

Abstract
The current state-of-the-art for drug-carrying biomedical devices is mostly limited to those that release a single drug. Yet there are many situations in which more than one therapeutic agent is needed. Also, most polyelectrolyte multilayer films intending for drug delivery are loaded with active molecules only during multilayer film preparation. In this paper, we present the integration of capsules as vehicles within polypeptide multilayer films for sustained release of multiple oppositely-charged drug molecules using layer-by-layer nanoassembly technology. Calcium carbonate (CaCO3) particles were impregnated with polyelectrolytes, shelled with polyelectrolyte multilayers, and then assembled onto polypeptide multilayer films using glutaraldehyde. Capsule-integrated polypeptide multilayer films were obtained after decomposition of CaCO3 templates. Two oppositely-charged drugs were loaded into capsules within polypeptide multilayer films post-preparation based on electrostatic interactions between the drugs and the polyelectrolytes impregnated within capsules. We determined that the developed innovative capsule-integrated polypeptide multilayer films could be used to load multiple drugs of very different properties (e.g. opposite charges) any time post-preparation (e.g. minutes before surgical implantation inside an operating room), and such capsule-integrated films allowed simultaneous delivery of two oppositely-charged drug molecules and a sustained (up to two weeks or longer) and sequential release was achieved.
Keywords: Post-preparation drug delivery, polyelectrolyte film, microcapsule, polypeptide, multiple drugs, co-delivery, sustained release
1. Introduction
Due to its high degree of control over film properties, flexible choice of assembly components, and ease of processing, layer-by-layer (LBL) nanoassembly has been widely used as a versatile technique for fabricating polyelectrolyte multilayer films and microcapsules with precisely controlled structures and compositions.1–10 The films and microcapsules are typically built by alternative adsorption of polymers with complementary functional groups based on electrostatic interactions,1,2,4–10 hydrogen bonding,3,6,11,12 hydrophilicity/hydrophobicity,13,14, and/or covalent interactions.15,16 Polyelectrolyte microcapsules have shown great potential application in drug delivery and polyelectrolyte films have applications in optics, electronics, and biomedical devices.3,17–22
Recently, increasing interest has been drawn to developing drug-carrying films at the implant/tissue interface to prevent device-associated infections.23–27 Millions of medical devices are implanted in patients every year28,29 and device-associated infections and related wound healing are significant clinical complications of almost all kinds of prosthetic medical devices.30–33 Drug-carrying films on implantable medical devices may play an important role in preventing device-associated infections and promoting wound healing; the current state-of-the-art for drug-carrying devices is mostly limited to those that release a single drug. Polyelectrolyte multilayer films carrying a single drug have been studied by assembly of charged drugs or drugconjugated polymers as film components, and by post-incorporation of drugs into the films.34–37 In addition, amphiphilic polymers, hydrogels, and micelles have been introduced in polyelectrolyte multilayer films for drug delivery control.38–42 However, there are many situations in which more than one therapeutic agent is needed. To give an example, drug-carrying implants that deliver multiple therapeutic drugs such as antibiotics and growth factors may not only prevent device-associated infections but also promote wound healing thereby achieving ideal treatment outcomes. As a result, a number of researchers have directly incorporated two or more drugs into polyelectrolyte multilayers and controlled release has been observed.43–46 For instance, Hammond and colleagues have engineered hydrolytically degradable multilayer films to release multiple drugs through control of the interlayer diffusion and/or the degradation of the multilayers.43,45 Functionalized nanoparticles have also recently been integrated within polyelectrolyte multilayer films.46 However, most of these polyelectrolyte multilayer films have been loaded with drugs only during film preparation. Finding a method to load multiple drugs in the films post-preparation is of great significance because it relieves the concerns over drug storage in the films and opens the opportunity for the surgeons to load the appropriate drugs minutes before surgical implantations.
In this study, we present the development of innovative capsule-integrated polypeptide multilayer films that allow for post-preparation loading and release of multiple therapeutic agents of different properties (e.g. opposite charges). Capsules impregnated with polyelectrolytes, serving as drug binding agents, were integrated as layers in polypeptide multilayer films. Charged drug molecules, both small and large, were tunably attracted (e.g. via incubation time) into capsules that were impregnated with oppositely-charged polyelectrolytes. Sustained release of two oppositely-charged drug molecules from one single polypeptide multilayer film was obtained.
2. Materials and methods
2.1. Materials
Poly-L-lysine hydrobromide (PLL, MW 150-300 kDa), poly-L-(glutamic acid) (PLGA, MW 50 kDa), glycine, fluorescein-isothiocyanate labeled bovine serum albumin (FITC-BSA), gentamicin sulfate, and glutaraldehyde (GA) were purchased from Sigma (St. Louis, MO). CaCO3 particles were purchased from PlasmaChem Gmbh (Rudower Chaussee, Berlin, Germany). Disodium ethylenediaminetetraacetic acid (EDTA) was obtained from Fisher (Fair Lawn, NJ). Quartz slides were purchased from Electron Microscopy Sciences (Hatfield, PA), cut into 1 × 2 cm2, and cleaned in a piranha solution (4:1 H2SO4/H2O2) followed by rinsing with deionized water. A 0.1 M phosphate buffered saline (PBS, pH 7.4) was prepared and used throughout this study. PLL and PLGA solutions were prepared by dissolving PLL and PLGA, respectively, in PBS. Fluorescein-isothiocyanate labeled PLL (FITC-PLL) and gentamicin (FITC-gentamicin) and Rhodamine B labeled PLGA (RhoB-PLGA) and gentamicin (RhoB-gentamicin) were prepared.
