventral pallidum; VTA = ventral tegmental area
Midbrain DA neurons exhibit two distinct modes of firing, referred to as “tonic” and “phasic” (Grace and Bunney, 1984). Tonic DA activity refers to steady-state firing generated by intrinsic pacemaker-like characteristics of DA neurons. Phasic activity—also known as “burst firing”—involves a rapid series of action potentials that induce a rapid rise in extracellular DA at terminal projection targets. As additionally outlined in figure 1, initiation of phasic activity requires excitatory signals from a variety of areas, including the prefrontal cortex, pedunculpontinetegmentum (PPt) and subthalamic nucleus (Floresco et al., 2003; Futami et al., 1995; Smith and Grace, 1992) as well as suppression of steady-state inhibitory signals arising from the NAcc and ventral pallidum (VP) (Sesack and Grace).
A key function of DA is to modulate the sensitivity of post-synaptic neurons to other types of input. In the striatum—the largest recipient of DA projections—DA may modulate the sensitivity of medium spiny neurons (MSN) to excitatory glutamatergic projections from prefrontal and limbic regions. As shown in figure 2, DA acts primarily on one of 5 post-synaptic G-protein coupled receptors, labeled D1–D5 (Cooper, 2003). These receptors are grouped into two “families”, described as D1-like (including D1 and D5 receptors) and D2-like (D2, D3 and D4 receptors). Upon receptor stimulation, Both D1-like and D2-like receptors interact with adenylate cyclase (AC) (Surmeier et al., 2007). D1-like receptor stimulation increases AC activity through coupling with either G alpha S or (Gα-s) G alpha olfactory (Gα-olf), which results in increased activation of protein kinase A (PKA) and subsequent phosphorylation of various intracellular targets. Recent evidence suggests that this intracellular pathway can result in increased responsiveness of MSNs to sustained release of glutamate, generating “up-states” (Surmeier et al., 2007). In contrast, D2-like receptor binding results in decreased AC activity, thereby reducing the responsiveness of MSNs (“down states”) (Hernandez-Lopez et al., 2000). Of note, due to their higher affinity for DA as well as their more centralized location on the post-synaptic membrane, D2-like receptors are often stimulated by tonic levels of DA release, whereas D1-like receptors are stimulated primarily during phasic DA release (Goto et al., 2007).
Figure 2.

Schematic illustration of dopamine synapse on striatal medium spiny neuron. DA stimulation of D1-like receptors increases the activity of adenylate cyclase, while stimulation of D2-like receptors suppresses adenylate cyclase activity. DA may be removed from the synapse either by reuptake via the DA transporter or degradation by monoamine oxidase, resulting in the DA metabolite of homovanillic acid. Psychostimulants increase synaptic DA by blocking DAT function, while monaomine oxidase inhibitors block MAO activity and pramipexole inhibits DA autoreceptors. AC = adenylate cyclase; DAT = DA transporter; DOPA = 3,4-dihydroxyphenylalanine; HVA = homovanillic acid; MAO = monoamine oxidase; MAOI = monoamine oxidase inhibitor; MSN = medium spiny neuron; TH = tyrosine hydroxlase
Initial evidence for the role of DA in mediating reward ‘wanting’ comes from the fact that 6-OHDA lesions of NAcc DA synapses do not impair hedonic liking expressions in rats (Berridge and Robinson, 1998). Similar effects have been found following the systemic administration of neuroleptic drugs—acting primarily on DAergic sites— which also failed to alter liking responses (Kaczmarek and Kiefer, 2000; Parker and Leeb, 1994; Pecina et al., 1997). Finally, DA burst-firing—which commonly occurs in response to unexpected rewards—ceases after the previously unexpected reward becomes predicted, despite the fact that the hedonic value of the predicted reward is presumably intact (Berridge, 2007; Schultz, 2006; Schultz et al., 1997). Even more striking evidence comes from studies using mice that have been genetically engineered to be incapable of endogenous DA synthesis without the aid of daily L-DOPA administration (Zhou and Palmiter, 1995). Suspension of these L-DOPA administrations for a single day can result in the near-total depletion of DA in the brain. However, even these highly DA-depleted mice still favor sucrose-water over regular water, and demonstrated a morphine-induced conditioned place-preference (Cannon and Palmiter, 2003; Hnasko et al., 2005). Finally, studies have found that increasing DA shows no effect on liking behavior. Genetically modified mice that exhibit a knockdown of the Dopamine Transporter (DAT) gene, thereby resulting in increased extracellular DA, showed no alterations in liking responses (Pecina et al., 1997). In sum, these findings provide clear evidence that DA function is neither necessary nor sufficient for hedonic liking responses to occur.
A second line of work has sought to demonstrate a pivotal role for DA in the motivation to pursue rewards, as indexed by overcoming response costs (Salamone et al., 2007). As shown in figure 3, Salamone and colleagues developed experimental paradigms that evaluate an animal’s willingness to work for a given reward. These paradigms, described herein as “effort-based decision-making” paradigms, include concurrent-choice tasks and progressive ratio tasks (Assadi et al., 2009). Initial studies employed a T-maze design, in which rats enter a T-shaped maze and made a choice between one arm of the maze containing a readily available food reward (Low-Cost/Low Reward, “LC/LR”), and another arm containing a larger food reward that was available only after climbing a barrier (High-Cost/High-Reward, “HC/HR”). Using this choice-paradigm, it was demonstrated that while control rats prefer the HC/HR option, rats with NAcc DA lesions or blockade of striatal D2 receptors show increased preference for the LC/LR option (Correa et al., 2002; Cousins et al., 1996; Cousins and Salamone, 1994; Denk et al., 2005; Salamone et al., 2007).
Figure 3.

Schematic diagram of effort-based decision-making paradigms. Animals may choose between a smaller food reward that is readily available (LC/LR option) or a greater food reward that can only be obtained after climbing over a barrier (HC/HR option). Control rats choose the HC/HR option approximately 90% of the time, while DA depleted rats show a strong preference for the LC/LR option.
Convergent evidence was found during an operant response concurrent-choice task, where rats must choose between eating freely-available, unpalatable “lab chow” (LC/LR option) or pressing a lever several times in order to receive a preferred food reward (HC/HR option). As with the T-maze paradigm, blockade of NAcc DA through either lesions of DA projection terminals with 6-OHDA will result in a reduced preference for the HR choice (Aberman and Salamone, 1999; Correa et al., 2002; Cousins and Salamone, 1996; Hamill et al., 1999; Salamone et al., 2005; Salamone et al., 1991). Additional studies have found that global blockade of DA using selective D1 or D2 receptor antagonists may also impair effort-based decision-making (Bardgett et al., 2009; Walton et al., 2009), though selective impairment of phasic DA release does not (Zweifel et al., 2009).
A key aspect of these paradigms for translational psychopathology research is the fact that control animals choose HC/HR options approximately 90% of the time, thereby suggesting that experimentally-induced increases in LC/LR choices can reasonably be interpreted as pathological in nature, rather than a minor shift in normative preferences. In addition, multiple control experiments have been performed to rule out possible confounding factors, such as alterations in the ability to engage in voluntary movement, or diminished understanding of reward contingencies. For example, in conditions where reward is removed entirely from the LC/LR option, or the paradigm is modified so that both LC/LR and HC/HR options require equal effort, NAcc DA depleted rats cease to differ from control animals (Denk et al., 2005; Salamone, 1996). Additionally, one recent study confirmed that NAcc DA influences effort-expenditure preferences even when controlling for differences in reward delay, as HC/HR options often require extra time to complete (Floresco et al., 2008). These additional studies suggest that experimentally-induced preferences for LC/LR options are; 1) sufficiently abnormal to be construed as a pathological deficit in motivation, and 2) do not result from impaired understanding of choice contingencies, physical inability, or temporal delay.
Taken together, these findings provide strong evidence for the role in DA as encoding the motivational aspects of reward processing, while being relatively uninvolved in the hedonic experience. In stating this, we do not intend to suggest that alterations in DA are the only way to produce motivational deficits (see section 4), but rather that alterations in DA functioning are sufficient to produce such deficits.
3. 2 Motivational Anhedonia: Dopamine and Basal Ganglia Function in Depression
Given that DA has been theorized to play a role in MDD since at least the 1970’s, it may seem surprising that it has received relatively less attention than either norepinephrine (NE), or serotonin (5HT). This in part reflects the known serotonergic and noradrenergic properties of many antidepressants. The focus on 5HT and NE further intensified with the discovery in the 1980’s that tricyclic antidepressant medications (TCAs) act primarily by blocking reuptake of NE and 5HT, and the subsequent success of SSRIs therapies in treating MDD. However, as Dunlop and Nemeroff have observed, the well-documented temporal delay between reuptake inhibition and clinical effects suggests that the therapeutic action of these drugs may involve downstream mechanisms, including alterations in DA function (Dunlop and Nemeroff, 2007). Additionally, it is worth noting that these medications may be more effective in treating the anxiogenic aspects of depression, as suggested by their efficacy in the treatment of anxiety disorders (Davis et al., 2006; Ninan, 2003; Sheehan and Mao, 2003; Vaswani et al., 2003). A DA role in anhedonia may also explain why 5HT and/or NE antidepressant medications do not adequately address this symptom (Dunlop and Nemeroff, 2007; Shelton and Tomarken, 2001).
Another challenge to uncovering the role of DA in MDD is that many studies have employed group designs with heterogeneous patient samples that are not limited to patients with anhedonic symptoms, much less specific motivational and consummatory subtypes. Such heterogeneity may mask group differences in DA function, as well as specific within-group associations between DA and anhedonia. This problem is worsened by the fact that assessment instruments for anhedonia are heavily weighted towards pleasure responses, which are unlikely to be strongly associated with DA function. Nevertheless, multiple studies support the hypothesis that there are abnormalities in DA in MDD, which is a desiderata if DA plays a specific role in one or more of the symptoms of the disorder.
Initial data supporting a role of DA in MDD comes from studies of DA turnover, which observed that individuals with MDD have decreased cerebrospinal fluid (CSF) levels of homovanillic acid (HVA), the primary metabolite of DA (Berger et al., 1980; Lambert et al., 2000; van Praag et al., 1973; Willner, 1983a). These studies suggest the presence of lowered basal DAergic tone in MDD. Additionally, pharmacological interventions that block or deplete DA can induce or deepen depressive symptoms in currently depressed or remitted individuals (Bremner et al., 2003; Hasler et al., 2008; Ruhe et al., 2007), further implicating DA dysfunction in MDD.
In animal models of depression, several lines of evidence also support the role of DA dysfunction. The Flinders sensitive line (FSL), a genetic animal model of MDD, exhibit reduced basal concentrations of DA in the NAcc and slower rates of DA release in the NAcc as compared to Sprague-Dawley (SD) rats (Zangen et al., 2001). One contributing cause of reduced extracellular DA concentrations in DA neuron terminal regions is altered firing patterns of midbrain DA neurons themselves. Consistent with this explanation, FSL rats have been observed to exhibit marked impairment in phasic burst firing (Friedman et al., 2007) (for a review, see (Yadid and Friedman, 2008)). Another animal model of depression that implicates DA function is the post-psychostimulant withdrawal model (Barr and Markou, 2005; Barr et al., 2002). This model is particularly relevant for research on DA in MDD, as it produces a significant number of symptoms associated with MDD (Markou et al., 1998) and results from direct manipulation of the DA system. Consistent with effort-expenditure deficits observed by Salamone and colleagues following NAcc DA blockade, psychostimulant withdrawal has been shown to reduce both NAcc extracellular DA levels (Weiss et al., 1992) and effort-expenditure for sucrose rewards during a progressive ratio task (Barr and Phillips, 1999).
DA acting drugs, particularly D2 agonists, have antidepressant properties in animal models of depression (for a review see (Gershon et al., 2007)). Indeed, a large number of studies have demonstrated that chronic administration of various classes of antidepressant medication show a common effect of increasing D2-like receptor binding or sensitivity in the NAcc, and increased psychomotor responses to psychostimulants (D'Aquila et al., 2000; Gershon et al., 2007). Such effects are observed following chronic treatment with both tricyclic antidepressants and SSRIs, even though the acute effects of these agents are primarily mediated through serotonergic and noradrenergic mechanisms. Notably, however, the antidepressant effects of these agents can be blocked entirely by D2-like receptor antagonists. Finally, the selective serotonin-reuptake enhancer (SSRE) tianeptine has been shown to have robust antidepressant properties, a finding contrary to what would be expected if MDD were associated with a specific deficit in 5HT signaling (Kasper and McEwen, 2008). While the mechanisms that underlie the antidepressant properties of tianeptine are unclear, it is noteworthy that this compound has been shown to increase NAcc extracellular DA levels as well as DA turnover in rodents (Invernizzi et al., 1992).
In humans, pharmacological enhancement of DA signaling provides at least temporary antidepressant effects, and has been seen with DA agonists such as bromocriptine, piribedil, ropinirole and pramipexole (Bouras and Bridges, 1982; Cassano et al., 2005; Shopsin and Gershon, 1978; Sitland-Marken et al., 1990; Vale et al., 1971; Waehrens and Gerlach, 1981). DAT inhibitors nomifensine (Kapur and Mann, 1992), methylphenidate (El-Mallakh, 2000), amineptine and bupropion also exhibit varying degrees of antidepressant effects, further highlighting the possible role DA in MDD (see section 5 for further review) (Kapur and Mann, 1992; Stahl, 2000).
Human neuroimaging studies of DA synthesis capacity have shown reduced L-DOPA uptake in MDD (Agren and Reibring, 1994). Moreover, studies exploring different sub-groups have found that L-DOPA alterations in the striatum are present in depressed individuals with flat affect or psychomotor slowing, but not depressed individuals without these symptoms (Bragulat et al., 2007; Martinot et al., 2001). Patients with reduced DA synthesis in Parkinson’s disease also show increased rates of MDD (Koerts et al., 2007). These data suggest that reduced DA synthesis capacity may be linked to specific symptoms in MDD.
Additional evidence of altered DA function in MDD comes from Positron Emission Tomography (PET) and Single Photon Emission Computed Tomography (SPECT) imaging of the DA transporter (DAT), where depression has been associated with both lower (Meyer et al., 2001) and higher (Amsterdam and Newberg, 2007; Laasonen-Balk et al., 1999; Yang et al., 2008) DAT binding potential in the striatum.. Of note, however, the one study that restricted its MDD patient sample to individuals with anhedonic symptoms reported decreased DAT binding (Sarchiapone et al., 2006). Monoamine oxidase A, a metabolizing enzyme of DA and other monoamines has been shown to be elevated in MDD across multiple brain regions, suggesting one possible mechanism through which observed decreases in monoamine transmission may occur during a depressive episode (Meyer et al., 2006). Heightened activity of MAOA in MDD may partially explain the efficacy of MAO inhibitors, which likely lead to the increased availability of monoamines—including DA—by returning MAOA activity to normative levels.
Studies of DA receptor availability in MDD have to date produced mixed results. In some cases, increased striatal D2/D3 receptor binding has been shown to occur in heterogeneous depressed samples (D'Haenen and Bossuyt, 1994; Shah et al., 1997), as well as in patient samples with specific symptoms of psychomotor slowing (Meyer et al., 2006). This increase in D2/D3 receptor availability would appear to contradict animal data in which antidepressant responses are associated with increased D2-like binding in the striatum. The source of this discrepancy is unclear, but it may be noted that the patients in human studies were not medication-naïve. Other studies using medication naïve or medication-free patients have failed to identify group differences in striatal receptor binding (Hirvonen et al., 2008; Parsey et al., 2001), while one additional small study showed variable changes in D2-like binding following treatment with SSRIs, with those showing increased binding showing more clinical improvement than those who did not (Klimke et al., 1999). Taken together, these studies suggest a possible role of D2-like receptors in downstream effects of antidepressant treatment. However, the precise nature of the effect and how alterations in D2-like receptor availability may relate to DA function as a whole remains unclear. Moreover the use of heterogeneous samples, and limited exploration of specific symptoms, has precluded examination of specific relationships between D2-like function and motivational anhedonia.
As for D1-like receptors, a recent study of D1 receptors using PET-[11C]NNC-112 found reduced D1-like receptor binding in the striatum bilaterally in a sample of individuals with MDD (Cannon et al., 2009). Anhedonia as assessed by a sub-scale of the IDS-C was not correlated with change in binding potential in the MDD group. However, as with other commonly used assessments of anhedonia, the anhedonia subscale from the Inventory of Depressive Symptoms-Clinician-rated primarily emphasizes consummatory, rather than motivational aspects.
It is additionally worth noting that a proposed role for DA dysfunction in the pathophysiology of MDD is consistent with current etiological models that highlight interactions between genetic risk factors and stressful life events in the onset, maintenance and relapse of MDD (Caspi et al., 2003; Hammen, 2005; Kendler et al., 1999; Kessler, 1997). Genetic studies have identified several polymorphisms related to DAergic function that increase risk for the development of depression. The most reliable of these findings is allelic variations in the DRD4 gene (Lopez-Leon et al., 2005) and D3 receptor gene in both unipolar and bipolar depression (Chiaroni et al., 2000; Dikeos et al., 1999). Additionally, the effects of chronic and acute stress are well known to have significant consequences on the DA system. Stress has been shown to increase glucocorticoid signaling (Holsboer, 2000), precipitate neuronal degeneration in the hippocampus (Sapolsky, 2000) and medial prefrontal cortex (McEwen, 2005; Radley et al., 2006), decrease the availability of brain-derived neurotrophic factor (BDNF) (Duman, 2009), and increase levels of pro-inflammatory cytokines in the brain (Dowlati et al.; Maier and Watkins, 1998). Importantly, all of these modulations have direct influence on DA function. Glucocorticoids modulate firing of DA neurons (Piazza et al., 1996a; Piazza et al., 1996b), and regions that suffer glucocorticoid-mediated atrophy are key regulators of mesolimbic and mesocortical DA projection pathways (Arnsten, 2009; Lisman and Grace, 2005). BDNF has been shown to regulate VTA DA neurons (Conner et al., 1997), and alterations in BDNF can influence mesolimbic DA responses to reward and resiliency to stress (Berton et al., 2006; Cordeira et al.). Finally, increases in proinflammatory cytokines can impact both the metabolism and synthesis of DA (Anisman et al., 2008) so as to result in reduced DA availability.
Taken together, the above studies provide evidence that 1) MDD is associated with compromised DA function, 2) manipulations of the DA system contribute to the actions of antidepressants and 3) alterations of DA function are often a downstream consequence of genetic and environmental risk factors, such as exposure to stress. These positive findings are qualified by the presence of null findings, as well as the difficulty in interpretation associated with some of the studies. Notably, some of the findings appear specific to sub-populations of depressed individuals defined by the presence or absence of specific symptoms. This observation is consistent with the central claim that rigorous phenotypic description is crucial for reliable results with biological measures.
