

Abstract
Several chiral primary amines, mainly those derived from the cinchona alkaloids, were evaluated as the organocatalysts for the asymmetric Biginelli reaction. With the quinine-derived amine catalyst 1 and after extensive optimization of the reaction conditions, 3,4-dihydropyrimidin-2(1H)-ones were obtained in moderate to good yields and 51–78% ee from a three-component reaction of aryl and aliphatic aldehydes, urea, and acetoacetate.
Keywords: Biginelli reaction, Catalysis, Enantioselective, Dihydroprimidinone, Multi-component reaction
Introduction
3,4-Dihydropyrimidin-2(1H)-one derivatives (DHPMs) are very important pharmacologically active molecules and have found applications as calcium channel modulators, α1a adrenoceptor-selective antagonists, and inhibitors of the kinesin motor protein and the HIV.[1] Biginelli reaction,[2] which uses a three-component reaction of an aromatic aldehyde, urea, and acetoacetate, is the most efficient method for the assembly of these biologically significant hetereocyclic compounds. Because it has been found that individual enantiomers of a given DHPM may exhibit totally different or even opposite pharmaceutical activities,[3] there has been a lot of interest in developing highly enantioselective syntheses of these DHPM derivatives in recent years.[4] For examples, Zhu and co-workers reported the first highly enantioselective synthesis of DHPMs using a chiral ytterbium Lewis acid catalyst in 2005.[5b] Soon afterwards, Gong and co-workers successfully developed an asymmetric synthesis of these compounds with excellent enantiocontrol using a BINOL-derived Brønsted acid.[5c,d] Besides Lewis acids[5a,b] and Brønsted acid,[5c,d] asymmetric induction in this reaction may also be achieved by using secondary amines as the catalyst and a suitable acid as the cocatalyst.[5e–i] The reported catalysts are mainly proline derivatives.[5e–h] In contrast, although primary amines are also known to participate as a catalyst in reactions that involve the enamine intermediate,[6] to our knowledge, there is no report on using primary amines as the catalyst for the enantioselective synthesis of DHPMs, except for the bisfunctional primary amine thioureas reported most recently by Chen and co-workers[5i] during the progress of our current work. Herein we wish to disclose our study of using primary amines, mainly those derived from the cinchona alkaloids, as the catalyst for the asymmetric synthesis of DHPMs using a three-component Biginelli reaction.
Results and Discussion
According to the previous reports by Feng,[5e] Juaristi,[5f] Wang,[5g] and Lee,[5h] the observed stereoselectivities in the asymmetric synthesis of DHPMs catalyzed by secondary amine catalysts were explained by the enamine activation mechanism.[5e–h] Since primary amines, especially those derived from the cinchona alkaloids, are also highly efficient catalysts for organic reactions involving the enamine intermediates,[6] we reasoned that these compounds should also be good catalysts for the asymmetric synthesis of DHPMs. Thus, we screened some readily available chial primary amine catalysts (1–7, Figure 1) in the three-component reaction of benzaldehyde (8a), urea (9), and ethyl acetoacetate (10) for the asymmetric synthesis of the DHPM derivative 11a. The results of the screening are summarized in Table 1.
Figure 1.
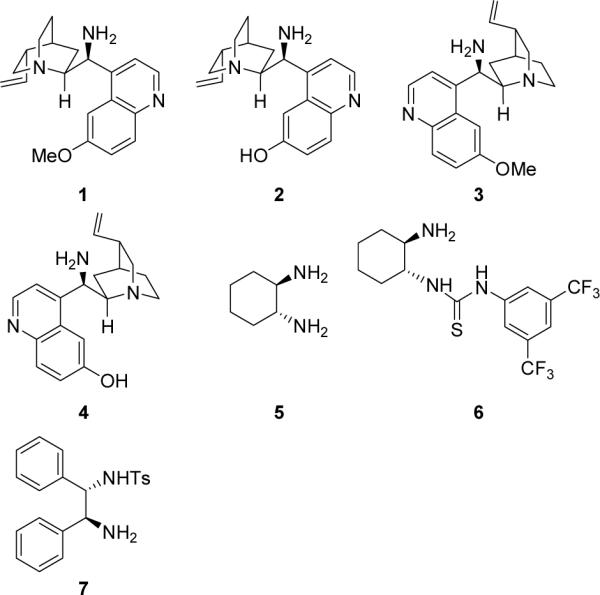
Catalysts screened for the three-component synthesis of DHPM 11a
Table 1.