2.2. Impregnation of polyelectrolytes in microparticles and subsequent nanoassembly of multilayered shells on polyelectrolyte-impregnated microparticles
Polyelectrolytes (i.e. PLL or PLGA) were impregnated into CaCO3 particles. Briefly, CaCO3 colloidal particles (200 mg) were immersed in 1 mg/ml, 2 mg/ml, or 5 mg/ml PLL or PLGA solution and incubated for 30 min in a vacuum chamber (3 torr). After centrifugation, the supernatant was removed and the particles were dried at 60°C under vacuum (380 torr) overnight. Polypeptides (i.e. PLL and PLGA) were then alternatively used to form a shell on these particles using LBL nanoassembly to form shelled CaCO3 particles. Briefly, polyelectrolyte-impregnated CaCO3 particles (200 mg) were incubated in 1 mg/mL PLL solution for 20 min, washed three times by immersing them in a glycine solution (pH 7.4) for 1 min followed by centrifugation at 5,000 rpm for 0.5 min. The microparticles were then incubated in 1 mg/mL PLGA solution for 20 min and washed three times. The deposition of PLL and PLGA layers was repeated three times followed by an additional assembling of PLL as the outermost layer. As a result, (PLL/PLGA)3.5 was formed on the CaCO3 particles. These shelled CaCO3 particles were then subjected to cross-linking by incubating them in a GA solution at ambient temperature for 2 h and as a result, the surfaces of CaCO3 particles were enabled with aldehyde groups due to the use of excessive GA (see Supplementary Data). The particles were finally washed three times in deionized water and kept at 4°C for further use; these PLL and PLGA impregnated CaCO3 particles with aldehyde groups on the surfaces were designated as CaCO3PLL and CaCO3PLGA, respectively. To form capsules, 0.1 g of CaCO3PLL or CaCO3PLGA particles was incubated in a 0.1 M EDTA solution for 1 h followed by centrifugation and removal of supernatants. Such incubation was repeated three times to dissolve the CaCO3 templates.
To quantify the impregnated PLL or PLGA within CaCO3 particles, FITC-PLL and RhoB-PLGA were used to prepare CaCO3FITC-PLL and CaCO3RhoB-PLGA particles, and unlabelled PLL and PLGA were used to form (PLL/PLGA)3.5 layers on the particles. The CaCO3 templates were decomposed and the particles were sonicated for 20 min. FITC-PLL and RhoB-PLGA were quantified by measuring their peak absorbance at 480 and 560 nm, respectively, using UV-vis spectrometry (BioMate 3 UV-Vis Spectrophotometer, NY, US). Standard curves were obtained by measuring the absorbance of known concentrations of FITC-PLL and RhoB-PLGA. In addition, the impregnated PLL or PLGA were also quantified by measuring the supernatant polymer concentrations before and after incubation with CaCO3 particles. No significant differences were observed between the two quantification approaches.
2.3. Formation of capsule-integrated polypeptide multilayer films
Polypeptide multilayer films were prepared using a dipping-machine (Riegler & Kirstein Gmbh, Germany). The procedure to form capsule-integrated multilayer films was as follows (see Fig. 1): (i) Substrate (i.e. quartz slide) was dipped into a PLL solution for 10 min, washed with PBS for 3 min, followed by air drying. Next the substrate was dipped in a PLGA solution, washed, and dried. The process was repeated to form (PLL/PLGA)3.5 layers on the substrate (Fig. 1a). (ii) The substrate was then dipped into a suspension (1 ml) of shelled CaCO3PLL particles with aldehyde groups for 60 min at a particle density of 2.0 × 108/ml, which was estimated using hemocytometry, and washed with deionized water three times for 3 min (Fig. 1b). (iii) Subsequent deposition of (PLL/PLGA)10.5 was conducted using LBL nanoassembly as described in step (i) (Fig. 1c). (iv) The substrate was next immersed into a suspension (1 ml) of shelled CaCO3PLGA particles for 60 min, washed and dried (Fig. 1d). (v) A final (PLL/PLGA)10.5 was assembled to obtain a multilayer film of (PLL/PLGA)3.5/CaCO3PLL/(PLL/PLGA)10.5/CaCO3PLGA/(PLL/PLGA)10.5 (Fig. 1e). The specimen was immersed in a 0.1 M EDTA solution for 1 h; the immersion was repeated three times to dissolve the CaCO3 templates in the film (Fig. 1f). The resulting capsule-integrated polypeptide multilayer film was denoted as (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5/CapsulePLGA/(PLL/PLGA)10.5. Similarly, polypeptide multilayer films of (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5 and (PLL/PLGA)3.5/CapsulePLGA/(PLL/PLGA)10.5 were prepared. Note that the assembly of PLL/PLGA multilayers was due to electrostatic interactions between PLL and PLGA, and the integration of CapsulePLL and CapsulePLGA was due to the interactions between the aldehyde groups on the capsule surfaces and the amino groups of PLL (see Supplementary Data). The morphology of capsule-integrated films was examined using scanning electron microscopy (SEM, Hitachi S-4700, Tokyo, Japan) operated under a voltage of 5 kV. Energy dispersive X-ray (EDX) analysis was carried out to confirm the decomposition of CaCO3 particles.