As previously noted, the DA system is only one aspect of the reward circuit involved in motivation. The striatum, particularly its ventral aspects, is also heavily implicated in reward motivation, and has exhibited functional and structural abnormalities in patients with MDD. Structural MRI studies have reported reduced grey-matter volume within regions of the striatum (Kim et al., 2008; Matsuo et al., 2008; Pizzagalli et al., 2009; Wacker et al., 2009). Functional MRI studies have revealed diminished responses to receipt of reward (McCabe et al., 2009; Pizzagalli et al., 2009; Smoski et al., 2009; Wacker et al., 2009), to anticipation of reward (Forbes et al., 2009; Smoski et al., 2009), exposure to DA-releasing agents (Tremblay et al., 2005) and to reward prediction errors (Kumar et al., 2008; Steele et al., 2007). It is also of note that MDD has been associated with a failure to develop a reward-dependent response-bias (Pizzagalli et al., 2008), a phenomenon that has been directly linked to striatal neuron function (Lauwereyns et al., 2002).
These findings provide strong evidence that structural and functional changes in the striatum are associated with depression, but further interpretation is difficult as most of the differences in neural activity occur in the absence of between-group differences in behavior. Additionally, only a small number of studies have sought to dissociate anticipatory/motivational and consummatory aspects of reward processing; these studies found mixed evidence for altered neural responses during reward anticipation and receipt. Using the Monetary Incentive Delay (MID) task, a well-validated reward-system probe that discriminates between anticipatory and consummatory aspects of reward, two groups failed to find group differences during reward anticipation (Knutson et al., 2008; Pizzagalli et al., 2009). This is contrary to what would be expected given that reward anticipation is most closely linked to DA function. Further, in healthy controls, the anticipation phase of the MID has been found to correlate with striatal DA release induced by amphetamine (Buckholtz et al. 2010) and extended performance of the MID itself (Schott et al., 2008). Do these findings challenge the hypothesis that altered DA function may mediate motivational anhedonia in MDD?
Several reasons suggest that while MID is a useful assay of motivation in healthy subjects, this interpretation may be less straightforward for individuals with MDD. First, studies using stimulation paradigms other than the MID have found group differences in striatal activation during reward anticipation (Forbes et al., 2009; Smoski et al., 2009), thereby suggesting the possibility that design elements unique to the MID may be at play. One issue is that the period of “reward anticipation” in the MID is a period of preparation to make a speeded-manual response. This is in contrast to the gambling tasks used by Smoski and colleagues or Forbes and colleagues, for which the anticipation period is passive and immediately precedes the potential reward as opposed to preceding a response. Interestingly, a recent study by Bar-Haim and colleagues used a modified version of the MID where for some trials reward anticipation was passive, while in others a speeded response was required. In a sample of adolescents with a history of inhibited temperament, Bar-Haim et al. reported a group by anticipation condition interaction in the ventral striatum, such that individuals with a history of inhibited temperament showed increased responses during active—but not passive—anticipation (Bar-Haim et al., 2009). This finding suggests that active reward anticipation may actually increase ventral striatal responses in anxiety-prone individuals, perhaps reflecting a DA stress response. After incorporating these additional results, it would appear that the anticipatory phase of the MID may not be a pure measure of appetitive motivation, and may capture heightened motor-preparatory responses in populations with anxiety symptoms, whose motivation may result less from reward-wanting than from a hypersensitivity to perceived failure (punishment motivation). Given the enhanced negative affectivity associated with MDD (Watson et al., 1995a), this could mask important group differences in reward-related ventral striatal activity. If this interpretation is correct than it may be necessary to devise probes that tap appetitive reward in the absence of potential confounds from other motivational features.
3.3 Reward “Liking”: Opioids, Amygdala and Ventromedial Prefrontal Cortex
The primary neurochemicals involved in pleasurable hedonic experience are endogenous opioids. Endogenous opioids include multiple families of neuropeptides, including endorphins, enkephalins, dynorphins and orphanin FG as well as their various receptor subtypes (μ, δ, κ and ORL 1) (Cooper et al., 2003). Functionally these peptides have been shown to play a significant role in the subjective experience of euphoria. Opioid receptors (μ, δ, κ) are widely expressed in the ventral striatum (particularly the shell of the NAcc). Stimulation of these receptors is believed to underlie hedonic responses to food, and possibly other natural rewards (Pecina et al., 2006). In humans, μ-opioid receptors in both the striatum and medial prefrontal cortex have been closely linked to affective responses (Liberzon et al., 2002; Zubieta et al., 2005; Zubieta et al., 2003; Zubieta et al., 2001).
Accruing evidence suggests that that opioid systems play a pivotal role in reward ‘liking’. To demonstrate this, researchers have shown that opioids are both necessary and sufficient for affective facial responses following the receipt of natural rewards (e.g., sucrose) (Pecina et al., 2006). Regions that result in increased “liking” responses following microinjection of μ-opioid receptor agonists have been dubbed “hedonic hotspots”. Two primary sties of these hotspots have been identified: the shell of the NAcc (Pecina and Berridge, 2005) and the VP (Smith et al., 2009). These two regions (with the possibility of others that have yet to be identified) are proposed to be the primary causal centers of affective pleasure responses.
Opioid function has also been proposed as the primary mechanism through which DAergic drugs of abuse, such as amphetamine and cocaine, induce subjective euphoria. Specifically, it has been shown that co-administration of opioid antagonists significantly attenuate the reported subjective effects of psychostimulants (Jayaram-Lindstrom et al., 2004). Further, co-administration of opioid agonists and psychostimulants result in significantly greater reported euphoria than psychostimulant administration alone (Mello et al., 2005). These data suggest that despite apparent correlations between stimulant-induced DA release and subjective euphoria (Drevets et al., 2001), this result is mediated by downstream effects on opioid function, and is unlikely to be a direct consequence of increased extracellular DA (Volkow et al., 1996).
In addition to these two subcortical “hedonic hotspots” that respond to opioid stimulation, multiple subregions of ventromedial prefrontal cortex (vmPFC) are also involved in processing the hedonic impact of rewarding stimuli, including the orbital frontal cortex (OFC) and the anterior cingulate cortex (ACC). The OFC provides substantial input into the ventral striatum (Zald and Kim, 1996a, b). Of primary importance for reward processing, the OFC has been shown to play a role in stimulus evaluation, and the association of stimuli with rewards (Zald and Kim, 1996a). Single-neuron recordings of the OFC suggest that the OFC may code the relative value of a reward (Tremblay and Schultz, 1999), although some coding also appears invariant to the presence or absence of other rewards (Padoa-Schioppa and Assad, 2008). Activations in the OFC are present both during the anticipation or prediction phase of a reward, and often precedes reward-expectancy activity in the striatum (Schoenbaum et al., 2003a; Schoenbaum et al., 2003; Setlow et al., 2003). OFC activations are also common during reward receipt and have been suggested to encode the affective value (and valence) of reward receipt (Kringelbach et al., 2004; O'Doherty et al., 2002; Rolls and Xiang, 2005). Moreover, some studies suggest that the magnitude of OFC responses is monotonically related to the magnitude of a given reward (Gottfried, 2006). Similarly, the rostral ACC also plays a role in encoding the receipt of reward (Knutson et al., 2001; Knutson et al., 2005; Sanfey et al., 2003). More unique to the ACC is the specialized role of making reward choices, particularly during more complex decisions that require the integration of both cost and benefits (Kennerley et al., 2009; Kennerley et al., 2006; Rushworth and Behrens, 2008; Walton et al., 2006).
Finally, the amygdala also plays a role in evaluation of positive stimuli (Balleine and Killcross, 2006; Baxter and Murray, 2002; Murray, 2007). Studies of amygdala lesions in rats show that damage to this structure may impair approach behavior (Everitt et al., 2003; Hiroi and White, 1991). In particular, studies have demonstrated a role for the amygdala in reinforcer evaluation. Amygdala lesions in monkeys significantly impair the animal’s ability to select an object based on the current value of a food reward (Baxter and Murray, 2002).
3.4 Consummatory Anhedonia: Opioid, Ventromedial Prefrontal Cortex and Amygdala Function in Depression
Initial interest in the possibility that depression might reflect opioid deficiency began in the early 1980’s, when two studies reported a temporary remission of depressive symptoms following injections of β-endorphin, a non-selective endogenous opioid receptor agonist (Catlin et al., 1982; Pickar et al., 1981). However, the ensuing two decades have largely produced equivocal findings (see (Hegadoren et al., 2009) for a review). Despite their clear role in mediating hedonic responses, research on the role of opioids in the pathophysiology of depression has focused primarily on the relationships between depression, stress and analgesic responses. Interestingly, psychological pain has been suggested to induce release of endogenous opioids. Consistent with this possibility, Kennedy et al. report increased μ-opioid activity in the ACC in response to a prolonged sadness induction paradigm in women with MDD (Kennedy et al., 2006). However, to date no studies have specifically evaluated opioid systems in reward liking or other aspects of reward processing in MDD.
In MDD, both OFC and ACC have shown a variety of alterations in gross morphology, neuronal structure, function, connectivity and neurochemistry (Botteron et al., 2002; Caetano et al., 2006; Chana et al., 2003; Coryell et al., 2005; Cotter et al., 2001; Drevets et al., 2002; Fitzgerald et al., 2008; Hastings et al., 2004; Koolschijn et al., 2009; Manji et al., 2001; Mayberg et al., 1997; Mayberg et al., 1999; Ongur et al., 1998; Treadway et al., 2009b; Yucel et al., 2009; Yucel et al., 2008). In studies of monetary reward processing, altered dorsal ACC and paracingulate activation has been observed in depressed patients when compared to controls (Knutson et al., 2008; Smoski et al., 2009). The most replicated finding in PFC processing of rewarding stimuli in MDD has been increased activity in rostral ACC and surrounding vmPFC in response to positively-valenced affective stimuli in MDD as compared to controls (Keedwell et al., 2005a, b; Mitterschiffthaler et al., 2003).
In contrast, amygdala responses to positive stimuli are often blunted in MDD. Beesdo et al., found reduced amygdala responses when passively viewing happy facial expressions in individuals with MDD as compared to control subjects or individuals with anxiety disorders (Beesdo et al., 2009). In the same study, both the MDD and anxiety groups showed increased activation of the amygdala relative to healthy controls when viewing fear faces. A second study using a longitudinal design reported that successful treatment with citalopram increased amygdala responses to happy faces (Norbury et al., 2009). Given the clear role of the amygdala in modulating striatal responses to rewarding stimuli, the possibility of hyporesponsivity in this region may underline some of the reported deficits in reward processing. However, as few imaging studies have directly tested this hypothesis, further research will be required before this hypothesis may be evaluated.
4. Wanting, Liking, Choosing: The Case for Decisional Anhedonia
Up to this point, we have considered anhedonia as a steady-state, mood-like phenomenon, in which individuals exhibit a general tendency to feel unmotivated or lack experienced pleasure. However, this conceptualization presents limitations when attempting to link anhedonic symptoms to underlying neurobiological mechanisms, as preclinical neurobiological studies typically examine behavior within a significantly narrower temporal window than is captured by self-report measures in humans. Indeed, preclinical studies of reward processing almost universally rely on aggregating discrete decisions, rather than more general temporally-extended measures. This emphasis on decision-making in the animal literature is not necessarily at odds with a steady-state conceptualization. For example, one could describe a motivational anhedonia as either a general feeling that it’s not worth getting out of bed, or a string of finite choices between staying in bed and some other activity in which the patient consistently chooses the former. From this perspective, the clinical phenomenon of anhedonia can be seen to emerge from a succession of individual decisions.
Using the preclinical literature’s focus on decision making as a guide, it may prove useful to explicitly characterize anhedonia in terms of abnormal reward-based decision making. Consequently, we propose that one manifestation of anhedonic symptoms may result in an impaired ability for normative decision-making. We term this “decisional anhedonia”, wherein the ability to balance costs and benefits when selecting among multiple options is impaired. We emphasize that decisional anhedonia is independent from cognitive or reasoning ability; making poor choices about complex financial instruments does not imply decisional anhedonia. Rather, we suggest that decisional anhedonia occurs when reward-based decision-making has 1) changed from a pre-morbid state and, 2) results in choices that substantially differ from normative decisions about potential cost-benefit choices. Importantly, we are not suggesting that decisional anhedonia is necessarily orthogonal to motivational or consummatory anhedonia. Rather, we suggest that the general, steady-state aspects of either motivational or consummatory anhedonia (or both), may lead to distinct decision-making impairments such that individuals overestimate future costs, underestimate future benefits, or simply fail to integrate cost/benefit information in a consistent manner, leading to erratic choice behavior. The critical benefit of focusing on a decisional anhedonia is that it provides clear behavior hypotheses and can be more readily linked to animal models.
One key component of decisional anhedonia may be an overestimation of costs associated with gaining different types of rewards. This is broadly consistent with a thoroughly documented negative response bias in MDD, a hypersensitivity to punishment, and a tendency to remember negative information (Gotlib et al., 2004a; Gotlib et al., 2004b; Joormann et al., 2006; Surguladze et al., 2004). Although we are aware of no direct investigations of cost/benefit decision-making in MDD, several studies provide initial support for the decisional anhedonia hypothesis. For instance, a recent study found that depressed individuals have difficulty integrating probabilistic reward cues (Forbes et al., 2006). Similarly, another study found that depressed individuals made less coherent choices on an intertemporal choice task, possibly suggesting a deficit in the ability to represent the value of future rewards in a consistent manner (Takahashi et al., 2008). A second intertemporal choice experiment reported that individuals with MDD showed a greater tendency to pick the delayed option, possibly reflecting a diminished sensitivity to reward magnitude of the immediate option (Lempert and Pizzagalli, 2010). Two other reports found altered patterns of reward decision-making using the Iowa Gambling Task in depressed individuals (Must et al., 2006; Smoski et al., 2008). Finally, some additional support for the concept of decisional anhedonia comes from schizophrenia, in which negative symptoms can include motivational anhedonia. The severity of negative symptoms in schizophrenia is inversely correlated with delay discounting (Heerey et al., 2007), a finding that is consistent with the MDD findings discussed above. Taken together, these data provide preliminary data to suggest that anhedonia in mood disorders may be associated with an impaired ability to accurately represent future costs and benefits during decision-making.
4.2 Neural Mechanisms of Decisional Anhedonia
The key advantage of adopting a focus on decision-making in understanding the neural substrates of anhedonia is that this approach may help us move beyond the identification of a biomarker to the development of mechanistic hypotheses. Based on preclinical models, deficits in cost/benefit decision making may be a consequence of functional impairments in a network comprised of the NAcc, ACC, amygdala and mesolimbic DA, each of which has been shown to be necessary for cost/benefit decision making, as depicted in figure 4.
Figure 4.
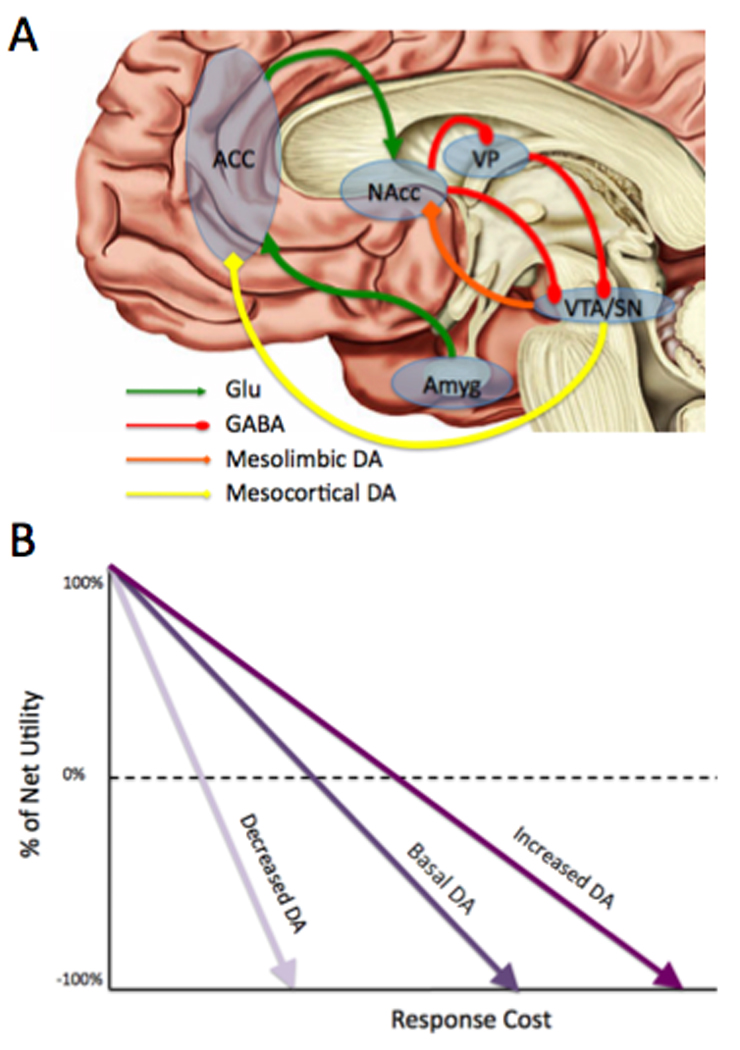
Neural network model of normative effort-based decision-making. (A) ACC and amygdala estimate of costs and benefits of potential options. This information is relayed to NAcc, where it may be modulated by NAcc DA. Upon excitation, NAcc sends GABAergic signals that relay through VP to VTA, resulting in burst firing. (B) Theoretical model of how mesolimbic DA may help organisms overcome response costs adapted from (Phillips et al., 2007). The solid black line represents the cost/benefit calculation determined by ACC under normal DA levels. The dotted gray line shows how increases in striatal DA results in an increase in perceived net-value, despite increasing response costs. Conversely, the solid gray line shows how this value is significantly diminished under conditions of reduced DA striatal DA, particularly as response costs increase. ACC = anterior cingulate cortex; Amyg = amygdala; DA = dopamine projections; GABA = GABAergic projections; Glu = glutamatergic projections; NAcc = nucleus accumbens; VP = ventral pallidum; VTA = ventral tegmental area.
As with the effects of NAcc DA depletion, ablation of rat ACC—but not neighboring infralimbic cortex—results in a behavioral shift towards LC/LR options during effort-based decision-making (Schweimer and Hauber, 2005; Schweimer et al., 2005; Walton et al., 2003; Walton et al., 2002; Walton et al., 2005; Walton et al., 2004; Walton et al., 2009; Walton et al., 2006; Walton et al., 2007). D1 receptor blockade within the ACC also produces similar effects on choice behavior. In seeking to integrate these findings with existing data on mesolimbic DA function, it has been suggested that the ACC is primarily involved in evaluating the costs and benefits of a given set of options. This role is consistent with a large number of studies that indicate that activity in the ACC is associated with processing future and current costs, including probability, pain and effort (Croxson et al., 2009; Knutson et al., 2005; Rushworth and Behrens, 2008; Talmi et al., 2009).
After its initial cost/benefit assessment, the ACC then relays this information via glutamatergic afferents to NAcc medium spiny neurons. Disconnection of ACC-NAcc afferent fibers also results in a shift away from HC/HR options (Hauber and Sommer, 2009). Additionally, disconnection between the amygdala and ACC produce a similar shift, consistent with the notion that the amygdala plays an important role in the initial appraisal of reward, which may modulate ACC input to the NAcc (McGinty and Grace, 2009a, b).
In the NAcc, MSN receiving prefrontal cost-benefit information may be modulated by mesolimbic DA, which is able to help “motivate” the animal toward the HC/HR option by orienting the NAcc to the reward side of the equation. In so doing, DA reduces the cost side of the cost/benefit ratio, thereby providing a means through which DA can help the organism overcome response costs. Evidence for this comes from the fact that when the difference in work requirements are between HC/HR and LC/LR options are low (e.g., one lever-press), lesions of NAcc DA terminals show little effect, but become more severe as response costs increase (Mingote et al., 2005; Phillips et al., 2007). More recently, it has been shown that NAcc measured in vivo using fast-scan cyclic voltammetry exhibits a monotonic relationship with changes in reward magnitude but not for changes in responses costs (Gan et al., 2009). Similarly in humans, it has been reported that increasing DA availability via L-Dopa administration can increase reports of expected gain from future rewards, consistent with the idea that DA helps overcome possible costs by emphasizing the value of potential benefits (Sharot et al., 2009).
Given the importance of the NAcc, ACC, amygdala and mesolimbic DA function in promoting normative cost/benefit decision-making, it is noteworthy that this network is heavily impacted in MDD, as described above. To date, however, only two neuroimaging studies have directly assessed reward-based decision-making in MDD, both of which used the wheel-of-fortune task (Ernst et al., 2004), in which separate events are modeled for reward decision, reward anticipation and reward feedback. In the first study, it was found that depressed individuals showed reduced ACC activation compared to controls during reward decision-making (Smoski et al., 2009), suggesting a possible failure to appropriately engage this region. The second study was a follow-up to the first, in which MDD subjects were scanned again after having received a brief behavioral treatment. Remitted individuals showed increased activation in ACC during reward-decision-making relative to their baseline scan (Dichter et al., 2009). Taken together, these data provide initial support for alterations in ACC during reward decision-making in MDD, which is consistent with the role of the ACC in calculating costs and benefits.
This decrease in ACC BOLD signal in MDD patients during cost-benefit decision-making appears inconsistent with previously mentioned studies that reported increased ACC activation in MDD patients. Indeed, challenges remain in translating animal findings using electrophysiology and lesion methods with human fMRI studies, let alone patient populations. One reason may be that different subregions of ACC may show exhibit differential patterns of altered function in MDD. For example, increases in ACC in MDD have been primarily localized to subgenual and rostral regions during resting-state, passive affective stimulus viewing and cognitive control tasks (Drevets et al., 2002; Keedwell et al., 2005a, b; Wagner et al., 2008), while more dorsal regions ACC appear critical for cost-benefit decision-making (Croxson et al., 2009; Rushworth, 2004). Nevertheless, two general findings emerge that provide initial support the hypothesis that ACC dysfunction may underlie decisional anhedonia. First, MDD patients consistently exhibit an altered pattern of BOLD responses multiple portions of the ACC across a variety of tasks. Second, animal studies demonstrate that this region (and its connections to the striatum) is critical for proper cost/benefit decision-making. Consequently, failure to appropriately engage the reward-processing relevant sub-regions of the ACC may contribute to alterations in reward decision-making in MDD. Future studies will be required to further elucidate the possible role of the ACC in decisional anhedonia.
5. Anhedonia Reconsidered
The fundamental goal of this paper has been to emphasize the crucial importance of developing symptom definitions that are aligned with both their clinical presentation and biological basis. In the case of anhedonia in depression, we have noted that while behavioral studies of anhedonia in MDD have largely focused on positively-valenced affective stimuli, biological studies have focused on neural substrates that are more closely involved in motivation and decision-making. This mismatch between symptom and substrate is a critical obstacle for uncovering neurobiological mechanisms of anhedonia. Consequently, we recommend the use of two sub-categories: motivational anhedonia and consummatory anhedonia. In this final section, we describe some of the implications of this proposal for future research and treatment.
5.1 Implications for Future Research
We suggest that future studies—particularly those using biological measures—would benefit by measuring not only the presence and absence of anhedonia, but also subdividing this criterion into motivational or hedonic elements. As a first step we would propose modifying psychiatric interviews such as the SCID to frame and code questions to clearly dissociate between these two facets of anhedonia. The adoption of specific terms “motivational anhedonia” and “consummatory anhedonia” are recommended as a means of refining the description of anhedonic symptoms. This has already begun to occur in the schizophrenia literature (Gard et al., 2007; Gold JM, 2008) and should be extended to other disorders that feature alterations in reward processing.
Further exploration of a possible “decisional anhedonia” may prove difficult to assess via interview or self-report, and it is likely that more objective and quantitative measures will be necessary. Recently, several experiment have been devised to explore separable aspects of motivation and decision-making (Heerey et al., 2007; Treadway et al., 2009a). In our own lab, we developed the Effort-Expenditure for Rewards Task (EEfRT or “effort”), which was adapted from animal paradigms of effort-based decision-making. Performance on this measure correlates with trait anhedonia, suggesting its potential utility as an assessment tool.
In addition to studies of motivational and decisional anhedonia, it is also recommended that researchers continue to explore the neurobiology of consummatory anhedonia. Growing evidence suggests that endogenous opioids playing a key role in pleasure responses. Remarkably, we were unable to identify any studies exploring the possibility of opioid-mediated consummatory anhedonia in clinical populations. In addition to opioids, animal research has highlighted a specific role for the ventral pallidum in the experience of subjective pleasure (Pecina et al., 2006). As such, it is tempting to speculate that dysfunction of the ventral pallidum could be a key mechanism in the clinical expression of consummatory anhedonia. Unfortunately, there is a dearth of clinical studies addressing this possibility. However, two case studies of patients with bilateral ventral pallidal damage appear consistent with the animal data, as both cases showed decreased self-reported responses to food or drug rewards (Miller et al., 2006; Vijayaraghavan et al., 2008). Given the strong projections of the NAcc to the ventral pallidum, it seems plausible that severe disruptions of the NAcc (or NAcc DA) could cause a secondary impact on the ventral pallidum, leading to the simultaneous presentation of both motivational and consummatory anhedonia. In theory it is thus possible that selective disruption of either the NAcc or ventral pallidum could result in just one component of anhedonia, whereas the full symptom picture including both consummatory and motivational components would only emerge with dysfunction of both regions.
5.2 Implications for Treatment
The overall goal of improving our understanding of neurobiological mechanisms is to improve treatment. If the assessment of “motivational anhedonia” is improved, this could potentially serve as a key predictor of treatment response to specific types of behavioral or biological therapies shown to alter motivational systems. We do not believe that these treatments will necessarily work for all cases of depression, but suggest that they may be particularly effective for treatment-resistant depressions involving significant motivational anhedonia. This form of tailored treatment is the primary means of utilizing our enhanced knowledge of neurobiology to improve clinical outcomes, but it is dependent on detailed phenotypic description to be successful.
Behavioral activation (BA) provides a potential example of a specific psychotherapeutic technique that might be particularly appropriate in cases with motivational anhedonia. Initially developed as a component of Cognitive Behavioral Therapy (CBT), Behavioral Activation (BA) differs primarily in its conceptualization of patient cognitions as a ruminative behavior (Dimidjian et al., 2006). The goal of treatment is to help the patient identify when they are engaging in rewarding and non-rewarding behaviors, and to help the patient make behavioral choices that are likely to increase exposure to positively reinforcing experiences.
Initial evidence suggests that by emphasizing an increase in motivated behaviors, BA may surpass CBT, particularly with clients diagnosed with co-morbid personality disorders (Coffman et al., 2007). Moreover, BA also includes specific techniques that address symptoms of decisional anhedonia. In one such technique, the therapist encourages the patient not to wait until the patient “feels like” engaging in a reward activity, thereby circumventing MDD-related impairments in reward decision-making due to lack of motivation (Martell et al., 2001).
Recent evidence from neuroimaging studies also suggests that BA may specifically target the reward system. Whereas fMRI studies have shown that treatment response to CBT results in a progressive decrease in amygdala sensitivity to negative stimuli (Siegle et al., 2006), successful treatment with BA led to increased BOLD responses of the striatum during reward anticipation (Dichter et al., 2009). Additionally, specific techniques used in BA treatments also address components of decisional anhedonia.
In terms of pharmacological treatments, the exploration of tailored treatments for individuals experiencing motivational anhedonia using DA-active pharmacotherapies is recommended. This includes psychostimulants, DA agonists, and the NE/DA reuptake inhibitor bupropion. Of the current FDA approved antidepressant drugs with DA-acting properties, bupropion is the most widely used in clinical practice. However, the pharmacological profile of bupropion is complex, and its effects on reward processing in animals and humans may rely on a variety of mechanisms, some of which are still not entirely known.
It is well established that bupropion has little direct effect on 5HT function (Stahl et al., 2004). Several studies exploring bupropion occupancy of DAT at clinical doses have reported occupancy rates ranging from 14%–26% in the striatum (Kugaya et al., 2003; Learned-Coughlin et al., 2003; Meyer et al., 2002), which are relatively low as compared to standard SERT occupancy rates of SSRIs (80%) or DAT occupancy of reinforcing psychostimulants (>50%) (Volkow et al., 1995; Volkow et al., 1997; Volkow et al., 1998). These findings suggest that bupropion’s direct ability to increase synaptic DA levels through blockade of DAT may account for only some of its antidepressant effects. However, more recent work has also shown that bupropion increases the activity of the intracellular vesicular monoamine transporter 2 (VMAT2) protein, which may enhance extracellular DA by increasing available DA in presynaptic pools (Rau et al., 2005). Bupropion may also exert regionally-specific influence over DA function through its action as an inhibitor of the norepinephrine transporter (NET), which is the primary transporter of DA in prefrontal regions. Finally, more recent work has suggested that bupropion decreases the activity of nicotinic acetylcholine receptors, which play a role in the effects of bupropion on psychomotor symptoms in MDD (See Dwoskin et al., 2006 for a review).
Preclinical studies have suggested that bupropion may be a superior treatment for symptoms of motivational anhedonia. Rats treated with bupropion demonstrate decreased immobility time during the forced swim test and tail suspension tests (Cryan et al., 2001; Cryan et al., 2004) and showed greater willingness to work for food rewards during a progressive ratio task (Bruijnzeel and Markou, 2003). Moreover, the influence of bupropion was blocked via administration of both D1-like and D2-like receptor antagonists, suggesting that effects of bupropion were partially mediated through DAergic mechanisms (Paterson and Markou, 2007). Additionally, rats treated with either chronic or acute doses of bupropion show a reduced threshold for intracranial self-stimulation of the posterior lateral hypothalamus (Paterson, 2009; Paterson et al., 2007). Similarly, bupropion enhanced responding to a conditioned reinforcer (Palmatier et al., 2009), although a separate study reported a bupropion-induced decrease in responding for sucrose (Reichel et al., 2008). The latter result is contrary to what would be expected, given the findings of (Bruijnzeel and Markou, 2003) and highlights the complex effects of the bupropion on reward processing. Interestingly, bupropion-mediated enhancement of conditioned reinforcers in the study by Palmatier et al. was ameliorated by Prazosin, an α2-NE receptor antagonist, suggesting that bupropion’s effects on reinforcement may also rely on noradrenergic mechanisms.
In addition to bupropion, psychostimulants, including dexamphetamine, methylphenidate and modafinil, have also been explored as both monotherapy and adjunctive treatment options for MDD. Results from these studies have not been encouraging (particularly in the case of monotherapy), although the majority of studies using psychostimulants were conducted several decades ago, before either DSM criteria or the Feighner criteria were in place (for reviews, see (Orr and Taylor, 2007) and (Candy et al., 2008)), and fail to meet current methodological standards for clinical trials. More recently, however, interest has reemerged in the utility of psychostimulants as an adjunctive therapy for specialized populations. In patients with advanced terminal illness, where tolerance and abuse potential are less of a concern, psychostimulants have shown a positive response, though few of these studies were placebo-controlled (Orr and Taylor, 2007). Similarly, in elderly populations, which often show less responsiveness to traditional antidepressants (Paykel et al., 1995; Reynolds et al., 1999) and exhibit higher rates of suicidality (Lebowitz et al., 1997), citalopram augmentation with methylphenidate produced a positive and rapid treatment response (Lavretsky and Kumar, 2001). Finally, DA agonists such as bromocriptine, ropinirole and pramipexole also exhibit antidepressant properties (Cassano et al., 2005; Corrigan et al., 2000; Sitland-Marken et al., 1990)). In addition to treating depressed patients, pramipexole has also been shown to be successful in treating anhedonic and depressive symptoms in patients with Parkinson’s disease, an illness associated with both loss of DA function and elevated rates of depressive illness (45%) (Lemke, 2008; Lemke et al., 2005).
Overall, head-to-head clinical trials between DA-acting agents and other pharmacotherapies have revealed strikingly similar response rates in the case of bupropion and pramipexole, (Chouinard, 1983; Coleman et al., 1999; Corrigan et al., 2000; Croft et al., 1999; Kavoussi et al., 1997; Mendels et al., 1983; Thase et al., 2005; Weihs et al., 2000; Weisler et al., 1994). For psychostimulants, response rates are usually significantly worse than other alternatives (Candy et al., 2008; Taylor & Orr, 2007). However, the potential role of DA-acting drugs as a superior treatment for anhedonic symptoms has received some empirical support. Bupropion has shown to be more effective at treating symptoms related to motivational and consummatory anhedonia (Bodkin et al., 1997; Tomarken et al., 2004). In a large-sample review of treatment records of 910 patients receiving outpatient pharmacotherapy for depression, Jamerson and colleagues (2003) reported that patients receiving bupropion sustained release (SR) showed significant improvement of symptoms related to reduced interest, energy and loss of libido as compared to placebo (Jamerson et al., 2003). Additionally, bupropion is often used to counter-act specific side effects of SSRIs (Nutt et al., 2007), which may include reduced responsiveness to rewards and positive experience (McCabe et al., 2009; Price et al., 2009; Shelton and Tomarken, 2001). A recent meta-analysis of DA-acting antidepressant treatments suggests that they enhance overall quality-of-life in individuals with MDD (IsHak et al., 2009). These findings are not only promising in terms of treatment options, they also further underscore the importance of tailoring DA-acting treatments to specific symptoms.
Although we have emphasized the role of DA function in the pathophysiology and treatment of motivational anhedonia, DA represents only one component of a highly complex and integrated circuit. Indeed, more recent developments in novel compounds for treatment of depression have focused less on monoamine systems directly, and more on systems that may regulate and/or interact with monoamines, such as glutamate. Recent evidence suggests that depression may be associated with reduced glutamate function in prefrontal regions including the ACC (Hasler et al., 2007), especially in individuals with anhedonic symptoms (Walter et al., 2009). Glutamatergic projections from the ACC to the NAcc appear to be critical for effort-based decision-making behavior in animals (Hauber and Sommer, 2009), and therefore may play a role in motivational and/or decisional anhedonia. Additionally, studies exploring the effects of glutamatergic compounds in the treatment of depression show promising initial results (McEwen et al. 2010; Rakofsky et al., 2009). Future research will be required to determine whether monoaminergic alterations in MDD are downstream consequences of glutamatergic dysfunction.
5.3 Anhedonia as a Case Study for Biologically-based Clinical Definitions
When Roy Wise and others first observed that neuroleptics reduced reward-dependent lever-pressing behavior in rats, it was natural for him to interpret this finding in terms of pleasure, stating at the time that neuroleptics “appear to…take the ‘goodness’ out of normally rewarding food.” (Wise et al., 1978). In everyday experience, we often assume that we want what we like and like what we want. The widespread acceptance of Wise’s conclusion most likely reflected the fact that at the time, there was insufficient reason to assume that wanting and liking would be neurally dissociable. Indeed, one could even have argued that such a proposition would have violated the law of parsimony. However, the fault lines of neurobiology do not necessarily match the intuitive understanding of our phenomenological experience.
The same can be said of clinical disorders and symptoms. While we have focused on a specific symptom in a specific disorder, we believe that anhedonia may serve as a useful case study regarding the inherent difficulties in linking clinical constructs to neural substrates. Many of the early approaches to biological and neuroimaging methods adopted a medical model of diagnosis, which emphasizes identification of specific biomarkers of a disorder. This approach has been very productive to date, but its potential for future advances may be limited by the vagueness of clinical definitions. In particular, the presence of multiple forms of heterogeneity at the level of co-morbidity, symptoms and etiology may hinder the detection of important neurobiological markers.
One strategy for meeting this challenge is the use of preclinical models as a means of identifying those cases of symptom, disorder or etiological heterogeneity that are likely to impact different neural substrates. While we believe that this approach is essential, a limitation should be acknowledged. Namely, it requires clinical researchers to “look where that light is”, and focus on those symptoms for which a sufficiently extensive animal literature exists. Indeed, the arguments presented in this review were made possible by the monumental efforts of basic science researchers over the last few decades. At the current time, availing oneself of the preclinical literature would produce far less benefit if one were attempting a similar strategy for other symptoms in MDD, such as guilt. Additionally, by focusing on specific symptoms, this approach may be limited in its ability to explain the larger constellations of symptoms that characterize many of our current categorical diagnoses. Arguments for this translational approach to examining specific symptom classes will be most powerful when there is a strong case to be made for the uniqueness of a class of symptoms (for instance due to differential genetics or treatment response), but such an approach may prove more difficult to support in the absence of such evidence.
6. Conclusion
In the present review we have argued that studies of anhedonia in MDD should be informed by animal models of reward processing. Specifically, we suggest that current clinical definitions of anhedonia are too broad. As the processes of reward wanting and liking are found to rely on separate neural systems, depression research must attend to these distinctions in order to develop specific neurobiological models and novel treatment targets. As a multifaceted construct, anhedonia requires a more thorough characterization than is currently provided in the DSM or common self-report assessments. We propose that clinical symptoms of anhedonia be divided into motivational and consummatory anhedonia in order to closely parallel the animal literature on reward. Additionally, we propose that an alternative way to conceptualize anhedonia is by focusing on decision-making, rather than general mood, as this strategy lends itself particularly well to translational research approaches.
When Ribot first defined anhedonia, the field of neuroscience was still in its infancy. In the ensuing 113 years, understanding of the neurobiological systems involved in the processing of rewards has grown exponentially, while the construct of anhedonia remains relatively unaltered. We believe that in order to fulfill the promise of sophisticated neurobiological research methods, we must adopt equally sophisticated behavioral measures and clinical definitions.
Research Highlights
Anhedonia includes motivational and consummatory aspects of reward processing.
Current research often fails to discriminate between these two aspects.
Preclinical data suggest that these two aspects are neurobiologically dissociable.
Consequently, we propose motivational and consummatory anhedonia.