Fig. 1.

Schematic diagram presenting the formation of polypeptide multilayer films sandwiched with capsules that are impregnated with positively- and negatively-charged polyelectrolytes. (a) Formation of (PLL/PLGA)3.5 multilayer films using LBL nanoassembly; (b) Deposition of a layer of CaCO3PLL microparticles; (c) Formation of (PLL/PLGA)10.5; (d) Deposition of a layer of CaCO3PLGA microparticles; (e) Formation of an additional (PLL/PLGA)10.5; and (f) Capsule formation via dissolution of CaCO3 templates.
2.4. Drug loading in capsule-integrated polypeptide multilayer films
Drug loading was conducted by incubating polypeptide multilayer films into 1 mg/ml FITC-BSA or RhoB-gentamicin solutions (pH 7.4, PBS buffered). The sample was washed in PBS for a few seconds to remove the loosely adsorbed drug molecules. To load FITC-BSA and RhoB-gentamicin in one film, polypeptide multilayer films of (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5/CapsulePLGA/(PLL/PLGA)10.5 were immersed in the FITC-BSA solution for 15 min followed by the RhoB-gentamicin solution. The loading of FITC-BSA and RhoB-gentamicin in the capsules integrated within polypeptide multilayer films was confirmed using confocal laser scanning microscopy (CLSM, LSM 510, Zeiss, Thornwood, NY).
2.5. Drug release from capsule-integrated polypeptide multilayer films
A polypeptide multilayer film was incubated in a cuvette containing 2 ml PBS (pH 7.4) in a water bath (37°C). At predetermined time periods, 0.4 ml of the PBS medium was taken for UV-vis measurements at 480 or 560 nm to quantify the release of FITC-BSA and RhoB-gentamicin, respectively. An equal volume of fresh PBS was added to keep a constant volume of release medium. All measurements were conducted in triplicate.
3. Results
3.1. Preparation of multilayered microparticles and capsules
CaCO3 particles, approximately 7 μm in diameter, were used to prepare multilayered or shelled microparticles and capsules. The CaCO3 particles had a nanoporous structure (Fig. 2a inset), and the pore size became smaller after impregnation of the polymers (e.g. PLGA; Fig. 2b). PLL or PLGA was successfully impregnated into the CaCO3 particles. The amount of PLL or PLGA impregnated within CaCO3 particles increased with increasing concentrations of PLL or PLGA solutions and reached saturation at about 2 mg/ml (Fig. 3). At the same concentration, more PLGA than PLL was impregnated into the particles. For instance, at 2 mg/ml, approximately 3.3 ng/particle of PLGA and 2.0 ng/particle of PLL were impregnated. After impregnation of PLGA or PLL inside CaCO3 particles, PLL and PLGA were alternatively deposited to form a shell of (PLL/PLGA)3.5 onto the particles (Fig. 2c). The CaCO3 templates could be decomposed using EDTA, and the decomposition of CaCO3 was confirmed using EDX which showed the disappearance of the Ca peak (Fig. 2e). PLGA or PLL impregnated capsules (i.e. CapsulePLGA or CapsulePLL) were obtained (Fig. 2d).
Fig. 2.

SEM images of (a) CaCO3 particles, (b) CaCO3PLGA particles, (c) (PLL/PLGA)3.5 shelled CaCO3PLGA particles, (d) CapsulePLGA, and (e) EDX spectra of CaCO3 particle (dotted line) and CapsulePLGA (solid line). Scale bar: 5 μm.
Fig. 3.

Impregnation of PLGA or PLL within CaCO3 particles to form CaCO3PLGA or CaCO3PLL particles as a function of the corresponding polyelectrolyte concentrations. Incubation time was 30 min.
3.2. Assembly of microparticle- and capsule-integrated polypeptide multilayer films
The density of shelled CaCO3 particles assembled in polypeptide multilayer films was found to depend on the incubation time (Fig. 4). Higher particle density was obtained with increasing incubation time from 0 to 60 min, at which the assembly of CaCO3 particles almost reached a plateau (1.5–1.7×106 particles/cm2).
Fig. 4.
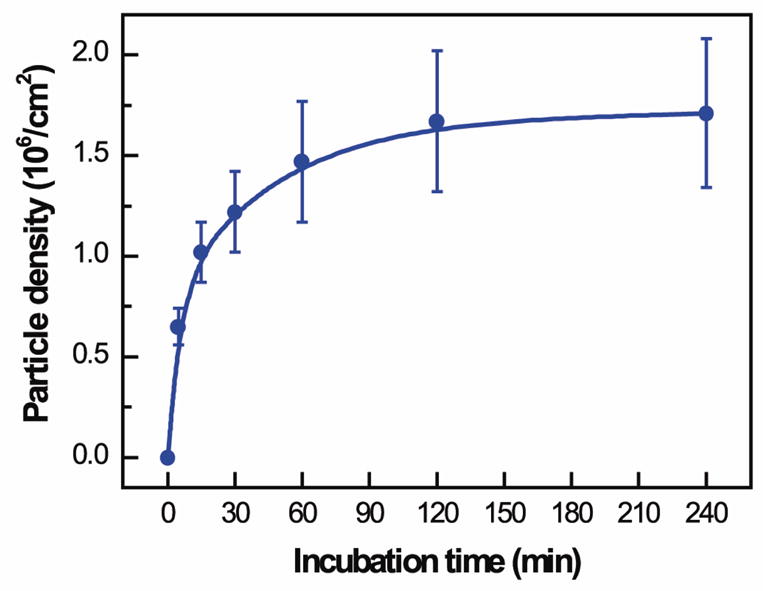
Density of (PLL/PLGA)3.5 shelled CaCO3 particles assembled in polypeptide multilayer films vs. incubation time in CaCO3 particle suspension (2.0 × 108 particles/ml).