We also introduce “decisional anhedonia” to address anhedonic effects on decisions.
Acknowledgements
This manuscript was produced with the support of the National Institute of Mental Health (F31MH087015) to MTT and the National Institute on Drug Abuse (R01DA019670) to DHZ. The authors report no conflict of interest. The authors wish to thank Joshua Buckholtz for thoughtful commentary, and Gabriel Dichter, Andrew Tomarken, Steven Hollon and Richard Shelton for points of inspiration.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aberman JE, Salamone JD. Nucleus accumbens dopamine depletions make rats more sensitive to high ratio requirements but do not impair primary food reinforcement. Neuroscience. 1999;92:545–552. doi: 10.1016/s0306-4522(99)00004-4. [DOI] [PubMed] [Google Scholar]
- Agren H, Reibring L. PET studies of presynaptic monoamine metabolism in depressed patients and healthy volunteers. Pharmacopsychiatry. 1994;27:2–6. doi: 10.1055/s-2007-1014265. [DOI] [PubMed] [Google Scholar]
- Allen NB, Trinder J, Brennan C. Affective startle modulation in clinical depression: preliminary findings. Biol. Psychiatry. 1999;46:542–550. doi: 10.1016/s0006-3223(99)00025-6. [DOI] [PubMed] [Google Scholar]
- Amsterdam JD, Newberg AB. A preliminary study of dopamine transporter binding in bipolar and unipolar depressed patients and healthy controls. Neuropsychobiology. 2007;55:167–170. doi: 10.1159/000106476. [DOI] [PubMed] [Google Scholar]
- Amsterdam JD, Settle RG, Doty RL, Abelman E, Winokur A. Taste and smell perception in depression. Biol. Psychiatry. 1987;22:1481–1485. doi: 10.1016/0006-3223(87)90108-9. [DOI] [PubMed] [Google Scholar]
- Andreasen NC. Negative symptoms in schizophrenia. Definition and reliability. Arch. Gen. Psychiatry. 1982;39:784–788. doi: 10.1001/archpsyc.1982.04290070020005. [DOI] [PubMed] [Google Scholar]
- Anisman H, Merali Z, Hayley S. Neurotransmitter, peptide and cytokine processes in relation to depressive disorder: comorbidity between depression and neurodegenerative disorders. Prog. Neurobiol. 2008;85:1–74. doi: 10.1016/j.pneurobio.2008.01.004. [DOI] [PubMed] [Google Scholar]
- APA. Practice guideline for the treatment of patients with major depressive disorder (revision) Am. J. Psychiatry. 2000;157:1–45. [PubMed] [Google Scholar]
- Arnsten AF. Toward a new understanding of attention-deficit hyperactivity disorder pathophysiology: an important role for prefrontal cortex dysfunction. CNS Drugs. 2009;23 Suppl 1:33–41. doi: 10.2165/00023210-200923000-00005. [DOI] [PubMed] [Google Scholar]
- Assadi SM, Yucel M, Pantelis C. Dopamine modulates neural networks involved in effort-based decision-making. Neurosci. Biobehav. Rev. 2009;33:383–393. doi: 10.1016/j.neubiorev.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Balleine BW, Killcross S. Parallel incentive processing: an integrated view of amygdala function. Trends Neurosci. 2006;29:272–279. doi: 10.1016/j.tins.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Bar-Haim Y, Fox NA, Benson B, Guyer AE, Williams A, Nelson EE, Perez-Edgar K, Pine DS, Ernst M. Neural correlates of reward processing in adolescents with a history of inhibited temperament. Psychol. Sci. 2009;20:1009–1018. doi: 10.1111/j.1467-9280.2009.02401.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardgett ME, Depenbrock M, Downs N, Points M, Green L. Dopamine modulates effort-based decision making in rats. Behav. Neurosci. 2009;123:242–251. doi: 10.1037/a0014625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr AM, Markou A. Psychostimulant withdrawal as an inducing condition in animal models of depression. Neurosci. Biobehav. Rev. 2005;29:675–706. doi: 10.1016/j.neubiorev.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Barr AM, Markou A, Phillips AG. A 'crash' course on psychostimulant withdrawal as a model of depression. Trends Pharmacol. Sci. 2002;23:475–482. doi: 10.1016/s0165-6147(02)02086-2. [DOI] [PubMed] [Google Scholar]
- Barr AM, Phillips AG. Withdrawal following repeated exposure to d-amphetamine decreases responding for a sucrose solution as measured by a progressive ratio schedule of reinforcement. Psychopharmacology (Berl.) 1999;141:99–106. doi: 10.1007/s002130050812. [DOI] [PubMed] [Google Scholar]
- Baxter MG, Murray EA. The amygdala and reward. Nat. Rev. Neurosci. 2002;3:563–573. doi: 10.1038/nrn875. [DOI] [PubMed] [Google Scholar]
- Beck AT, Steer RA, Ball R, Ranieri W. Comparison of Beck Depression Inventories -IA and -II in psychiatric outpatients. J. Pers. Assess. 1996;67:588–597. doi: 10.1207/s15327752jpa6703_13. [DOI] [PubMed] [Google Scholar]
- Beesdo K, Lau JY, Guyer AE, McClure-Tone EB, Monk CS, Nelson EE, Fromm SJ, Goldwin MA, Wittchen HU, Leibenluft E, Ernst M, Pine DS. Common and distinct amygdala-function perturbations in depressed vs anxious adolescents. Arch. Gen. Psychiatry. 2009;66:275–285. doi: 10.1001/archgenpsychiatry.2008.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenbaum H. Posed facial expressions of emotion in schizophrenia and depression. Psychol. Med. 1992a;22:929–937. doi: 10.1017/s0033291700038502. [DOI] [PubMed] [Google Scholar]
- Berenbaum H, Oltmanns TF. Emotional experience and expression in schizophrenia and depression. J. Abnorm. Psychol. 1992b;101:37–44. doi: 10.1037//0021-843x.101.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger PA, Faull KF, Kilkowski J, Anderson PJ, Kraemer H, Davis KL, Barchas JD. CSF monoamine metabolites in depression and schizophrenia. Am. J. Psychiatry. 1980;137:174–180. doi: 10.1176/ajp.137.2.174. [DOI] [PubMed] [Google Scholar]
- Berlin I, Givry-Steiner L, Lecrubier Y, Puech AJ. Measures of anhedonia and hedonic responses to sucrose in depressive and scizophrenic patients in comparison with healthy subjects. European Psychiatry. 1998;13:303–309. doi: 10.1016/S0924-9338(98)80048-5. [DOI] [PubMed] [Google Scholar]
- Berridge KC. The debate over dopamine's role in reward: the case for incentive salience. Psychopharmacology (Berl.) 2007;191:391–431. doi: 10.1007/s00213-006-0578-x. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res. Brain Res. Rev. 1998;28:309–369. doi: 10.1016/s0165-0173(98)00019-8. [DOI] [PubMed] [Google Scholar]
- Berridge KC, Robinson TE. Parsing reward. Trends Neurosci. 2003;26:507–513. doi: 10.1016/S0166-2236(03)00233-9. [DOI] [PubMed] [Google Scholar]
- Berton O, McClung CA, Dileone RJ, Krishnan V, Renthal W, Russo SJ, Graham D, Tsankova NM, Bolanos CA, Rios M, Monteggia LM, Self DW, Nestler EJ. Essential role of BDNF in the mesolimbic dopamine pathway in social defeat stress. Science. 2006;311:864–868. doi: 10.1126/science.1120972. [DOI] [PubMed] [Google Scholar]
- Bodkin JA, Lasser RA, Wines JD, Jr, Gardner DM, Baldessarini RJ. Combining serotonin reuptake inhibitors and bupropion in partial responders to antidepressant monotherapy. J. Clin. Psychiatry. 1997;58:137–145. doi: 10.4088/jcp.v58n0401. [DOI] [PubMed] [Google Scholar]
- Botteron KN, Raichle ME, Drevets WC, Heath AC, Todd RD. Volumetric reduction in left subgenual prefrontal cortex in early onset depression. Biol. Psychiatry. 2002;51:342–344. doi: 10.1016/s0006-3223(01)01280-x. [DOI] [PubMed] [Google Scholar]
- Bouras N, Bridges PK. Bromocriptine in depression. Curr. Med. Res. Opin. 1982;8:150–153. doi: 10.1185/03007998209112376. [DOI] [PubMed] [Google Scholar]
- Bragulat V, Paillere-Martinot ML, Artiges E, Frouin V, Poline JB, Martinot JL. Dopaminergic function in depressed patients with affective flattening or with impulsivity: [18F]fluoro-L-dopa positron emission tomography study with voxel-based analysis. Psychiatry Res. 2007;154:115–124. doi: 10.1016/j.pscychresns.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Bremner JD, Vythilingam M, Ng CK, Vermetten E, Nazeer A, Oren DA, Berman RM, Charney DS. Regional brain metabolic correlates of alphamethylparatyrosine-induced depressive symptoms: implications for the neural circuitry of depression. JAMA. 2003;289:3125–3134. doi: 10.1001/jama.289.23.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruijnzeel AW, Markou A. Characterization of the effects of bupropion on the reinforcing properties of nicotine and food in rats. Synapse. 2003;50:20–28. doi: 10.1002/syn.10242. [DOI] [PubMed] [Google Scholar]
- Buckby JA, Cotton SM, Cosgrave EM, Killackey EJ, Yung AR. A factor analytic investigation of the Tripartite model of affect in a clinical sample of young Australians. BMC Psychiatry. 2008;8:79. doi: 10.1186/1471-244X-8-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckholtz JW, Treadway MT, Cowan RL, Woodward ND, Benning SD, Li R, Ansari MS, Baldwin RM, Schwartzman AN, Shelby ES, Smith CE, Cole D, Kessler RM, Zald DH. Mesolimbic dopamine reward system hypersensitivity in individuals with psychopathic traits. Nat. Neurosci. 13:419–421. doi: 10.1038/nn.2510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns DD, Eidelson RJ. Why are depression and anxiety correlated? A test of the tripartite model. J. Consult. Clin. Psychol. 1998;66:461–473. doi: 10.1037//0022-006x.66.3.461. [DOI] [PubMed] [Google Scholar]
- Bylsma LM, Morris BH, Rottenberg J. A meta-analysis of emotional reactivity in major depressive disorder. Clin. Psychol. Rev. 2008;28:676–691. doi: 10.1016/j.cpr.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Caetano SC, Kaur S, Brambilla P, Nicoletti M, Hatch JP, Sassi RB, Mallinger AG, Keshavan MS, Kupfer DJ, Frank E, Soares JC. Smaller cingulate volumes in unipolar depressed patients. Biol. Psychiatry. 2006;59:702–706. doi: 10.1016/j.biopsych.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Candy M, Jones L, Williams R, Tookman A, King M. Psychostimulants for depression. Cochrane Database Syst. Rev. 2008 doi: 10.1002/14651858.CD006722.pub2. CD006722. [DOI] [PubMed] [Google Scholar]
- Cannon CM, Palmiter RD. Reward without dopamine. J. Neurosci. 2003;23:10827–10831. doi: 10.1523/JNEUROSCI.23-34-10827.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon DM, Klaver JM, Peck SA, Rallis-Voak D, Erickson K, Drevets WC. Dopamine type-1 receptor binding in major depressive disorder assessed using positron emission tomography and [11C]NNC-112. Neuropsychopharmacology. 2009;34:1277–1287. doi: 10.1038/npp.2008.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspi A, Sugden K, Moffitt TE, Taylor A, Craig IW, Harrington H, McClay J, Mill J, Martin J, Braithwaite A, Poulton R. Influence of life stress on depression: moderation by a polymorphism in the 5-HTT gene. Science. 2003;301:386–389. doi: 10.1126/science.1083968. [DOI] [PubMed] [Google Scholar]
- Cassano P, Lattanzi L, Fava M, Navari S, Battistini G, Abelli M, Cassano GB. Ropinirole in treatment-resistant depression: a 16-week pilot study. Can. J. Psychiatry. 2005;50:357–360. doi: 10.1177/070674370505000612. [DOI] [PubMed] [Google Scholar]
- Catlin DH, Gorelick DA, Gerner RH. Clinical pharmacology of beta-endorphin in depression and schizophrenia. Ann. N. Y. Acad. Sci. 1982;398:434–447. doi: 10.1111/j.1749-6632.1982.tb39515.x. [DOI] [PubMed] [Google Scholar]
- Chana G, Landau S, Beasley C, Everall IP, Cotter D. Two-dimensional assessment of cytoarchitecture in the anterior cingulate cortex in major depressive disorder, bipolar disorder, and schizophrenia: evidence for decreased neuronal somal size and increased neuronal density. Biol. Psychiatry. 2003;53:1086–1098. doi: 10.1016/s0006-3223(03)00114-8. [DOI] [PubMed] [Google Scholar]
- Chapman LJ, Chapman JP, Raulin ML. Scales for physical and social anhedonia. J. Abnorm. Psychol. 1976;85:374–382. doi: 10.1037//0021-843x.85.4.374. [DOI] [PubMed] [Google Scholar]
- Chiaroni P, Azorin JM, Dassa D, Henry JM, Giudicelli S, Malthiery Y, Planells R. Possible involvement of the dopamine D3 receptor locus in subtypes of bipolar affective disorder. Psychiatr. Genet. 2000;10:43–49. doi: 10.1097/00041444-200010010-00008. [DOI] [PubMed] [Google Scholar]
- Chouinard G. Bupropion and amitriptyline in the treatment of depressed patients. J. Clin. Psychiatry. 1983;44:121–129. [PubMed] [Google Scholar]
- Cloninger CR. A systematic method for clinical description and classification of personality variants. A proposal. Arch. Gen. Psychiatry. 1987;44:573–588. doi: 10.1001/archpsyc.1987.01800180093014. [DOI] [PubMed] [Google Scholar]
- Coffman SJ, Martell CR, Dimidjian S, Gallop R, Hollon SD. Extreme nonresponse in cognitive therapy: can behavioral activation succeed where cognitive therapy fails? J. Consult. Clin. Psychol. 2007;75:531–541. doi: 10.1037/0022-006X.75.4.531. [DOI] [PubMed] [Google Scholar]
- Coleman CC, Cunningham LA, Foster VJ, Batey SR, Donahue RM, Houser TL, Ascher JA. Sexual dysfunction associated with the treatment of depression: a placebo-controlled comparison of bupropion sustained release and sertraline treatment. Ann. Clin. Psychiatry. 1999;11:205–215. doi: 10.1023/a:1022309428886. [DOI] [PubMed] [Google Scholar]
- Conner JM, Lauterborn JC, Yan Q, Gall CM, Varon S. Distribution of brain-derived neurotrophic factor (BDNF) protein and mRNA in the normal adult rat CNS: evidence for anterograde axonal transport. J. Neurosci. 1997;17:2295–2313. doi: 10.1523/JNEUROSCI.17-07-02295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JC, Bloom FE, Roth RH. The Biochemical Basis of Neuropharmacology. Eigth ed. New York: Oxford University Press; 2003. [Google Scholar]
- Cordeira JW, Frank L, Sena-Esteves M, Pothos EN, Rios M. Brain-derived neurotrophic factor regulates hedonic feeding by acting on the mesolimbic dopamine system. J. Neurosci. 30:2533–2541. doi: 10.1523/JNEUROSCI.5768-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correa M, Carlson BB, Wisniecki A, Salamone JD. Nucleus accumbens dopamine and work requirements on interval schedules. Behav. Brain Res. 2002;137:179–187. doi: 10.1016/s0166-4328(02)00292-9. [DOI] [PubMed] [Google Scholar]
- Corrigan MH, Denahan AQ, Wright CE, Ragual RJ, Evans DL. Comparison of pramipexole, fluoxetine, and placebo in patients with major depression. Depress. Anxiety. 2000;11:58–65. doi: 10.1002/(sici)1520-6394(2000)11:2<58::aid-da2>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Coryell W, Nopoulos P, Drevets W, Wilson T, Andreasen NC. Subgenual prefrontal cortex volumes in major depressive disorder and schizophrenia: diagnostic specificity and prognostic implications. Am. J. Psychiatry. 2005;162:1706–1712. doi: 10.1176/appi.ajp.162.9.1706. [DOI] [PubMed] [Google Scholar]
- Cotter D, Mackay D, Landau S, Kerwin R, Everall I. Reduced glial cell density and neuronal size in the anterior cingulate cortex in major depressive disorder. Arch. Gen. Psychiatry. 2001;58:545–553. doi: 10.1001/archpsyc.58.6.545. [DOI] [PubMed] [Google Scholar]
- Cousins MS, Atherton A, Turner L, Salamone JD. Nucleus accumbens dopamine depletions alter relative response allocation in a T-maze cost/benefit task. Behav. Brain Res. 1996;74:189–197. doi: 10.1016/0166-4328(95)00151-4. [DOI] [PubMed] [Google Scholar]
- Cousins MS, Salamone JD. Nucleus accumbens dopamine depletions in rats affect relative response allocation in a novel cost/benefit procedure. Pharmacol. Biochem. Behav. 1994;49:85–91. doi: 10.1016/0091-3057(94)90460-x. [DOI] [PubMed] [Google Scholar]
- Cousins MS, Salamone JD. Skilled motor deficits in rats induced by ventrolateral striatal dopamine depletions: behavioral and pharmacological characterization. Brain Res. 1996;732:186–194. doi: 10.1016/0006-8993(96)00519-7. [DOI] [PubMed] [Google Scholar]
- Croft H, Settle E, Jr, Houser T, Batey SR, Donahue RM, Ascher JA. A placebo-controlled comparison of the antidepressant efficacy and effects on sexual functioning of sustained-release bupropion and sertraline. Clin. Ther. 1999;21:643–658. doi: 10.1016/S0149-2918(00)88317-4. [DOI] [PubMed] [Google Scholar]
- Croxson PL, Walton ME, O'Reilly JX, Behrens TE, Rushworth MF. Effort-based cost-benefit valuation and the human brain. J. Neurosci. 2009;29:4531–4541. doi: 10.1523/JNEUROSCI.4515-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cryan JF, Dalvi A, Jin SH, Hirsch BR, Lucki I, Thomas SA. Use of dopamine-beta-hydroxylase-deficient mice to determine the role of norepinephrine in the mechanism of action of antidepressant drugs. J. Pharmacol. Exp. Ther. 2001;298:651–657. [PubMed] [Google Scholar]
- Cryan JF, O'Leary OF, Jin SH, Friedland JC, Ouyang M, Hirsch BR, Page ME, Dalvi A, Thomas SA, Lucki I. Norepinephrine-deficient mice lack responses to antidepressant drugs, including selective serotonin reuptake inhibitors. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8186–8191. doi: 10.1073/pnas.0401080101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Aquila PS, Collu M, Gessa GL, Serra G. The role of dopamine in the mechanism of action of antidepressant drugs. Eur. J. Pharmacol. 2000;405:365–373. doi: 10.1016/s0014-2999(00)00566-5. [DOI] [PubMed] [Google Scholar]
- D'Haenen HA, Bossuyt A. Dopamine D2 receptors in depression measured with single photon emission computed tomography. Biol. Psychiatry. 1994;35:128–132. doi: 10.1016/0006-3223(94)91202-5. [DOI] [PubMed] [Google Scholar]
- Davis LL, Frazier EC, Williford RB, Newell JM. Long-term pharmacotherapy for post-traumatic stress disorder. CNS Drugs. 2006;20:465–476. doi: 10.2165/00023210-200620060-00003. [DOI] [PubMed] [Google Scholar]
- Denk F, Walton ME, Jennings KA, Sharp T, Rushworth MF, Bannerman DM. Differential involvement of serotonin and dopamine systems in cost-benefit decisions about delay or effort. Psychopharmacology (Berl.) 2005;179:587–596. doi: 10.1007/s00213-004-2059-4. [DOI] [PubMed] [Google Scholar]
- Depue RA, Collins PF. Neurobiology of the structure of personality: dopamine, facilitation of incentive motivation, and extraversion. Behav. Brain Sci. 1999;22:491–517. doi: 10.1017/s0140525x99002046. discussion 518002D469. [DOI] [PubMed] [Google Scholar]
- Depue RA, Iacono WG. Neurobehavioral aspects of affective disorders. Annu. Rev. Psychol. 1989;40:457–492. doi: 10.1146/annurev.ps.40.020189.002325. [DOI] [PubMed] [Google Scholar]
- Dichter GS, Felder JN, Petty C, Bizzell J, Ernst M, Smoski MJ. The effects of psychotherapy on neural responses to rewards in major depression. Biol. Psychiatry. 2009;66:886–897. doi: 10.1016/j.biopsych.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichter GS, Smoski MJ, Kampov-Polevoy AB, Gallop R, Garbutt JC. Unipolar depression does not moderate responses to the Sweet Taste Test. Depress. Anxiety. doi: 10.1002/da.20690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dichter GS, Tomarken AJ, Shelton RC, Sutton SK. Early- and late-onset startle modulation in unipolar depression. Psychophysiology. 2004;41:433–440. doi: 10.1111/j.1469-8986.00162.x. [DOI] [PubMed] [Google Scholar]
- Dikeos DG, Papadimitriou GN, Avramopoulos D, Karadima G, Daskalopoulou EG, Souery D, Mendlewicz J, Vassilopoulos D, Stefanis CN. Association between the dopamine D3 receptor gene locus (DRD3) and unipolar affective disorder. Psychiatr. Genet. 1999;9:189–195. doi: 10.1097/00041444-199912000-00005. [DOI] [PubMed] [Google Scholar]
- Dimidjian S, Hollon SD, Dobson KS, Schmaling KB, Kohlenberg RJ, Addis ME, Gallop R, McGlinchey JB, Markley DK, Gollan JK, Atkins DC, Dunner DL, Jacobson NS. Randomized trial of behavioral activation, cognitive therapy, and antidepressant medication in the acute treatment of adults with major depression. J. Consult. Clin. Psychol. 2006;74:658–670. doi: 10.1037/0022-006X.74.4.658. [DOI] [PubMed] [Google Scholar]
- Dowlati Y, Herrmann N, Swardfager W, Liu H, Sham L, Reim EK, Lanctot KL. A meta-analysis of cytokines in major depression. Biol. Psychiatry. 67:446–457. doi: 10.1016/j.biopsych.2009.09.033. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Bogers W, Raichle ME. Functional anatomical correlates of antidepressant drug treatment assessed using PET measures of regional glucose metabolism. Eur. Neuropsychopharmacol. 2002;12:527–544. doi: 10.1016/s0924-977x(02)00102-5. [DOI] [PubMed] [Google Scholar]
- Drevets WC, Gautier C, Price JC, Kupfer DJ, Kinahan PE, Grace AA, Price JL, Mathis CA. Amphetamine-induced dopamine release in human ventral striatum correlates with euphoria. Biol. Psychiatry. 2001;49:81–96. doi: 10.1016/s0006-3223(00)01038-6. [DOI] [PubMed] [Google Scholar]
- Duffy VB, Bartoshuk LM. Food acceptance and genetic variation in taste. J. Am. Diet. Assoc. 2000;100:647–655. doi: 10.1016/S0002-8223(00)00191-7. [DOI] [PubMed] [Google Scholar]
- Duman RS. Neuronal damage and protection in the pathophysiology and treatment of psychiatric illness: stress and depression. Dialogues Clin. Neurosci. 2009;11:239–255. doi: 10.31887/DCNS.2009.11.3/rsduman. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlop BW, Nemeroff CB. The role of dopamine in the pathophysiology of depression. Arch. Gen. Psychiatry. 2007;64:327–337. doi: 10.1001/archpsyc.64.3.327. [DOI] [PubMed] [Google Scholar]
- Dunn BD, Dalgleish T, Lawrence AD, Cusack R, Ogilvie AD. Categorical and dimensional reports of experienced affect to emotion-inducing pictures in depression. J. Abnorm. Psychol. 2004;113:654–660. doi: 10.1037/0021-843X.113.4.654. [DOI] [PubMed] [Google Scholar]
- Dwoskin LP, Rauhut AS, King-Pospisil KA, Bardo MT. Review of the pharmacology and clinical profile of bupropion, an antidepressant and tobacco use cessation agent. CNS Drug Rev. 2006;12:178–207. doi: 10.1111/j.1527-3458.2006.00178.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Mallakh RS. An open study of methylphenidate in bipolar depression. Bipolar Disord. 2000;2:56–59. doi: 10.1034/j.1399-5618.2000.020108.x. [DOI] [PubMed] [Google Scholar]
- Ernst M, Nelson EE, McClure EB, Monk CS, Munson S, Eshel N, Zarahn E, Leibenluft E, Zametkin A, Towbin K, Blair J, Charney D, Pine DS. Choice selection and reward anticipation: an fMRI study. Neuropsychologia. 2004;42:1585–1597. doi: 10.1016/j.neuropsychologia.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Cardinal RN, Parkinson JA, Robbins TW. Appetitive behavior: impact of amygdala-dependent mechanisms of emotional learning. Ann. N. Y. Acad. Sci. 2003;985:233–250. [PubMed] [Google Scholar]
- Everitt BJ, Robbins TW. Neural systems of reinforcement for drug addiction: from actions to habits to compulsion. Nat. Neurosci. 2005;8:1481–1489. doi: 10.1038/nn1579. [DOI] [PubMed] [Google Scholar]
- Fawcett J, Clark DC, Scheftner WA, Hedeker D. Differences between anhedonic and normally hedonic depressive states. Am. J. Psychiatry. 1983;140:1027–1030. doi: 10.1176/ajp.140.8.1027. [DOI] [PubMed] [Google Scholar]
- Feighner JP,RE, Guze SB, Woodruff RA, Jr, Winokur G, Munoz R. Diagnostic criteria for use in psychiatric research. Arch. Gen. Psychiatry. 1972;26:57–63. doi: 10.1001/archpsyc.1972.01750190059011. [DOI] [PubMed] [Google Scholar]
- Fitzgerald PB, Laird AR, Maller J, Daskalakis ZJ. A meta-analytic study of changes in brain activation in depression. Hum. Brain Mapp. 2008;29:683–695. doi: 10.1002/hbm.20426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floresco SB, St Onge JR, Ghods-Sharifi S, Winstanley CA. Cortico-limbic-striatal circuits subserving different forms of cost-benefit decision making. Cogn. Affect. Behav. Neurosci. 2008;8:375–389. doi: 10.3758/CABN.8.4.375. [DOI] [PubMed] [Google Scholar]
- Floresco SB, West AR, Ash B, Moore H, Grace AA. Afferent modulation of dopamine neuron firing differentially regulates tonic and phasic dopamine transmission. Nat. Neurosci. 2003;6:968–973. doi: 10.1038/nn1103. [DOI] [PubMed] [Google Scholar]
- Forbes EE. Where's the fun in that? Broadening the focus on reward function in depression. Biol. Psychiatry. 2009;66:199–200. doi: 10.1016/j.biopsych.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes EE, Dahl RE. Neural systems of positive affect: relevance to understanding child and adolescent depression? Dev. Psychopathol. 2005;17:827–850. doi: 10.1017/S095457940505039X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forbes EE, Hariri AR, Martin SL, Silk JS, Moyles DL, Fisher PM, Brown SM, Ryan ND, Birmaher B, Axelson DA, Dahl RE. Altered striatal activation predicting real-world positive affect in adolescent major depressive disorder. Am. J. Psychiatry. 2009;166:64–73. doi: 10.1176/appi.ajp.2008.07081336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman A, Deri I, Friedman Y, Dremencov E, Goutkin S, Kravchinsky E, Mintz M, Levi D, Overstreet DH, Yadid G. Decoding of dopaminergic mesolimbic activity and depressive behavior. J. Mol. Neurosci. 2007;32:72–79. doi: 10.1007/s12031-007-0016-5. [DOI] [PubMed] [Google Scholar]
- Fu CH, Williams SC, Cleare AJ, Brammer MJ, Walsh ND, Kim J, Andrews CM, Pich EM, Williams PM, Reed LJ, Mitterschiffthaler MT, Suckling J, Bullmore ET. Attenuation of the neural response to sad faces in major depression by antidepressant treatment: a prospective, event-related functional magnetic resonance imaging study. Arch. Gen. Psychiatry. 2004;61:877–889. doi: 10.1001/archpsyc.61.9.877. [DOI] [PubMed] [Google Scholar]
- Futami T, Takakusaki K, Kitai ST. Glutamatergic and cholinergic inputs from the pedunculopontine tegmental nucleus to dopamine neurons in the substantia nigra pars compacta. Neurosci. Res. 1995;21:331–342. doi: 10.1016/0168-0102(94)00869-h. [DOI] [PubMed] [Google Scholar]
- Gan JO, Walton ME, Phillips PE. Dissociable cost and benefit encoding of future rewards by mesolimbic dopamine. Nat. Neurosci. 2009 doi: 10.1038/nn.2460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gard DE, Gard MG, Kring AM, John OP. Anticipatory and consummatory components of the experience of pleasure: A scale development study. Journal of Reserch in Personality. 2006;40:1086–1102. [Google Scholar]
- Gard DE, Kring AM, Gard MG, Horan WP, Green MF. Anhedonia in schizophrenia: distinctions between anticipatory and consummatory pleasure. Schizophr. Res. 2007;93:253–260. doi: 10.1016/j.schres.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehricke J, Shapiro D. Reduced facial expression and social context in major depression: discrepancies between facial muscle activity and self-reported emotion. Psychiatry Res. 2000;95:157–167. doi: 10.1016/s0165-1781(00)00168-2. [DOI] [PubMed] [Google Scholar]
- Gershon AA, Vishne T, Grunhaus L. Dopamine D2-like receptors and the antidepressant response. Biol. Psychiatry. 2007;61:145–153. doi: 10.1016/j.biopsych.2006.05.031. [DOI] [PubMed] [Google Scholar]
- Gold JM,WJ, Prentice KJ, Morris SE, Heerey EA. Reward processing in schizophrenia: a deficit in the representation of value. Schizophr. Bull. 2008;34:835–847. doi: 10.1093/schbul/sbn068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotlib IH, Kasch KL, Traill S, Joormann J, Arnow BA, Johnson SL. Coherence and specificity of information-processing biases in depression and social phobia. J. Abnorm. Psychol. 2004a;113:386–398. doi: 10.1037/0021-843X.113.3.386. [DOI] [PubMed] [Google Scholar]
- Gotlib IH, Krasnoperova E, Yue DN, Joormann J. Attentional biases for negative interpersonal stimuli in clinical depression. J. Abnorm. Psychol. 2004b;113:121–135. doi: 10.1037/0021-843X.113.1.121. [DOI] [PubMed] [Google Scholar]
- Goto Y, Otani S, Grace AA. The Yin and Yang of dopamine release: a new perspective. Neuropharmacology. 2007;53:583–587. doi: 10.1016/j.neuropharm.2007.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottfried JA, Small DM, Zald DH. Chemosensory processing. In: Zald DH, Rauch SL, editors. The Orbitofrontal Cortex. Oxford, U.K.: Oxford University Press; 2006. [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: burst firing. J. Neurosci. 1984;4:2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray JA. The Neuropsychology of Anxiety: An Enquiry into the Functions of the Septo-Hippocampal System. New York, NY: Oxford Univerisity Press; 1987. [Google Scholar]
- Hamill S, Trevitt JT, Nowend KL, Carlson BB, Salamone JD. Nucleus accumbens dopamine depletions and time-constrained progressive ratio performance: effects of different ratio requirements. Pharmacol. Biochem. Behav. 1999;64:21–27. doi: 10.1016/s0091-3057(99)00092-1. [DOI] [PubMed] [Google Scholar]
- Hamilton M. A rating scale for depression. J. Neurol. Neurosurg. Psychiatry. 1960;23:56–62. doi: 10.1136/jnnp.23.1.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammen C. Stress and depression. Annu Rev Clin Psychol. 2005;1:293–319. doi: 10.1146/annurev.clinpsy.1.102803.143938. [DOI] [PubMed] [Google Scholar]
- Hasler G, Fromm S, Carlson PJ, Luckenbaugh DA, Waldeck T, Geraci M, Roiser JP, Neumeister A, Meyers N, Charney DS, Drevets WC. Neural response to catecholamine depletion in unmedicated subjects with major depressive disorder in remission and healthy subjects. Arch. Gen. Psychiatry. 2008;65:521–531. doi: 10.1001/archpsyc.65.5.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasler G, van der Veen JW, Tumonis T, Meyers N, Shen J, Drevets WC. Reduced prefrontal glutamate/glutamine and gamma-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Arch. Gen. Psychiatry. 2007;64:193–200. doi: 10.1001/archpsyc.64.2.193. [DOI] [PubMed] [Google Scholar]
- Hastings RS, Parsey RV, Oquendo MA, Arango V, Mann JJ. Volumetric analysis of the prefrontal cortex, amygdala, and hippocampus in major depression. Neuropsychopharmacology. 2004;29:952–959. doi: 10.1038/sj.npp.1300371. [DOI] [PubMed] [Google Scholar]
- Hauber W, Sommer S. Prefrontostriatal Circuitry Regulates Effort-Related Decision Making. Cereb. Cortex. 2009 doi: 10.1093/cercor/bhn241. [DOI] [PubMed] [Google Scholar]
- Heerey EA, Robinson BM, McMahon RP, Gold JM. Delay discounting in schizophrenia. Cogn Neuropsychiatry. 2007;12:213–221. doi: 10.1080/13546800601005900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegadoren KM, O'Donnell T, Lanius R, Coupland NJ, Lacaze-Masmonteil N. The role of beta-endorphin in the pathophysiology of major depression. Neuropeptides. 2009 doi: 10.1016/j.npep.2009.06.004. [DOI] [PubMed] [Google Scholar]
- Heim C, Plotsky PM, Nemeroff CB. Importance of studying the contributions of early adverse experience to neurobiological findings in depression. Neuropsychopharmacology. 2004;29:641–648. doi: 10.1038/sj.npp.1300397. [DOI] [PubMed] [Google Scholar]
- Henriques JB, Glowacki JM, Davidson RJ. Reward fails to alter response bias in depression. J. Abnorm. Psychol. 1994;103:460–466. doi: 10.1037//0021-843x.103.3.460. [DOI] [PubMed] [Google Scholar]
- Hernandez-Lopez S, Tkatch T, Perez-Garci E, Galarraga E, Bargas J, Hamm H, Surmeier DJ. D2 dopamine receptors in striatal medium spiny neurons reduce L-type Ca2+ currents and excitability via a novel PLC[beta]1-IP3-calcineurin-signaling cascade. J. Neurosci. 2000;20:8987–8995. doi: 10.1523/JNEUROSCI.20-24-08987.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroi N, White NM. The lateral nucleus of the amygdala mediates expression of the amphetamine-produced conditioned place preference. J. Neurosci. 1991;11:2107–2116. doi: 10.1523/JNEUROSCI.11-07-02107.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirvonen J, Karlsson H, Kajander J, Markkula J, Rasi-Hakala H, Nagren K, Salminen JK, Hietala J. Striatal dopamine D2 receptors in medication-naive patients with major depressive disorder as assessed with [11C]raclopride PET. Psychopharmacology (Berl.) 2008;197:581–590. doi: 10.1007/s00213-008-1088-9. [DOI] [PubMed] [Google Scholar]
- Hnasko TS, Sotak BN, Palmiter RD. Morphine reward in dopamine-deficient mice. Nature. 2005;438:854–857. doi: 10.1038/nature04172. [DOI] [PubMed] [Google Scholar]
- Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501. doi: 10.1016/S0893-133X(00)00159-7. [DOI] [PubMed] [Google Scholar]
- Insel TR. Translating scientific opportunity into public health impact: a strategic plan for research on mental illness. Arch. Gen. Psychiatry. 2009;66:128–133. doi: 10.1001/archgenpsychiatry.2008.540. [DOI] [PubMed] [Google Scholar]
- Insel TR, Cuthbert BN. Endophenotypes: bridging genomic complexity and disorder heterogeneity. Biol. Psychiatry. 2009;66:988–989. doi: 10.1016/j.biopsych.2009.10.008. [DOI] [PubMed] [Google Scholar]
- Invernizzi R, Pozzi L, Garattini S, Samanin R. Tianeptine increases the extracellular concentrations of dopamine in the nucleus accumbens by a serotonin-independent mechanism. Neuropharmacology. 1992;31:221–227. doi: 10.1016/0028-3908(92)90171-k. [DOI] [PubMed] [Google Scholar]
- IsHak WW, Davis M, Jeffrey J, Balayan K, Pechnick RN, Bagot K, Rapaport MH. The role of dopaminergic agents in improving quality of life in major depressive disorder. Curr. Psychiatry Rep. 2009;11:503–508. doi: 10.1007/s11920-009-0076-z. [DOI] [PubMed] [Google Scholar]
- Jamerson BD, Krishnan KR, Roberts J, Krishen A, Modell JG. Effect of bupropion SR on specific symptom clusters of depression: analysis of the 31-item Hamilton Rating Scale for depression. Psychopharmacol. Bull. 2003;37:67–78. [PubMed] [Google Scholar]
- Jayaram-Lindstrom N, Wennberg P, Hurd YL, Franck J. Effects of naltrexone on the subjective response to amphetamine in healthy volunteers. J. Clin. Psychopharmacol. 2004;24:665–669. doi: 10.1097/01.jcp.0000144893.29987.e5. [DOI] [PubMed] [Google Scholar]
- Joormann J, Dkane M, Gotlib IH. Adaptive and maladaptive components of rumination? Diagnostic specificity and relation to depressive biases. Behav. Ther. 2006;37:269–280. doi: 10.1016/j.beth.2006.01.002. [DOI] [PubMed] [Google Scholar]