The assembly of shelled CaCO3 particles in polypeptide multilayer films was examined using CLSM and SEM (Fig. 5). The fluorescence of RhoB-PLGA (Figs. 5a1 and 5b1) had the same pattern as the CaCO3 particles (Fig. 5a2) and RhoB-PLGA was impregnated within CaCO3 particles (Fig. 5a1). Decomposition of CaCO3 was subsequently achieved after treating with EDTA; the images under transmittance mode confirmed the decomposition of CaCO3 particles (Figs. 5a2 vs. 5b2). The SEM image shows the morphology of the developed capsule-integrated polypeptide multilayer films (Fig. 5c). It seems that CaCO3 particles tended to aggregate within polypeptide multilayer films and a unique pattern of capsules within polypeptide multilayer films was observed (Fig. 5c).
Fig. 5.

In-situ observation of (PLL/PLGA)3.5/CaCO3RhoB-PLGA/(PLL/PLGA)10.5 films. CLSM images (a1, b1) under fluorescence mode and (a2, b2) under transmittance mode. (c) SEM image of (PLL/PLGA)3.5/CapsuleRhoB-PLGA/(PLL/PLGA)10.5 film. (a1, a2) before and (b1, b2) after CaCO3 template removal. RhoB-PLGA served as a drug binding agent and was impregnated into CaCO3 particles.
3.3. Drug loading into capsule-integrated polypeptide multilayer films
Charged drug molecules were successfully loaded into polyelectrolyte-impregnated capsules within polypeptide multilayer films and the drug loading was tunable, for instance, via incubation time. FITC-gentamicin, a positively-charged drug model, was loaded into capsule-integrated polypeptide multilayer films. The green fluorescent spots (i.e. FITC-gentamicin; Figs. 6a1, b1, and c1) overlapped with the red fluorescent ones (i.e. RhoB-PLGA; Figs. 6a2, b2, and c2); this indicated the successful loading of FITC-gentamicin within (RhoB-PLGA)-impregnated capsules in (PLL/PLGA)3.5/CapsuleRhoB-PLGA/(PLL/PLGA)10.5 films. In-situ observations further showed that the loading of FITC-gentamicin was incubation time-dependent since its fluorescence intensity increased with incubation time ranging from 3 to 15 min (Figs. 6a1, 6b1, and 6c1). Loading of FITC-gentamicin reached a maximum upon 15 min incubation; no further increase in its fluorescent intensity was observed with further increasing incubation time (data not shown). Similarly, FITC-BSA was loaded into (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5 films (Fig. 7).
Fig. 6.

Polypeptide multilayer films of (PLL/PLGA)3.5/CapsuleRhoB-PLGA/(PLL/PLGA)10.5 loaded with FITC-gentamicin for (a) 3, (b) 8, and (c) 15 min. FITC-gentamicin and RhoB-PLGA were visualized at (a1, b1, and c1) 480 and (a2, b2 and c2) 560 nm, respectively.
Fig. 7.

Polypeptide multilayer films of (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5 loaded with FITC-BSA for (a) 3 and (b) 15 min under (a1, b1) fluorescence and (a2, b2) transmittance modes.
Loading of two oppositely-charged drug molecules within one polypeptide multilayer film was subsequently achieved. FITC-BSA was loaded into polypeptide multilayer films containing both CapsulePLL and CapsulePLGA (Figs. 8a1 and 8b1 top sections) while very little FITC-BSA was loaded into polypeptide multilayer films containing CapsulePLGA alone (Figs. 8a1 and 8b1 bottom sections). Moreover, both FITC-BSA and RhoB-gentamicin were loaded into polypeptide multilayer films containing both CapsulePLGA and CapsulePLL (Figs. 8a3 and 8b2 top sections) while only RhoB-gentamicin was loaded into polypeptide multilayer films containing only CapsulePLGA (Figs. 8a3 and 8b2 bottom sections).
Fig. 8.

In-situ CLSM images of polypeptide multilayer films after incubating in (a1, b1) FITC-BSA (green) for 15 min followed by incubating in (a2, b2) RhoB-gentamicin (red) for 15 min. (a3) Combined pictures of (a1) and (a2). (b) Z-stack images of two angle views (i.e. 90° and 20°) of polypeptide film in the film thickness direction. Top half: (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5/CapsulePLGA/(PLL/PLGA)10.5, containing both CapsulePLL and CapsulePLGA; Bottom half: (PLL/PLGA)3.5/CapsulePLGA/(PLL/PLGA)10.5, containing only CapsulePLGA.