- Kaczmarek HJ, Kiefer SW. Microinjections of dopaminergic agents in the nucleus accumbens affect ethanol consumption but not palatability. Pharmacol. Biochem. Behav. 2000;66:307–312. doi: 10.1016/s0091-3057(00)00182-9. [DOI] [PubMed] [Google Scholar]
- Kapur S, Mann JJ. Role of the dopaminergic system in depression. Biol. Psychiatry. 1992;32:1–17. doi: 10.1016/0006-3223(92)90137-o. [DOI] [PubMed] [Google Scholar]
- Kasper S, McEwen BS. Neurobiological and clinical effects of the antidepressant tianeptine. CNS Drugs. 2008;22:15–26. doi: 10.2165/00023210-200822010-00002. [DOI] [PubMed] [Google Scholar]
- Kaviani H, Gray JA, Checkley SA, Raven PW, Wilson GD, Kumari V. Affective modulation of the startle response in depression: influence of the severity of depression, anhedonia, and anxiety. J. Affect. Disord. 2004;83:21–31. doi: 10.1016/j.jad.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Kavoussi RJ, Segraves RT, Hughes AR, Ascher JA, Johnston JA. Double-blind comparison of bupropion sustained release and sertraline in depressed outpatients. J. Clin. Psychiatry. 1997;58:532–537. doi: 10.4088/jcp.v58n1204. [DOI] [PubMed] [Google Scholar]
- Kazes M, Danion JM, Grange D, Pradignac A, Simon C, Burrus-Mehl F, Schlienger JL, Singer L. Eating behaviour and depression before and after antidepressant treatment: a prospective, naturalistic study. J. Affect. Disord. 1994;30:193–207. doi: 10.1016/0165-0327(94)90080-9. [DOI] [PubMed] [Google Scholar]
- Keedwell PA, Andrew C, Williams SC, Brammer MJ, Phillips ML. A double dissociation of ventromedial prefrontal cortical responses to sad and happy stimuli in depressed and healthy individuals. Biol. Psychiatry. 2005a;58:495–503. doi: 10.1016/j.biopsych.2005.04.035. [DOI] [PubMed] [Google Scholar]
- Keedwell PA, Andrew C, Williams SC, Brammer MJ, Phillips ML. The neural correlates of anhedonia in major depressive disorder. Biol. Psychiatry. 2005b;58:843–853. doi: 10.1016/j.biopsych.2005.05.019. [DOI] [PubMed] [Google Scholar]
- Kendler KS, Karkowski LM, Prescott CA. Causal relationship between stressful life events and the onset of major depression. Am. J. Psychiatry. 1999;156:837–841. doi: 10.1176/ajp.156.6.837. [DOI] [PubMed] [Google Scholar]
- Kennedy SE, Koeppe RA, Young EA, Zubieta JK. Dysregulation of endogenous opioid emotion regulation circuitry in major depression in women. Arch. Gen. Psychiatry. 2006;63:1199–1208. doi: 10.1001/archpsyc.63.11.1199. [DOI] [PubMed] [Google Scholar]
- Kennerley SW, Dahmubed AF, Lara AH, Wallis JD. Neurons in the frontal lobe encode the value of multiple decision variables. J. Cogn. Neurosci. 2009;21:1162–1178. doi: 10.1162/jocn.2009.21100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennerley SW, Walton ME, Behrens TE, Buckley MJ, Rushworth MF. Optimal decision making and the anterior cingulate cortex. Nat. Neurosci. 2006;9:940–947. doi: 10.1038/nn1724. [DOI] [PubMed] [Google Scholar]
- Kessler RC. The effects of stressful life events on depression. Annu. Rev. Psychol. 1997;48:191–214. doi: 10.1146/annurev.psych.48.1.191. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, Rush AJ, Walters EE, Wang PS. The epidemiology of major depressive disorder: results from the National Comorbidity Survey Replication (NCS-R) JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- Kiernan G, Laurent J, Joiner TE, Jr, Catanzaro SJ, MacLachlan M. Cross-cultural examination of the tripartite model with children: data from the Barretstown studies. J. Pers. Assess. 2001;77:359–379. doi: 10.1207/S15327752JPA7702_15. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Hamilton JP, Gotlib IH. Reduced caudate gray matter volume in women with major depressive disorder. Psychiatry Res. 2008;164:114–122. doi: 10.1016/j.pscychresns.2007.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein DN. Depression and Anhedonia. In: Clark DC, Fawcett J, editors. Anhedonia and Affect Deficit States. New York: PMA Publishing Corporation; 1987. [Google Scholar]
- Klimke A, Larisch R, Janz A, Vosberg H, Muller-Gartner HW, Gaebel W. Dopamine D2 receptor binding before and after treatment of major depression measured by [123I]IBZM SPECT. Psychiatry Res. 1999;90:91–101. doi: 10.1016/s0925-4927(99)00009-8. [DOI] [PubMed] [Google Scholar]
- Knutson B, Bhanji JP, Cooney RE, Atlas LY, Gotlib IH. Neural responses to monetary incentives in major depression. Biol. Psychiatry. 2008;63:686–692. doi: 10.1016/j.biopsych.2007.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knutson B, Fong GW, Adams CM, Varner JL, Hommer D. Dissociation of reward anticipation and outcome with event-related fMRI. Neuroreport. 2001;12:3683–3687. doi: 10.1097/00001756-200112040-00016. [DOI] [PubMed] [Google Scholar]
- Knutson B, Taylor J, Kaufman M, Peterson R, Glover G. Distributed neural representation of expected value. J. Neurosci. 2005;25:4806–4812. doi: 10.1523/JNEUROSCI.0642-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koerts J, Leenders KL, Koning M, Portman AT, van Beilen M. Striatal dopaminergic activity (FDOPA-PET) associated with cognitive items of a depression scale (MADRS) in Parkinson's disease. Eur. J. Neurosci. 2007;25:3132–3136. doi: 10.1111/j.1460-9568.2007.05580.x. [DOI] [PubMed] [Google Scholar]
- Koolschijn PC, van Haren NE, Lensvelt-Mulders GJ, Hulshoff Pol HE, Kahn RS. Brain volume abnormalities in major depressive disorder: A meta-analysis of magnetic resonance imaging studies. Hum. Brain Mapp. 2009 doi: 10.1002/hbm.20801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kringelbach ML, de Araujo IE, Rolls ET. Taste-related activity in the human dorsolateral prefrontal cortex. Neuroimage. 2004;21:781–788. doi: 10.1016/j.neuroimage.2003.09.063. [DOI] [PubMed] [Google Scholar]
- Kugaya A, Seneca NM, Snyder PJ, Williams SA, Malison RT, Baldwin RM, Seibyl JP, Innis RB. Changes in human in vivo serotonin and dopamine transporter availabilities during chronic antidepressant administration. Neuropsychopharmacology. 2003;28:413–420. doi: 10.1038/sj.npp.1300036. [DOI] [PubMed] [Google Scholar]
- Kumar P, Waiter G, Ahearn T, Milders M, Reid I, Steele JD. Abnormal temporal difference reward-learning signals in major depression. Brain. 2008;131:2084–2093. doi: 10.1093/brain/awn136. [DOI] [PubMed] [Google Scholar]
- Laasonen-Balk T, Kuikka J, Viinamaki H, Husso-Saastamoinen M, Lehtonen J, Tiihonen J. Striatal dopamine transporter density in major depression. Psychopharmacology (Berl.) 1999;144:282–285. doi: 10.1007/s002130051005. [DOI] [PubMed] [Google Scholar]
- Lambert G, Johansson M, Agren H, Friberg P. Reduced brain norepinephrine and dopamine release in treatment-refractory depressive illness: evidence in support of the catecholamine hypothesis of mood disorders. Arch. Gen. Psychiatry. 2000;57:787–793. doi: 10.1001/archpsyc.57.8.787. [DOI] [PubMed] [Google Scholar]
- Lauwereyns J, Watanabe K, Coe B, Hikosaka O. A neural correlate of response bias in monkey caudate nucleus. Nature. 2002;418:413–417. doi: 10.1038/nature00892. [DOI] [PubMed] [Google Scholar]
- Lavretsky H, Kumar A. Methylphenidate augmentation of citalopram in elderly depressed patients. Am. J. Geriatr. Psychiatry. 2001;9:298–303. [PubMed] [Google Scholar]
- Learned-Coughlin SM, Bergstrom M, Savitcheva I, Ascher J, Schmith VD, Langstrom B. In vivo activity of bupropion at the human dopamine transporter as measured by positron emission tomography. Biol. Psychiatry. 2003;54:800–805. doi: 10.1016/s0006-3223(02)01834-6. [DOI] [PubMed] [Google Scholar]
- Lebowitz BD, Pearson JL, Schneider LS, Reynolds CF, 3rd, Alexopoulos GS, Bruce ML, Conwell Y, Katz IR, Meyers BS, Morrison MF, Mossey J, Niederehe G, Parmelee P. Diagnosis and treatment of depression in late life. Consensus statement update. JAMA. 1997;278:1186–1190. [PubMed] [Google Scholar]
- Lemke MR. Dopamine agonists in the treatment of non-motor symptoms of Parkinson's disease: depression. Eur. J. Neurol. 2008;15 Suppl 2:9–14. doi: 10.1111/j.1468-1331.2008.02213.x. [DOI] [PubMed] [Google Scholar]
- Lemke MR, Brecht HM, Koester J, Kraus PH, Reichmann H. Anhedonia, depression, and motor functioning in Parkinson's disease during treatment with pramipexole. J. Neuropsychiatry Clin. Neurosci. 2005;17:214–220. doi: 10.1176/jnp.17.2.214. [DOI] [PubMed] [Google Scholar]
- Lempert KM, Pizzagalli DA. Delay discounting and future-directed thinking in anhedonic individuals. J. Behav. Ther. Exp. Psychiatry. doi: 10.1016/j.jbtep.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leventhal AM, Chasson GS, Tapia E, Miller EK, Pettit JW. Measuring hedonic capacity in depression: a psychometric analysis of three anhedonia scales. J. Clin. Psychol. 2006;62:1545–1558. doi: 10.1002/jclp.20327. [DOI] [PubMed] [Google Scholar]
- Liberzon I, Zubieta JK, Fig LM, Phan KL, Koeppe RA, Taylor SF. mu-Opioid receptors and limbic responses to aversive emotional stimuli. Proc. Natl. Acad. Sci. U. S. A. 2002;99:7084–7089. doi: 10.1073/pnas.102174799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Lopez Leon S, Croes EA, Sayed-Tabatabaei FA, Claes S, Van Broeckhoven C, van Duijn CM. The dopamine D4 receptor gene 48-base-pair-repeat polymorphism and mood disorders: a meta-analysis. Biol. Psychiatry. 2005;57:999–1003. doi: 10.1016/j.biopsych.2005.01.030. [DOI] [PubMed] [Google Scholar]
- Maier SF, Watkins LR. Cytokines for psychologists: implications of bidirectional immune-to-brain communication for understanding behavior, mood, and cognition. Psychol. Rev. 1998;105:83–107. doi: 10.1037/0033-295x.105.1.83. [DOI] [PubMed] [Google Scholar]
- Manji HK, Drevets WC, Charney DS. The cellular neurobiology of depression. Nat. Med. 2001;7:541–547. doi: 10.1038/87865. [DOI] [PubMed] [Google Scholar]
- Markou A, Kosten TR, Koob GF. Neurobiological similarities in depression and drug dependence: a self-medication hypothesis. Neuropsychopharmacology. 1998;18:135–174. doi: 10.1016/S0893-133X(97)00113-9. [DOI] [PubMed] [Google Scholar]
- Martell CR, Addis ME, Jacobson NS. Depression in Context: Strategies for Guided Action. New York: W. W. Norton & Co., Inc.; 2001. [Google Scholar]
- Martinot M, Bragulat V, Artiges E, Dolle F, Hinnen F, Jouvent R, Martinot J. Decreased presynaptic dopamine function in the left caudate of depressed patients with affective flattening and psychomotor retardation. Am. J. Psychiatry. 2001;158:314–316. doi: 10.1176/appi.ajp.158.2.314. [DOI] [PubMed] [Google Scholar]
- Matsuo K, Rosenberg DR, Easter PC, MacMaster FP, Chen HH, Nicoletti M, Caetano SC, Hatch JP, Soares JC. Striatal volume abnormalities in treatment-naive patients diagnosed with pediatric major depressive disorder. J. Child Adolesc. Psychopharmacol. 2008;18:121–131. doi: 10.1089/cap.2007.0026. [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Brannan SK, Mahurin RK, Jerabek PA, Brickman JS, Tekell JL, Silva JA, McGinnis S, Glass TG, Martin CC, Fox PT. Cingulate function in depression: a potential predictor of treatment response. Neuroreport. 1997;8:1057–1061. doi: 10.1097/00001756-199703030-00048. [DOI] [PubMed] [Google Scholar]
- Mayberg HS, Liotti M, Brannan SK, McGinnis S, Mahurin RK, Jerabek PA, Silva JA, Tekell JL, Martin CC, Lancaster JL, Fox PT. Reciprocal limbic-cortical function and negative mood: converging PET findings in depression and normal sadness. Am. J. Psychiatry. 1999;156:675–682. doi: 10.1176/ajp.156.5.675. [DOI] [PubMed] [Google Scholar]
- McCabe C, Cowen PJ, Harmer CJ. Neural representation of reward in recovered depressed patients. Psychopharmacology (Berl.) 2009;205:667–677. doi: 10.1007/s00213-009-1573-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCabe CMZ, Cowen PJ, Harmer CJ. Diminished Neural Processing of Aversive and Rewarding Stimuli During Selective Serotonin Reuptake Inhibitor Treatment. Biol. Psychiatry. 2009 doi: 10.1016/j.biopsych.2009.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BS. Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism. 2005;54:20–23. doi: 10.1016/j.metabol.2005.01.008. [DOI] [PubMed] [Google Scholar]
- McEwen BS, Chattarji S, Diamond DM, Jay TM, Reagan LP, Svenningsson P, Fuchs E. The neurobiological properties of tianeptine (Stablon): from monoamine hypothesis to glutamatergic modulation. Mol. Psychiatry. 2010;15:237–249. doi: 10.1038/mp.2009.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland BR, Klein DN. Emotional reactivity in depression: diminished responsiveness to anticipated reward but not to anticipated punishment or to nonreward or avoidance. Depress. Anxiety. 2009;26:117–122. doi: 10.1002/da.20513. [DOI] [PubMed] [Google Scholar]
- McGinty VB, Grace AA. Activity-dependent depression of medial prefrontal cortex inputs to accumbens neurons by the basolateral amygdala. Neuroscience. 2009a;162:1429–1436. doi: 10.1016/j.neuroscience.2009.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinty VB, Grace AA. Timing-dependent regulation of evoked spiking in nucleus accumbens neurons by integration of limbic and prefrontal cortical inputs. J. Neurophysiol. 2009b;101:1823–1835. doi: 10.1152/jn.91162.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGlinchey JB, Zimmerman M, Young D, Chelminski I. Diagnosing major depressive disorder VIII: are some symptoms better than others? J. Nerv. Ment. Dis. 2006;194:785–790. doi: 10.1097/01.nmd.0000240222.75201.aa. [DOI] [PubMed] [Google Scholar]
- Mello NK, Mendelson JH, Sholar MB, Jaszyna-Gasior M, Goletiani N, Siegel AJ. Effects of the mixed mu/kappa opioid nalbuphine on cocaine-induced changes in subjective and cardiovascular responses in men. Neuropsychopharmacology. 2005;30:618–632. doi: 10.1038/sj.npp.1300631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendels J, Amin MM, Chouinard G, Cooper AJ, Miles JE, Remick RA, Saxena B, Secunda SK, Singh AN. A comparative study of bupropion and amitriptyline in depressed outpatients. J. Clin. Psychiatry. 1983;44:118–120. [PubMed] [Google Scholar]
- Meyer JH, Goulding VS, Wilson AA, Hussey D, Christensen BK, Houle S. Bupropion occupancy of the dopamine transporter is low during clinical treatment. Psychopharmacology (Berl.) 2002;163:102–105. doi: 10.1007/s00213-002-1166-3. [DOI] [PubMed] [Google Scholar]
- Meyer JH, Kruger S, Wilson AA, Christensen BK, Goulding VS, Schaffer A, Minifie C, Houle S, Hussey D, Kennedy SH. Lower dopamine transporter binding potential in striatum during depression. Neuroreport. 2001;12:4121–4125. doi: 10.1097/00001756-200112210-00052. [DOI] [PubMed] [Google Scholar]
- Meyer JH, McNeely HE, Sagrati S, Boovariwala A, Martin K, Verhoeff NP, Wilson AA, Houle S. Elevated putamen D(2) receptor binding potential in major depression with motor retardation: an [11C]raclopride positron emission tomography study. Am. J. Psychiatry. 2006;163:1594–1602. doi: 10.1176/ajp.2006.163.9.1594. [DOI] [PubMed] [Google Scholar]
- Miller G. Psychiatry. Beyond DSM: seeking a brain-based classification of mental illness. Science. 327:1437. doi: 10.1126/science.327.5972.1437. [DOI] [PubMed] [Google Scholar]
- Miller JM, Vorel SR, Tranguch AJ, Kenny ET, Mazzoni P, van Gorp WG, Kleber HD. Anhedonia after a selective bilateral lesion of the globus pallidus. Am. J. Psychiatry. 2006;163:786–788. doi: 10.1176/ajp.2006.163.5.786. [DOI] [PubMed] [Google Scholar]
- Mingote S, Weber SM, Ishiwari K, Correa M, Salamone JD. Ratio and time requirements on operant schedules: effort-related effects of nucleus accumbens dopamine depletions. Eur. J. Neurosci. 2005;21:1749–1757. doi: 10.1111/j.1460-9568.2005.03972.x. [DOI] [PubMed] [Google Scholar]
- Mitterschiffthaler MT, Kumari V, Malhi GS, Brown RG, Giampietro VP, Brammer MJ, Suckling J, Poon L, Simmons A, Andrew C, Sharma T. Neural response to pleasant stimuli in anhedonia: an fMRI study. Neuroreport. 2003;14:177–182. doi: 10.1097/00001756-200302100-00003. [DOI] [PubMed] [Google Scholar]
- Murray CJ, Lopez AD. Global mortality, disability, and the contribution of risk factors: Global Burden of Disease Study. Lancet. 1997;349:1436–1442. doi: 10.1016/S0140-6736(96)07495-8. [DOI] [PubMed] [Google Scholar]
- Murray EA. The amygdala, reward and emotion. Trends Cogn Sci. 2007;11:489–497. doi: 10.1016/j.tics.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Must A, Szabo Z, Bodi N, Szasz A, Janka Z, Keri S. Sensitivity to reward and punishment and the prefrontal cortex in major depression. J. Affect. Disord. 2006;90:209–215. doi: 10.1016/j.jad.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Ninan PT. Obsessive-compulsive disorder: implications of the efficacy of an SSRI, paroxetine. Psychopharmacol. Bull. 2003;37 Suppl 1:89–96. [PubMed] [Google Scholar]
- Norbury R, Taylor MJ, Selvaraj S, Murphy SE, Harmer CJ, Cowen PJ. Short-term antidepressant treatment modulates amygdala response to happy faces. Psychopharmacology (Berl.) 2009 doi: 10.1007/s00213-009-1597-1. [DOI] [PubMed] [Google Scholar]
- Nutt D, Demyttenaere K, Janka Z, Aarre T, Bourin M, Canonico PL, Carrasco JL, Stahl S. The other face of depression, reduced positive affect: the role of catecholamines in causation and cure. J Psychopharmacol. 2007;21:461–471. doi: 10.1177/0269881106069938. [DOI] [PubMed] [Google Scholar]
- O'Doherty JP, Deichmann R, Critchley HD, Dolan RJ. Neural responses during anticipation of a primary taste reward. Neuron. 2002;33:815–826. doi: 10.1016/s0896-6273(02)00603-7. [DOI] [PubMed] [Google Scholar]
- Ongur D, Drevets WC, Price JL. Glial reduction in the subgenual prefrontal cortex in mood disorders. Proc. Natl. Acad. Sci. U. S. A. 1998;95:13290–13295. doi: 10.1073/pnas.95.22.13290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr K, Taylor D. Psychostimulants in the treatment of depression : a review of the evidence. CNS Drugs. 2007;21:239–257. doi: 10.2165/00023210-200721030-00004. [DOI] [PubMed] [Google Scholar]
- Padoa-Schioppa C, Assad JA. The representation of economic value in the orbitofrontal cortex is invariant for changes of menu. Nat. Neurosci. 2008;11:95–102. doi: 10.1038/nn2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmatier MI, Levin ME, Mays KL, Donny EC, Caggiula AR, Sved AF. Bupropion and nicotine enhance responding for nondrug reinforcers via dissociable pharmacological mechanisms in rats. Psychopharmacology (Berl.) 2009;207:381–390. doi: 10.1007/s00213-009-1666-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker L, Leeb K. Amphetamine-induced modification of quinine palatability: analysis by the taste reactivity test. Pharmacol. Biochem. Behav. 1994;47:413–420. doi: 10.1016/0091-3057(94)90137-6. [DOI] [PubMed] [Google Scholar]
- Parsey RV, Oquendo MA, Zea-Ponce Y, Rodenhiser J, Kegeles LS, Pratap M, Cooper TB, Van Heertum R, Mann JJ, Laruelle M. Dopamine D(2) receptor availability and amphetamine-induced dopamine release in unipolar depression. Biol. Psychiatry. 2001;50:313–322. doi: 10.1016/s0006-3223(01)01089-7. [DOI] [PubMed] [Google Scholar]
- Paterson NE. Behavioural and pharmacological mechanisms of bupropion's anti-smoking effects: recent preclinical and clinical insights. Eur. J. Pharmacol. 2009;603:1–11. doi: 10.1016/j.ejphar.2008.12.009. [DOI] [PubMed] [Google Scholar]
- Paterson NE, Balfour DJ, Markou A. Chronic bupropion attenuated the anhedonic component of nicotine withdrawal in rats via inhibition of dopamine reuptake in the nucleus accumbens shell. Eur. J. Neurosci. 2007;25:3099–3108. doi: 10.1111/j.1460-9568.2007.05546.x. [DOI] [PubMed] [Google Scholar]
- Paterson NE, Markou A. Animal models and treatments for addiction and depression co-morbidity. Neurotox. Res. 2007;11:1–32. doi: 10.1007/BF03033479. [DOI] [PubMed] [Google Scholar]
- Paykel ES, Ramana R, Cooper Z, Hayhurst H, Kerr J, Barocka A. Residual symptoms after partial remission: an important outcome in depression. Psychol. Med. 1995;25:1171–1180. doi: 10.1017/s0033291700033146. [DOI] [PubMed] [Google Scholar]
- Pecina S, Berridge KC. Hedonic hot spot in nucleus accumbens shell: where do mu-opioids cause increased hedonic impact of sweetness? J. Neurosci. 2005;25:11777–11786. doi: 10.1523/JNEUROSCI.2329-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pecina S, Berridge KC, Parker LA. Pimozide does not shift palatability: separation of anhedonia from sensorimotor suppression by taste reactivity. Pharmacol. Biochem. Behav. 1997;58:801–811. doi: 10.1016/s0091-3057(97)00044-0. [DOI] [PubMed] [Google Scholar]
- Pecina S, Smith KS, Berridge KC. Hedonic hot spots in the brain. Neuroscientist. 2006;12:500–511. doi: 10.1177/1073858406293154. [DOI] [PubMed] [Google Scholar]
- Pelizza L, Ferrari A. Anhedonia in schizophrenia and major depression: state or trait? Ann Gen Psychiatry. 2009;8:22. doi: 10.1186/1744-859X-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips PE, Walton ME, Jhou TC. Calculating utility: preclinical evidence for cost-benefit analysis by mesolimbic dopamine. Psychopharmacology (Berl.) 2007;191:483–495. doi: 10.1007/s00213-006-0626-6. [DOI] [PubMed] [Google Scholar]
- Piazza PV, Barrot M, Rouge-Pont F, Marinelli M, Maccari S, Abrous DN, Simon H, Le Moal M. Suppression of glucocorticoid secretion and antipsychotic drugs have similar effects on the mesolimbic dopaminergic transmission. Proc. Natl. Acad. Sci. U. S. A. 1996a;93:15445–15450. doi: 10.1073/pnas.93.26.15445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piazza PV, Rouge-Pont F, Deroche V, Maccari S, Simon H, Le Moal M. Glucocorticoids have state-dependent stimulant effects on the mesencephalic dopaminergic transmission. Proc. Natl. Acad. Sci. U. S. A. 1996b;93:8716–8720. doi: 10.1073/pnas.93.16.8716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickar D, Davis GC, Schulz SC, Extein I, Wagner R, Naber D, Gold PW, van Kammen DP, Goodwin FK, Wyatt RJ, Li CH, Bunney WE., Jr Behavioral and biological effects of acute beta-endorphin injection in schizophrenic and depressed patients. Am. J. Psychiatry. 1981;138:160–166. doi: 10.1176/ajp.138.2.160. [DOI] [PubMed] [Google Scholar]
- Pizzagalli DA, Holmes AJ, Dillon DG, Goetz EL, Birk JL, Bogdan R, Dougherty DD, Iosifescu DV, Rauch SL, Fava M. Reduced caudate and nucleus accumbens response to rewards in unmedicated individuals with major depressive disorder. Am. J. Psychiatry. 2009;166:702–710. doi: 10.1176/appi.ajp.2008.08081201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzagalli DA, Iosifescu D, Hallett LA, Ratner KG, Fava M. Reduced hedonic capacity in major depressive disorder: evidence from a probabilistic reward task. J. Psychiatr. Res. 2008;43:76–87. doi: 10.1016/j.jpsychires.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzagalli DA, Jahn AL, O'Shea JP. Toward an objective characterization of an anhedonic phenotype: a signal-detection approach. Biol. Psychiatry. 2005;57:319–327. doi: 10.1016/j.biopsych.2004.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price J, Cole V, Goodwin GM. Emotional side-effects of selective serotonin reuptake inhibitors: qualitative study. Br. J. Psychiatry. 2009;195:211–217. doi: 10.1192/bjp.bp.108.051110. [DOI] [PubMed] [Google Scholar]
- Radley JJ, Rocher AB, Miller M, Janssen WG, Liston C, Hof PR, McEwen BS, Morrison JH. Repeated stress induces dendritic spine loss in the rat medial prefrontal cortex. Cereb. Cortex. 2006;16:313–320. doi: 10.1093/cercor/bhi104. [DOI] [PubMed] [Google Scholar]
- Rakofsky JJ, Holtzheimer PE, Nemeroff CB. Emerging targets for antidepressant therapies. Curr. Opin. Chem. Biol. 2009;13:291–302. doi: 10.1016/j.cbpa.2009.04.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rau KS, Birdsall E, Hanson JE, Johnson-Davis KL, Carroll FI, Wilkins DG, Gibb JW, Hanson GR, Fleckenstein AE. Bupropion increases striatal vesicular monoamine transport. Neuropharmacology. 2005;49:820–830. doi: 10.1016/j.neuropharm.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Reichel CM, Linkugel JD, Bevins RA. Bupropion differentially impacts acquisition of methamphetamine self-administration and sucrose-maintained behavior. Pharmacol. Biochem. Behav. 2008;89:463–472. doi: 10.1016/j.pbb.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renneberg B, Heyn K, Gebhard R, Bachmann S. Facial expression of emotions in borderline personality disorder and depression. J. Behav. Ther. Exp. Psychiatry. 2005;36:183–196. doi: 10.1016/j.jbtep.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Reynolds CF, 3rd, Frank E, Dew MA, Houck PR, Miller M, Mazumdar S, Perel JM, Kupfer DJ. Treatment of 70(+)-year-olds with recurrent major depression. Excellent short-term but brittle long-term response. Am. J. Geriatr. Psychiatry. 1999;7:64–69. [PubMed] [Google Scholar]
- Ribot T. La psychologie des sentiment [The psychology of feelings] Paris: Felix Alcan; 1896. [Google Scholar]
- Rolls ET, Xiang JZ. Reward-spatial view representations and learning in the primate hippocampus. J. Neurosci. 2005;25:6167–6174. doi: 10.1523/JNEUROSCI.1481-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottenberg J, Gross JJ, Gotlib IH. Emotion context insensitivity in major depressive disorder. J. Abnorm. Psychol. 2005;114:627–639. doi: 10.1037/0021-843X.114.4.627. [DOI] [PubMed] [Google Scholar]
- Rottenberg J, Kasch KL, Gross JJ, Gotlib IH. Sadness and amusement reactivity differentially predict concurrent and prospective functioning in major depressive disorder. Emotion. 2002;2:135–146. doi: 10.1037/1528-3542.2.2.135. [DOI] [PubMed] [Google Scholar]
- Ruhe HG, Mason NS, Schene AH. Mood is indirectly related to serotonin, norepinephrine and dopamine levels in humans: a meta-analysis of monoamine depletion studies. Mol. Psychiatry. 2007;12:331–359. doi: 10.1038/sj.mp.4001949. [DOI] [PubMed] [Google Scholar]
- Rushworth MF, Behrens TE. Choice, uncertainty and value in prefrontal and cingulate cortex. Nat. Neurosci. 2008;11:389–397. doi: 10.1038/nn2066. [DOI] [PubMed] [Google Scholar]
- Rushworth MFWM, Kennerley SW, Bannerman DM. Action sets and decisions in the medial frontal cortex. Trends Cogn Sci. 2004;8:410–417. doi: 10.1016/j.tics.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Salamone JD. The behavioral neurochemistry of motivation: methodological and conceptual issues in studies of the dynamic activity of nucleus accumbens dopamine. J. Neurosci. Methods. 1996;64:137–149. doi: 10.1016/0165-0270(95)00125-5. [DOI] [PubMed] [Google Scholar]
- Salamone JD, Correa M, Farrar A, Mingote SM. Effort-related functions of nucleus accumbens dopamine and associated forebrain circuits. Psychopharmacology (Berl.) 2007;191:461–482. doi: 10.1007/s00213-006-0668-9. [DOI] [PubMed] [Google Scholar]
- Salamone JD, Correa M, Mingote SM, Weber SM. Beyond the reward hypothesis: alternative functions of nucleus accumbens dopamine. Curr. Opin. Pharmacol. 2005;5:34–41. doi: 10.1016/j.coph.2004.09.004. [DOI] [PubMed] [Google Scholar]
- Salamone JD, Steinpreis RE, McCullough LD, Smith P, Grebel D, Mahan K. Haloperidol and nucleus accumbens dopamine depletion suppress lever pressing for food but increase free food consumption in a novel food choice procedure. Psychopharmacology (Berl.) 1991;104:515–521. doi: 10.1007/BF02245659. [DOI] [PubMed] [Google Scholar]
- Sanfey AG, Rilling JK, Aronson JA, Nystrom LE, Cohen JD. The neural basis of economic decision-making in the Ultimatum Game. Science. 2003;300:1755–1758. doi: 10.1126/science.1082976. [DOI] [PubMed] [Google Scholar]
- Sapolsky RM. Glucocorticoids and hippocampal atrophy in neuropsychiatric disorders. Arch. Gen. Psychiatry. 2000;57:925–935. doi: 10.1001/archpsyc.57.10.925. [DOI] [PubMed] [Google Scholar]
- Sarchiapone M, Carli V, Camardese G, Cuomo C, Di Giuda D, Calcagni ML, Focacci C, De Risio S. Dopamine transporter binding in depressed patients with anhedonia. Psychiatry Res. 2006;147:243–248. doi: 10.1016/j.pscychresns.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Schoenbaum G, Saddoris MP, Ramus SJ, Shaham Y, Setlow B. Cocaine-experienced rats exhibit learning deficits in a task sensitive to orbitofrontal cortex lesions. Eur. J. Neurosci. 2004;19:1997–2002. doi: 10.1111/j.1460-9568.2004.03274.x. [DOI] [PubMed] [Google Scholar]
- Schoenbaum G, Setlow B. Lesions of nucleus accumbens disrupt learning about aversive outcomes. J. Neurosci. 2003;23:9833–9841. doi: 10.1523/JNEUROSCI.23-30-09833.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenbaum G, Setlow B, Ramus SJ. A systems approach to orbitofrontal cortex reveals interactions with different learning systems. 2003a;146:19–209. doi: 10.1016/j.bbr.2003.09.013. [DOI] [PubMed] [Google Scholar]
- Schoenbaum G, Setlow B, Saddoris MP, Gallagher M. Encoding predicted outcome and acquired value in orbitofrontal cortex during cue sampling depends upon input from basolateral amygdala. Neuron. 2003;39:855–867. doi: 10.1016/s0896-6273(03)00474-4. [DOI] [PubMed] [Google Scholar]
- Schott BH, Minuzzi L, Krebs RM, Elmenhorst D, Lang M, Winz OH, Seidenbecher CI, Coenen HH, Heinze HJ, Zilles K, Duzel E, Bauer A. Mesolimbic functional magnetic resonance imaging activations during reward anticipation correlate with reward-related ventral striatal dopamine release. J. Neurosci. 2008;28:14311–14319. doi: 10.1523/JNEUROSCI.2058-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz W. Behavioral theories and the neurophysiology of reward. Annu. Rev. Psychol. 2006;57:87–115. doi: 10.1146/annurev.psych.56.091103.070229. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Schweimer J, Hauber W. Involvement of the rat anterior cingulate cortex in control of instrumental responses guided by reward expectancy. Learn. Mem. 2005;12:334–342. doi: 10.1101/lm.90605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweimer J, Saft V, Hauber W. Involvement of catecholamine neurotransmission in the rat anterior cingulate in effort-related decision making. Behav. Neurosci. 2005;119:1687–1692. doi: 10.1037/0735-7044.119.6.1687. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Grace AA. Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology. 35:27–47. doi: 10.1038/npp.2009.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Setlow B, Schoenbaum G, Gallagher M. Neural encoding in ventral striatum during olfactory discrimination learning. Neuron. 2003;38:625–636. doi: 10.1016/s0896-6273(03)00264-2. [DOI] [PubMed] [Google Scholar]
- Shah PJ, Ogilvie AD, Goodwin GM, Ebmeier KP. Clinical and psychometric correlates of dopamine D2 binding in depression. Psychol. Med. 1997;27:1247–1256. doi: 10.1017/s0033291797005382. [DOI] [PubMed] [Google Scholar]
- Sharot T, Shiner T, Brown AC, Fan J, Dolan RJ. Dopamine enhances expectation of pleasure in humans. Curr. Biol. 2009;19:2077–2080. doi: 10.1016/j.cub.2009.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehan DV, Mao CG. Paroxetine treatment of generalized anxiety disorder. Psychopharmacol. Bull. 2003;37 Suppl 1:64–75. [PubMed] [Google Scholar]
- Shelton RC, Tomarken AJ. Can recovery from depression be achieved? Psychiatr. Serv. 2001;52:1469–1478. doi: 10.1176/appi.ps.52.11.1469. [DOI] [PubMed] [Google Scholar]
- Shopsin B, Gershon S. Dopamine receptor stimulation in the treatment of depression: piribedil (ET-495) Neuropsychobiology. 1978;4:1–14. doi: 10.1159/000117615. [DOI] [PubMed] [Google Scholar]
- Siegle GJ, Carter CS, Thase ME. Use of FMRI to predict recovery from unipolar depression with cognitive behavior therapy. Am. J. Psychiatry. 2006;163:735–738. doi: 10.1176/ajp.2006.163.4.735. [DOI] [PubMed] [Google Scholar]
- Siegle GJ, Steinhauer SR, Thase ME, Stenger VA, Carter CS. Can't shake that feeling: event-related fMRI assessment of sustained amygdala activity in response to emotional information in depressed individuals. Biol. Psychiatry. 2002;51:693–707. doi: 10.1016/s0006-3223(02)01314-8. [DOI] [PubMed] [Google Scholar]
- Siegle GJ, Thompson W, Carter CS, Steinhauer SR, Thase ME. Increased amygdala and decreased dorsolateral prefrontal BOLD responses in unipolar depression: related and independent features. Biol. Psychiatry. 2007;61:198–209. doi: 10.1016/j.biopsych.2006.05.048. [DOI] [PubMed] [Google Scholar]
- Sigmon S, Nelson-Gray R. Sensitivity to aversive events in depression: Antecedent, concommitant, or consequent? Journal of Psycholopathology and Behavioral Assessment. 1992;14:225–246. [Google Scholar]
- Sitland-Marken PA, Wells BG, Froemming JH, Chu CC, Brown CS. Psychiatric applications of bromocriptine therapy. J. Clin. Psychiatry. 1990;51:68–82. [PubMed] [Google Scholar]
- Sloan DM, Strauss ME, Quirk SW, Sajatovic M. Subjective and expressive emotional responses in depression. J. Affect. Disord. 1997;46:135–141. doi: 10.1016/s0165-0327(97)00097-9. [DOI] [PubMed] [Google Scholar]
- Sloan DM, Strauss ME, Wisner KL. Diminished response to pleasant stimuli by depressed women. J. Abnorm. Psychol. 2001;110:488–493. doi: 10.1037//0021-843x.110.3.488. [DOI] [PubMed] [Google Scholar]
- Smith ID, Grace AA. Role of the subthalamic nucleus in the regulation of nigral dopamine neuron activity. Synapse. 1992;12:287–303. doi: 10.1002/syn.890120406. [DOI] [PubMed] [Google Scholar]