3.4. Drug release from capsule-integrated polypeptide multilayer films
Release of FITC-BSA and RhoB-gentamicin from polypeptide multilayer films with and without capsules was performed. A burst release (within 1 day) followed by a sustained release of FITC-BSA and RhoB-gentamicin was obtained in polypeptide multilayer films of (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5 and (PLL/PLGA)3.5/CapsulePLGA/(PLL/PLGA)10.5, respectively (Fig. 9a). Approximately 25 μg/cm2 of FITC-BSA was released from (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5 within the first day, and approximately 40 μg/cm2 was released within 14 days. Similarly, approximately 30 and 55 μg/cm2 of RhoB-gentamicin were released from (PLL/PLGA)3.5/CapsulePLGA/(PLL/PLGA)10.5 within 1 and 14 days, respectively. By contrast, very little loading and release (~5 μg/cm2) of FITC-BSA was observed in (PLL/PLGA)3.5/CapsulePLGA/(PLL/PLGA)10.5 films where both the drug BSA and the impregnated PLGA are negatively-charged (Fig. 9a). Similarly, very little loading and release of FITC-BSA or RhoB-gentamicin was found in polypeptide multilayer films without capsules (Fig. 9a).
Fig. 9.
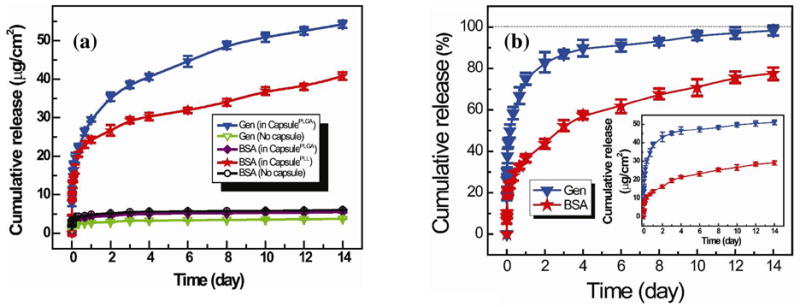
(a) Cumulative release profiles of FITC-BSA and RhoB-gentamicin from polypeptide multilayer films with (solid symbols) and without (empty symbols) CapsulePLL and/or CapsulePLGA. FITC-BSA was released from multilayer films of (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5, (PLL/PLGA)3.5/CapsulePLGA/(PLL/PLGA)10.5, and (PLL/PLGA)3.5/PLGA/(PLL/PLGA)10.5. RhoB-gentamicin was released from multilayer films of (PLL/PLGA)3.5/CapsulePLGA/(PLL/PLGA)10.5 and (PLL/PLGA)3.5/PLGA/(PLL/PLGA)10.5. (b) Cumulative release profiles in percentage of FITC-BSA and RhoB-gentamicin from polypeptide multilayer films, i.e. (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5/CapsulePLGA/(PLL/PLGA)10.5, containing both CapsulePLL and CapsulePLGA. Inset shows the cumulative release in actual amounts. All experiments were carried out in triplicate.
Release of FITC-BSA and RhoB-gentamicin from one single polypeptide multilayer film was determined. Sustained release of both FITC-BSA and RhoB-gentamicin was obtained (Fig. 9b) in polypeptide multilayer films of (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5/CapsulePLGA/(PLL/PLGA)10.5. The release of FITC-BSA was much slower than that of RhoB-gentamicin, as approximately 35% of FITC-BSA and 75% of RhoB-gentamicin were released within day 1 (Fig. 9b). At day 14, RhoB-gentamicin was completely released and approximately 25% of FITC-BSA was still remaining.
4. Discussion
We developed innovative capsule-integrated polypeptide multilayer films that allow for post-preparation loading of multiple drugs and simultaneous delivery of two oppositely-charged drug molecules in a sustained and sequential manner. The introduction of drug binding sites within capsules was obtained by impregnating polyelectrolytes inside porous colloidal templates (i.e. CaCO3 particles) followed by capsule formation. The amount of polyelectrolytes (e.g. PLGA) that could be impregnated inside capsules could be controlled by the concentration of polyelectrolytes (Fig. 3).
LBL nanoassembly has been studied to prepare polypeptide multilayers on CaCO3 particles47–49 and here it was applied to deposit multilayers of (PLL/PLGA)3.5 on CaCO3 particles impregnated with polypeptides. The shelled CaCO3 particles were treated with GA in excess, and the outermost layer (i.e. PLL) of the shelled CaCO3 particles was therefore conjugated with GA through the reaction between aldehyde groups of GA and the amino groups of PLL. The surface of the CaCO3 particles was consequently enabled with aldehyde groups49 (see Supplementary Data). These GA treated CaCO3 particles were deposited onto polypeptide multilayer films on quartz slides because their aldehyde groups react with the amino groups of the outermost layer (i.e. PLL) of polypeptide multilayer films49 (see Supplementary Data). The aldehyde groups also enabled the subsequent deposition of a PLL layer (Fig. 1c). As a result, shelled CaCO3 particles were assembled within polypeptide multilayer films. The use of a crosslinking agent such as GA in this study was necessary. Without GA crosslinking, little adsorption of shelled CaCO3 particles was observed in polypeptide multilayer films (see Supplementary Data). Moreover, direct assembly of the capsules as a layer, based on electrostatic attraction, in the multilayer films led to substantially fewer capsules that could be adsorbed compared to the use of GA (data not shown). In addition, the use of shelled CaCO3 particles instead of capsules is advantageous since the use of capsules will expose the impregnated polyelectrolytes to the subsequent LBL process. This will lead to potential interactions of the impregnated polyelectrolytes with oppositely-charged polyelectrolytes and therefore the impregnated polyelectrolytes will not function as drug binding sites. Similarly, direct assembly of drug-carrying capsules in the multilayer films will lead to (i) the release of drugs during the subsequent LBL process of the films and (ii) interactions between the drug loaded and the subsequent LBL solutions. Therefore, the use of shelled particles, decomposed only in the final film preparation process, provides the multilayer films with the capabilities to load and release two or more oppositely-charged drug molecules any time post-preparation. To our knowledge, such an approach has not been reported. In addition, by impregnating different polymers or other materials inside the porous particles, different types of drugs or other agents could be loaded and released.