- Smith KS, Tindell AJ, Aldridge JW, Berridge KC. Ventral pallidum roles in reward and motivation. Behav. Brain Res. 2009;196:155–167. doi: 10.1016/j.bbr.2008.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoski MJ, Felder J, Bizzell J, Green SR, Ernst M, Lynch TR, Dichter GS. fMRI of alterations in reward selection, anticipation, and feedback in major depressive disorder. J. Affect. Disord. 2009 doi: 10.1016/j.jad.2009.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smoski MJ, Lynch TR, Rosenthal MZ, Cheavens JS, Chapman AL, Krishnan RR. Decision-making and risk aversion among depressive adults. J. Behav. Ther. Exp. Psychiatry. 2008;39:567–576. doi: 10.1016/j.jbtep.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snaith RP, Hamilton M, Morley S, Humayan A, Hargreaves D, Trigwell P. A scale for the assessment of hedonic tone the Snaith-Hamilton Pleasure Scale. Br. J. Psychiatry. 1995;167:99–103. doi: 10.1192/bjp.167.1.99. [DOI] [PubMed] [Google Scholar]
- Spijker J, Bijl RV, de Graaf R, Nolen WA. Determinants of poor 1-year outcome of DSM-III-R major depression in the general population: results of the Netherlands Mental Health Survey and Incidence Study (NEMESIS) Acta Psychiatr. Scand. 2001;103:122–130. doi: 10.1034/j.1600-0447.2001.103002122.x. [DOI] [PubMed] [Google Scholar]
- Stahl SM. Placebo-controlled comparison of the selective serotonin reuptake inhibitors citalopram and sertraline. Biol. Psychiatry. 2000;48:894–901. doi: 10.1016/s0006-3223(00)00957-4. [DOI] [PubMed] [Google Scholar]
- Stahl SM, Pradko JF, Haight BR, Modell JG, Rockett CB, Learned-Coughlin S. A Review of the Neuropharmacology of Bupropion, a Dual Norepinephrine and Dopamine Reuptake Inhibitor. Prim Care Companion J Clin Psychiatry. 2004;6:159–166. doi: 10.4088/pcc.v06n0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele JD, Kumar P, Ebmeier KP. Blunted response to feedback information in depressive illness. Brain. 2007;130:2367–2374. doi: 10.1093/brain/awm150. [DOI] [PubMed] [Google Scholar]
- Surguladze S, Brammer MJ, Keedwell P, Giampietro V, Young AW, Travis MJ, Williams SC, Phillips ML. A differential pattern of neural response toward sad versus happy facial expressions in major depressive disorder. Biol. Psychiatry. 2005;57:201–209. doi: 10.1016/j.biopsych.2004.10.028. [DOI] [PubMed] [Google Scholar]
- Surguladze SA, Young AW, Senior C, Brebion G, Travis MJ, Phillips ML. Recognition accuracy and response bias to happy and sad facial expressions in patients with major depression. Neuropsychology. 2004;18:212–218. doi: 10.1037/0894-4105.18.2.212. [DOI] [PubMed] [Google Scholar]
- Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Oono H, Inoue T, Boku S, Kako Y, Kitaichi Y, Kusumi I, Masui T, Nakagawa S, Suzuki K, Tanaka T, Koyama T, Radford MH. Depressive patients are more impulsive and inconsistent in intertemporal choice behavior for monetary gain and loss than healthy subjects--an analysis based on Tsallis' statistics. Neuro Endocrinol Lett. 2008;29:351–358. [PubMed] [Google Scholar]
- Talmi D, Dayan P, Kiebel SJ, Frith CD, Dolan RJ. How humans integrate the prospects of pain and reward during choice. J. Neurosci. 2009;29:14617–14626. doi: 10.1523/JNEUROSCI.2026-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thase ME, Haight BR, Richard N, Rockett CB, Mitton M, Modell JG, VanMeter S, Harriett AE, Wang Y. Remission rates following antidepressant therapy with bupropion or selective serotonin reuptake inhibitors: a meta-analysis of original data from 7 randomized controlled trials. J. Clin. Psychiatry. 2005;66:974–981. doi: 10.4088/jcp.v66n0803. [DOI] [PubMed] [Google Scholar]
- Tomarken AJ, Dichter GS, Freid C, Addington S, Shelton RC. Assessing the effects of bupropion SR on mood dimensions of depression. J. Affect. Disord. 2004;78:235–241. doi: 10.1016/S0165-0327(02)00306-3. [DOI] [PubMed] [Google Scholar]
- Treadway MT, Buckholtz JW, Schwartzman AN, Lambert WE, Zald DH. Worth the 'EEfRT'? The effort expenditure for rewards task as an objective measure of motivation and anhedonia. PLoS ONE 4. 2009a:e6598. doi: 10.1371/journal.pone.0006598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treadway MT, Grant MM, Ding Z, Hollon SD, Gore JC, Shelton RC. Early adverse events, HPA activity and rostral anterior cingulate volume in MDD. PLoS ONE 4. 2009b:e4887. doi: 10.1371/journal.pone.0004887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay L, Schultz W. Relative reward preference in primate orbitofrontal cortex. Nature. 1999;398:704–708. doi: 10.1038/19525. [DOI] [PubMed] [Google Scholar]
- Tremblay LK, Naranjo CA, Graham SJ, Herrmann N, Mayberg HS, Hevenor S, Busto UE. Functional neuroanatomical substrates of altered reward processing in major depressive disorder revealed by a dopaminergic probe. Arch. Gen. Psychiatry. 2005;62:1228–1236. doi: 10.1001/archpsyc.62.11.1228. [DOI] [PubMed] [Google Scholar]
- Tremeau F, Malaspina D, Duval F, Correa H, Hager-Budny M, Coin-Bariou L, Macher JP, Gorman JM. Facial expressiveness in patients with schizophrenia compared to depressed patients and nonpatient comparison subjects. Am. J. Psychiatry. 2005;162:92–101. doi: 10.1176/appi.ajp.162.1.92. [DOI] [PubMed] [Google Scholar]
- Tsai JL, Pole N, Levenson RW, Munoz RF. The effects of depression on the emotional responses of Spanish-speaking Latinas. Cultur. Divers. Ethnic Minor. Psychol. 2003;9:49–63. doi: 10.1037/1099-9809.9.1.49. [DOI] [PubMed] [Google Scholar]
- Vale S, Espejel MA, Dominguez JC. Amantadine in depression. Lancet. 1971;2:437. doi: 10.1016/s0140-6736(71)90153-x. [DOI] [PubMed] [Google Scholar]
- van Praag HM, Korf J, Schut D. Cerebral monoamines and depression. An investigation with the Probenecid technique. Arch. Gen. Psychiatry. 1973;28:827–831. doi: 10.1001/archpsyc.1973.01750360053007. [DOI] [PubMed] [Google Scholar]
- Vaswani M, Linda FK, Ramesh S. Role of selective serotonin reuptake inhibitors in psychiatric disorders: a comprehensive review. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2003;27:85–102. doi: 10.1016/s0278-5846(02)00338-x. [DOI] [PubMed] [Google Scholar]
- Vijayaraghavan L, Vaidya JG, Humphreys CT, Beglinger LJ, Paradiso S. Emotional and motivational changes after bilateral lesions of the globus pallidus. Neuropsychology. 2008;22:412–418. doi: 10.1037/0894-4105.22.3.412. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Ding YS, Fowler JS, Wang GJ, Logan J, Gatley JS, Dewey S, Ashby C, Liebermann J, Hitzemann R, et al. Is methylphenidate like cocaine? Studies on their pharmacokinetics and distribution in the human brain. Arch. Gen. Psychiatry. 1995;52:456–463. doi: 10.1001/archpsyc.1995.03950180042006. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fischman MW, Foltin RW, Fowler JS, Abumrad NN, Vitkun S, Logan J, Gatley SJ, Pappas N, Hitzemann R, Shea CE. Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature. 1997;386:827–830. doi: 10.1038/386827a0. [DOI] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Gatley SJ, Ding YS, Logan J, Dewey SL, Hitzemann R, Lieberman J. Relationship between psychostimulant-induced "high" and dopamine transporter occupancy. Proc. Natl. Acad. Sci. U. S. A. 1996;93:10388–10392. doi: 10.1073/pnas.93.19.10388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkow ND, Wang GJ, Fowler JS, Gatley SJ, Logan J, Ding YS, Hitzemann R, Pappas N. Dopamine transporter occupancies in the human brain induced by therapeutic doses of oral methylphenidate. Am. J. Psychiatry. 1998;155:1325–1331. doi: 10.1176/ajp.155.10.1325. [DOI] [PubMed] [Google Scholar]
- Vythilingam M, Heim C, Newport J, Miller AH, Anderson E, Bronen R, Brummer M, Staib L, Vermetten E, Charney DS, Nemeroff CB, Bremner JD. Childhood trauma associated with smaller hippocampal volume in women with major depression. Am. J. Psychiatry. 2002;159:2072–2080. doi: 10.1176/appi.ajp.159.12.2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wacker J, Dillon DG, Pizzagalli DA. The role of the nucleus accumbens and rostral anterior cingulate cortex in anhedonia: integration of resting EEG, fMRI, and volumetric techniques. Neuroimage. 2009;46:327–337. doi: 10.1016/j.neuroimage.2009.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waehrens J, Gerlach J. Bromocriptine and imipramine in endogenous depression. A double-blind controlled trial in out-patients. J. Affect. Disord. 1981;3:193–202. doi: 10.1016/0165-0327(81)90044-6. [DOI] [PubMed] [Google Scholar]
- Wagner G, Koch K, Schachtzabel C, Reichenbach JR, Sauer H, Schlosser Md RG. Enhanced rostral anterior cingulate cortex activation during cognitive control is related to orbitofrontal volume reduction in unipolar depression. J. Psychiatry Neurosci. 2008;33:199–208. [PMC free article] [PubMed] [Google Scholar]
- Walter M, Henning A, Grimm S, Schulte RF, Beck J, Dydak U, Schnepf B, Boeker H, Boesiger P, Northoff G. The relationship between aberrant neuronal activation in the pregenual anterior cingulate, altered glutamatergic metabolism, and anhedonia in major depression. Arch. Gen. Psychiatry. 2009;66:478–486. doi: 10.1001/archgenpsychiatry.2009.39. [DOI] [PubMed] [Google Scholar]
- Walton ME, Bannerman DM, Alterescu K, Rushworth MF. Functional specialization within medial frontal cortex of the anterior cingulate for evaluating effort-related decisions. J. Neurosci. 2003;23:6475–6479. doi: 10.1523/JNEUROSCI.23-16-06475.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton ME, Bannerman DM, Rushworth MF. The role of rat medial frontal cortex in effort-based decision making. J. Neurosci. 2002;22:10996–11003. doi: 10.1523/JNEUROSCI.22-24-10996.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton ME, Croxson PL, Rushworth MF, Bannerman DM. The mesocortical dopamine projection to anterior cingulate cortex plays no role in guiding effort-related decisions. Behav. Neurosci. 2005;119:323–328. doi: 10.1037/0735-7044.119.1.323. [DOI] [PubMed] [Google Scholar]
- Walton ME, Devlin JT, Rushworth MF. Interactions between decision making and performance monitoring within prefrontal cortex. Nat. Neurosci. 2004;7:1259–1265. doi: 10.1038/nn1339. [DOI] [PubMed] [Google Scholar]
- Walton ME, Groves J, Jennings KA, Croxson PL, Sharp T, Rushworth MF, Bannerman DM. Comparing the role of the anterior cingulate cortex and 6-hydroxydopamine nucleus accumbens lesions on operant effort-based decision making. Eur. J. Neurosci. 2009;29:1678–1691. doi: 10.1111/j.1460-9568.2009.06726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton ME, Kennerley SW, Bannerman DM, Phillips PE, Rushworth MF. Weighing up the benefits of work: behavioral and neural analyses of effort-related decision making. Neural Netw. 2006;19:1302–1314. doi: 10.1016/j.neunet.2006.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walton ME, Rudebeck PH, Bannerman DM, Rushworth MF. Calculating the cost of acting in frontal cortex. Ann. N. Y. Acad. Sci. 2007;1104:340–356. doi: 10.1196/annals.1390.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson D, Clark LA, Weber K, Assenheimer JS, Strauss ME, McCormick RA. Testing a tripartite model: II. Exploring the symptom structure of anxiety and depression in student, adult, and patient samples. J. Abnorm. Psychol. 1995a;104:15–25. doi: 10.1037//0021-843x.104.1.15. [DOI] [PubMed] [Google Scholar]
- Watson D, Weber K, Assenheimer JS, Clark LA, Strauss ME, McCormick RA. Testing a tripartite model: I. Evaluating the convergent and discriminant validity of anxiety and depression symptom scales. J. Abnorm. Psychol. 1995b;104:3–14. doi: 10.1037//0021-843x.104.1.3. [DOI] [PubMed] [Google Scholar]
- Weihs KL, Settle EC, Jr, Batey SR, Houser TL, Donahue RM, Ascher JA. Bupropion sustained release versus paroxetine for the treatment of depression in the elderly. J. Clin. Psychiatry. 2000;61:196–202. doi: 10.4088/jcp.v61n0309. [DOI] [PubMed] [Google Scholar]
- Weisler RH, Johnston JA, Lineberry CG, Samara B, Branconnier RJ, Billow AA. Comparison of bupropion and trazodone for the treatment of major depression. J. Clin. Psychopharmacol. 1994;14:170–179. [PubMed] [Google Scholar]
- Weiss F, Markou A, Lorang MT, Koob GF. Basal extracellular dopamine levels in the nucleus accumbens are decreased during cocaine withdrawal after unlimited-access self-administration. Brain Res. 1992;593:314–318. doi: 10.1016/0006-8993(92)91327-b. [DOI] [PubMed] [Google Scholar]
- Wexler BE, Levenson L, Warrenburg S, Price LH. Decreased perceptual sensitivity to emotion-evoking stimuli in depression. Psychiatry Res. 1994;51:127–138. doi: 10.1016/0165-1781(94)90032-9. [DOI] [PubMed] [Google Scholar]
- Willner P. Dopamine and depression: a review of recent evidence. I. Empirical studies. Brain Res. 1983a;287:211–224. doi: 10.1016/0165-0173(83)90005-x. [DOI] [PubMed] [Google Scholar]
- Willner P. Dopamine and depression: a review of recent evidence. II. Theoretical approaches. Brain Res. 1983b;287:225–236. doi: 10.1016/0165-0173(83)90006-1. [DOI] [PubMed] [Google Scholar]
- Willner P. Dopamine and depression: a review of recent evidence. III. The effects of antidepressant treatments. Brain Res. 1983c;287:237–246. doi: 10.1016/0165-0173(83)90007-3. [DOI] [PubMed] [Google Scholar]
- Wise RA. The dopamine synapse and the notion of ‘pleasure centers’ in the brain. Trends Neurosci. 1980;3:91–95. [Google Scholar]
- Wise RA, Spindler J, deWit H, Gerberg GJ. Neuroleptic-induced "anhedonia" in rats: pimozide blocks reward quality of food. Science. 1978;201:262–264. doi: 10.1126/science.566469. [DOI] [PubMed] [Google Scholar]
- Yadid G, Friedman A. Dynamics of the dopaminergic system as a key component to the understanding of depression. Prog. Brain Res. 2008;172:265–286. doi: 10.1016/S0079-6123(08)00913-8. [DOI] [PubMed] [Google Scholar]
- Yang YK, Yeh TL, Yao WJ, Lee IH, Chen PS, Chiu NT, Lu RB. Greater availability of dopamine transporters in patients with major depression--a dual-isotope SPECT study. Psychiatry Res. 2008;162:230–235. doi: 10.1016/j.pscychresns.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Yucel K, M MCK, Chahal R, Taylor V, Macdonald K, Joffe R, Macqueen G. Increased subgenual prefrontal cortex size in remitted patients with major depressive disorder. Psychiatry Res. 2009;173:71–76. doi: 10.1016/j.pscychresns.2008.07.013. [DOI] [PubMed] [Google Scholar]
- Yucel K, McKinnon MC, Chahal R, Taylor VH, Macdonald K, Joffe R, MacQueen GM. Anterior cingulate volumes in never-treated patients with major depressive disorder. Neuropsychopharmacology. 2008;33:3157–3163. doi: 10.1038/npp.2008.40. [DOI] [PubMed] [Google Scholar]
- Zald DH, Kim SW. Anatomy and function of the orbital frontal cortex, I: anatomy, neurocircuitry; and obsessive-compulsive disorder. J. Neuropsychiatry Clin. Neurosci. 1996a;8:125–138. doi: 10.1176/jnp.8.2.125. [DOI] [PubMed] [Google Scholar]
- Zald DH, Kim SW. Anatomy and function of the orbital frontal cortex, II: Function and relevance to obsessive-compulsive disorder. J. Neuropsychiatry Clin. Neurosci. 1996b;8:249–261. doi: 10.1176/jnp.8.3.249. [DOI] [PubMed] [Google Scholar]
- Zangen A, Nakash R, Overstreet DH, Yadid G. Association between depressive behavior and absence of serotonin-dopamine interaction in the nucleus accumbens. Psychopharmacology (Berl.) 2001;155:434–439. doi: 10.1007/s002130100746. [DOI] [PubMed] [Google Scholar]
- Zhou QY, Palmiter RD. Dopamine-deficient mice are severely hypoactive, adipsic, and aphagic. Cell. 1995;83:1197–1209. doi: 10.1016/0092-8674(95)90145-0. [DOI] [PubMed] [Google Scholar]
- Zimmerman M, McGlinchey JB, Young D, Chelminski I. Diagnosing major depressive disorder introduction: an examination of the DSM-IV diagnostic criteria. J. Nerv. Ment. Dis. 2006;194:151–154. doi: 10.1097/01.nmd.0000202235.78441.53. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Bueller JA, Jackson LR, Scott DJ, Xu Y, Koeppe RA, Nichols TE, Stohler CS. Placebo effects mediated by endogenous opioid activity on mu-opioid receptors. J. Neurosci. 2005;25:7754–7762. doi: 10.1523/JNEUROSCI.0439-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubieta JK, Ketter TA, Bueller JA, Xu Y, Kilbourn MR, Young EA, Koeppe RA. Regulation of human affective responses by anterior cingulate and limbic mu-opioid neurotransmission. Arch. Gen. Psychiatry. 2003;60:1145–1153. doi: 10.1001/archpsyc.60.11.1145. [DOI] [PubMed] [Google Scholar]
- Zubieta JK, Smith YR, Bueller JA, Xu Y, Kilbourn MR, Jewett DM, Meyer CR, Koeppe RA, Stohler CS. Regional mu opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293:311–315. doi: 10.1126/science.1060952. [DOI] [PubMed] [Google Scholar]
- Zweifel LS, Parker JG, Lobb CJ, Rainwater A, Wall VZ, Fadok JP, Darvas M, Kim MJ, Mizumori SJ, Paladini CA, Phillips PE, Palmiter RD. Disruption of NMDAR-dependent burst firing by dopamine neurons provides selective assessment of phasic dopamine-dependent behavior. Proc. Natl. Acad. Sci. U. S. A. 2009;106:7281–7288. doi: 10.1073/pnas.0813415106. [DOI] [PMC free article] [PubMed] [Google Scholar]