Capsule-integrated polypeptide multilayer films were formed after decomposition of CaCO3 templates using EDTA (Figs. 1f and 5). Loading of charged drug molecules into polyelectrolyte-impregnated capsules within polypeptide multilayer films was observed in-situ (Figs. 6, 7, and 8). FITC-gentamicin (green spots) was loaded in the capsules (red spots) and the loading was time-dependent (Fig. 6). Large macromolecules and even nanoparticles were reported to be loaded into polyelectrolyte multilayer films and microcapsules.34,35,50,51 Similarly, in this study, biomacromolecules like BSA were loaded into capsule-integrated polypeptide multilayer films, and relatively more loading of smaller drug molecules (i.e. gentamicin) was observed. The loading of drugs in capsule-integrated polypeptide multilayer films was mainly due to electrostatic interactions between the oppositely-charged drugs and the polyelectrolytes impregnated inside the capsules. This is consistent with the observations that a significantly high amount (40 μg/cm2) of negatively-charged drug (i.e. FITC-BSA) was loaded into polypeptide multilayer films with PLL-impregnated capsules while very little (<5 μg/cm2 or less than 10%) non-specific loading of FITC-BSA was found in polypeptide multilayer films without capsules or with capsules impregnated with polyelectrolytes (i.e. PLGA) of the same charge as the drug (Figs. 8 and 9a). The minimal loading of drugs inside the multilayer films or capsule walls was probably because most of the polymers in the multilayer films or capsule walls interact with their oppositely-charged polymers during film buildup and had few net charges for interacting with the drug molecules.
Moreover, sustained release of RhoB-gentamicin and FITC-BSA from capsule-integrated polypeptide multilayer films was achieved (Figs. 9a and b). Drug concentration difference between the polypeptide multilayer film and its environment was the main driving force that led to the release of drugs. Two oppositely-charged drug molecules were successfully loaded into and released from one polypeptide multilayer film, i.e. (PLL/PLGA)3.5/CapsulePLL/(PLL/PLGA)10.5/CapsulePLGA/(PLL/PLGA)10.5 (Figs. 8 and 9b), and a sustained (up to two weeks or longer) drug release was observed (Fig. 9b). Compared to the release of gentamicin, the release of BSA was much slower, probably due to its larger molecular weight and its stronger hydrophobic interactions with PLL impregnated within the capsules. Gentamicin was completely released at day 14 while a substantial amount (i.e. 25%) of BSA still remained. This indicated that a sequential release of multiple drugs (e.g. release of two drugs within 14 days and one drug thereafter) was obtained in the developed capsule-integrated polypeptide multilayer films.
The concept of loading multiple therapeutic agents (especially agents with very different properties) within polyelectrolyte multilayer films after film preparation would be of real interest and has not been validated very well yet. It has been observed that certain combinations of polyelectrolytes allow for the completely irreversible loading of proteins52 and nanoparticles could be reversibly loaded into exponentially growing polyelectrolyte multilayer films.50 The significance of this study in finding a method to load multiple drugs post-preparation of film buildup include: (i) it would enable fast and universal preparation of the capsule-integrated films for a variety of applications, (ii) the polymer, intact post-preparation of film buildup, inside the capsules would enable flexible control over the types of drugs to be loaded, (iii) the use of polypeptides as film components would provide enormous potential for functionalization, and (iv) it would offer a universal approach to generate films with multiple functions on medical devices.
5. Conclusions
Advanced drug delivery systems were developed by integrating capsules within polypeptide multilayer films. Two oppositely-charged model drugs were successfully loaded into capsule-integrated polypeptide multilayer films by means of impregnating positively- or negatively-charged polyelectrolytes as drug-binding agents within the capsules. The loading of charged drugs was mainly based on electrostatic interactions between the drugs and drug-binding agents within capsules. Polypeptide multilayer films were able to load multiple drugs post-preparation of films and to deliver two or more oppositely-charged drugs at a sustained level (up to two weeks or longer) in a sequential manner. This provides an exciting new route to incorporate and deliver multiple drugs in a controlled manner, potentially at the implant/tissue interface, for medical applications (e.g. prevention of medical device-associated infections and stimulation of wound healing).
Supplementary Material
Acknowledgments
Financial support from the National Science Foundation (OISE-0737735), AO Foundation, Osteosynthesis and Trauma Care Foundation, and West Virginia University Research Corporation is acknowledged. Project S-07-43L was supported by the AO Research Fund of the AO Foundation. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the funding agencies. Microscope experiments and image analysis were performed in the West Virginia University Imaging Facility, which is supported in part by the Mary Babb Randolph Cancer Center and NIH grant P20 RR016440. We thank Karen Martin for assisting in confocal imaging and Suzanne Smith for proofreading.
Footnotes
Conflict of interest: None
Supporting Information Available. Additional characterizations of glutaraldehyde crosslinking, FTIR spectra, and FITC-BSA loading. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Decher G. Science. 1997;277:1232–1237. [Google Scholar]
- 2.Ai H, Fang M, Jones SA, Lvov YM. Biomacromolecules. 2002;3:560–564. doi: 10.1021/bm015659r. [DOI] [PubMed] [Google Scholar]
- 3.Stockton WB, Rubner MF. Macromolecules. 1997;30:2717–2725. [Google Scholar]
- 4.Laurent D, Schlenoff JB. Langmuir. 1997;13:1552–1557. [Google Scholar]
- 5.Kotov NA, Haraszti T, Turi L, Zavala G, Geer RE, Dekany I, Fendler JH. J Am Chem Soc. 1997;119:6821–6832. [Google Scholar]
- 6.Hammond PT. Adv Mater. 2004;16:1271–1293. [Google Scholar]
- 7.Picart C, Mutterer J, Richert L, Luo Y, Prestwich GD, Schaaf P, Voegel JC, Lavalle P. Proc Natl Acad Sci U S A. 2002;99:12531–12535. doi: 10.1073/pnas.202486099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gao CY, Liu XY, Shen JC, Möhwald H. Chem Commun. 2002;17:1928–1929. doi: 10.1039/b204583d. [DOI] [PubMed] [Google Scholar]
- 9.Li B, Haynie DT. Biomacromolecules. 2004;5:1667–1670. doi: 10.1021/bm0496155. [DOI] [PubMed] [Google Scholar]
- 10.De Geest BG, Sanders NN, Sukhorukov GB, Demeester J, De Smedt SC. Chem Soc Rev. 2007;36:636–649. doi: 10.1039/b600460c. [DOI] [PubMed] [Google Scholar]
- 11.Zhang HY, Wang ZQ, Zhang YQ, Zhang X. Langmuir. 2004;20:9366–9370. doi: 10.1021/la048685u. [DOI] [PubMed] [Google Scholar]
- 12.Kharlampieva E, Kozlovskaya V, Sukhishvili SA. Adv Mater. 2009;21(30):3053–3065. [Google Scholar]
- 13.Serizawa T, Kamimura S, Kawanishi N, Akashi M. Langmuir. 2002;18(22):8381–8385. [Google Scholar]
- 14.Quinn JF, Caruso F. Macromolecules. 2005;38:3414–3419. [Google Scholar]
- 15.Liu YL, Bruening ML, Bergbreiter DE, Crooks RM. Angew Chem Int Ed. 1997;36:2114–2116. [Google Scholar]
- 16.Feng ZQ, Gao CY, Shen JC. Macromol Chem Phys. 2009;210:1387–1393. doi: 10.1002/macp.200900025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lvov Y, Yamada S, Kunitake T. Thin Solid Films. 1997;300:107–112. [Google Scholar]
- 18.McShane MJ, Brown JQ, Guice KB, Lvov YM. J Nanosci Nanotechn. 2002;2:411–416. doi: 10.1166/jnn.2002.118. [DOI] [PubMed] [Google Scholar]
- 19.Sahoo SK, Parveen S, Panda JJ. Nanomedicine: NBM. 2007;3:20–31. doi: 10.1016/j.nano.2006.11.008. [DOI] [PubMed] [Google Scholar]
- 20.Xue W, Cui TH. J Nano Res. 2008;1:1–9. [Google Scholar]
- 21.Chong SF, Chandrawati R, Stadler B, Park J, Cho J, Wang Y, Jia Z, Bulmus V, Davis TP, Zelikin AN, Caruso F. Small. 2009;5:2601–2610. doi: 10.1002/smll.200900906. [DOI] [PubMed] [Google Scholar]
- 22.Ochs CJ, Such GeK, Yan Y, van Koeverden MP, Caruso F. ACS Nano. 2010;4:1653–1663. doi: 10.1021/nn9014278. [DOI] [PubMed] [Google Scholar]
- 23.Darouiche RO, Mansouri MD, Zakarevicz D, AlSharif A, Landon GC. J Bone Joint Surg Am. 2007;89A:792–797. doi: 10.2106/JBJS.F.00414. [DOI] [PubMed] [Google Scholar]
- 24.Jiang B, Li B. J Biomed Mater Res B Appl Biomater. 2009;88B:332–338. doi: 10.1002/jbm.b.31021. [DOI] [PubMed] [Google Scholar]
- 25.Li H, Ogle H, Jiang B, Hagar M, Li B. J Orthop Res. 2010;28:992–999. doi: 10.1002/jor.21115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moskowitz JS, Blaisse MR, Samuel RE, Hsu HP, Harris MB, Martin SD, Lee JC, Spector M, Hammond PT. Biomaterials. 2010;31:6019–6030. doi: 10.1016/j.biomaterials.2010.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shukla A, Fleming KE, Chuang HF, Chau TM, Loose CR, Stephanopoulos GN, Hammond PT. Biomaterials. 2010;31(8):2348–2357. doi: 10.1016/j.biomaterials.2009.11.082. [DOI] [PubMed] [Google Scholar]
- 28.Costerton B, Cook G, Shirtliff M, Stoodley P, Pasmore M. Biofilms, biomaterials, and device-related infections. In: Ratner BD, Hoffman AS, Schoen FJ, Lemons JE, editors. Biomaterials Science: An Introduction to Materials in Medicine. Elsevier Academic Press; California US: 2004. pp. 345–354. [Google Scholar]
- 29.Long PH. Toxicol Pathol. 2008;36:85–91. doi: 10.1177/0192623307310951. [DOI] [PubMed] [Google Scholar]
- 30.Dougherty SH. Rev Infect Dis. 1988;10:1102–1117. doi: 10.1093/clinids/10.6.1102. [DOI] [PubMed] [Google Scholar]
- 31.Wu P, Grainge DW. Biomaterials. 2006;27:2450–2467. doi: 10.1016/j.biomaterials.2005.11.031. [DOI] [PubMed] [Google Scholar]
- 32.Li B, Jiang B, Boyce BM, Lindsey BA. Biomaterials. 2009;30:2552–2558. doi: 10.1016/j.biomaterials.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li B, Jiang B, Dietz MJ, Smith ES, Clovis NB, Rao KMK. J Orthop Res. 2010;28:48–54. doi: 10.1002/jor.20939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jessel N, Atalar F, Lavalle P, Mutterer J, Decher G, Schaaf P, Voegel JC, Ogier J. Adv Mater. 2003;15:692–695. [Google Scholar]
- 35.Berg MC, Zhai L, Cohen RE, Rubner MF. Biomacromolecules. 2006;7:357–364. doi: 10.1021/bm050174e. [DOI] [PubMed] [Google Scholar]
- 36.Werner S, Huck O, Frisch B, Vautier D, Elkaim R, Voegel JC, Brunel G, Tenenbaum H. Biomaterials. 2009;30:2291–2301. doi: 10.1016/j.biomaterials.2009.01.004. [DOI] [PubMed] [Google Scholar]
- 37.Jiang B, Li B. Int J Nanomed. 2009;4:37–53. doi: 10.2147/ijn.s4970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Quinn JF, Caruso F. Langmuir. 2004;20:20–22. doi: 10.1021/la0360310. [DOI] [PubMed] [Google Scholar]
- 39.Qi B, Tong X, Zhao Y. Macromolecules. 2006;39(17):5714–5719. [Google Scholar]
- 40.Zhong Y, Whittington CF, Haynie DT. Chem Commun. 2007;14:1415–1417. doi: 10.1039/b615699a. [DOI] [PubMed] [Google Scholar]
- 41.Wang L, Wang X, Xu MF, Chen DD, Sun JQ. Langmuir. 2008;24:1902–1909. doi: 10.1021/la7031048. [DOI] [PubMed] [Google Scholar]
- 42.Kim BS, Park SW, Hammond PT. ACS Nano. 2008;2(2):386–392. doi: 10.1021/nn700408z. [DOI] [PubMed] [Google Scholar]
- 43.Wood KC, Chuang HF, Batten RD, Lynn DM, Hammond PT. Proc Natl Acad Sci U S A. 2006;103:10207–10212. doi: 10.1073/pnas.0602884103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu XH, Zhang JT, Lynn DM. Adv Mater. 2008;20(21):4148–4153. doi: 10.1002/adma.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim BS, Smith RC, Poon Z, Hammond PT. Langmuir. 2009;25(24):14086–14092. doi: 10.1021/la9017618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Soike T, Streff AK, Guan C, Ortega R, Tantawy M, Pino C, Shastri VP. Adv Mater. 2010;22(12):1392–1397. doi: 10.1002/adma.200903069. [DOI] [PubMed] [Google Scholar]
- 47.Volodkin DV, Petrov AI, Prevot M, Sukhorukov GB. Langmuir. 2004;20:3398–3406. doi: 10.1021/la036177z. [DOI] [PubMed] [Google Scholar]
- 48.Haynie DT, Palath N, Liu Y, Li B, Pargaonkar N. Langmuir. 2005;21:1136–1138. doi: 10.1021/la047833d. [DOI] [PubMed] [Google Scholar]
- 49.Tong WJ, Gao CY, Möhwald H. Chem Mater. 2005;17:4610–4616. [Google Scholar]
- 50.Srivastava S, Ball V, Podsiadlo P, Lee J, Ho P, Kotov NA. J Am Chem Soc. 2008;130:3748–3749. doi: 10.1021/ja7110288. [DOI] [PubMed] [Google Scholar]
- 51.Ball V, Bernsmann F, Werner S, Voegel JC, Piedra-Garza LF, Kortz U. Eur J Inorg Chem. 2009;34:5115–5124. [Google Scholar]
- 52.Salloum DS, Schlenoff JB. Biomacromolecules. 2004;5(3):1089–1096. doi: 10.1021/bm034522t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.