Screening of the catalysts and optimization of the reaction conditions[a]
| Entry | Catalyst | Acid | Solvent | Time [d] | Yield [%][b] | ee [%][c] |
|---|---|---|---|---|---|---|
| 1 | 1 | HCl | THF | 5 | 64 | 66 |
| 2 | 2 | HCl | THF | 5 | 63 | 50 |
| 3 | 3 | HCl | THF | 5 | 51 | 57[d] |
| 4 | 4 | HCl | THF | 8 | 21 | 40[d] |
| 5 | 5 | HCl | THF | 9 | trace | nd[e] |
| 6 | 6 | HCl | THF | 5 | 21 | 5 |
| 7 | 7 | HCl | THF | 6 | 56 | 3 |
| 8 | 1 | HCl | dioxane | 7 | 80 | 61 |
| 9 | 1 | HCl | CHCl3 | 5 | 60 | 59 |
| 10 | 1 | HCl | CH2Cl2 | 5 | 62 | 50 |
| 11 | 1 | HCl | toluene | 9 | 12 | 37 |
| 12 | 1 | HCl | TFE[f] | 5 | trace | nd[e] |
| 13 | 1 | HCl | CH3CN | 5 | 85 | 40 |
| 14 | 1 | HCl | DMSO | 5 | 83 | 17 |
| 15 | 1 | HCl | acetone | 5 | 43 | 46 |
| 16 | 1 | PhCO2H | THF | 15 | <5 | nd |
| 17 | 1 | 2-NBA[g] | THF | 15 | 13 | 0 |
| 18 | 1 | TFA | THF | 5 | 60 | 44 |
| 19 | 1 | p-TSA | THF | 8 | 75 | 55 |
| 20 | 1 | CF3SO3H | THF | 6 | 71 | 20 |
| 21[h] | 1 | HCl | THF | 7 | 51 | 52 |
| 22[i] | 1 | HCl | THF | 5 | 73 | 61 |
| 23[i,j] | 1 | HCl | THF | 5 | 91 | 62 |
| 24[i,k] | 1 | HCl | THF | 5 | 97 | 64 |
| 25[i,k,l] | 1 [m] | HCl | THF | 6 | 81 | 73 |
| 26[i,k,l] | 1[n,o] | HCl | THF | 6 | 76 | 72 |
Unless otherwise noted, all reactions were conducted with compounds 8a (0.25 mmol), 9 (0.25 mmol), and 10 (0.25 mmol) in the presence of the catalyst (0.025 mmol, 10 mol %) and the acid cocatalyst (0.025 mmol, 10 mol %) in the specified solvent (1.5 mL) at room temperature.
Yield of the isolated product after column chromatography.
Determined by the HPLC analysis on a ChiralCel OD-H column.
The S-enantiomer was obtained as the major product.
Not determined.
2,2,2-Trifluoroethanol.
2-Nitrobenzoic acid.
Conducted with 5 mol % of the catalyst and 5 mol % of the acid cocatalyst.
Conducted with 20 mol % of the catalyst and 20 mol % of the acid cocatalyst.
Conducted with compounds 8a (0.25 mmol), 9 (0.375 mmol), and 10 (0.75 mmol).
Conducted with compounds 8a (0.25 mmol), 9 (0.50 mmol), and 10 (1.25 mmol).
The reaction temperature was 0 °C.
Catalyst 1 was recovered in 93% yield after the reaction.
Carried out with the recovered catalyst 1.
Catalyst 1 was recovered in 98% yield after the reaction.
As shown by the results in Table 1, when the quinine-derived amine 1 was used as the catalyst and HCl as the acid cocatalyst (10 mol % each), the reaction of benzaldehyde (8a), urea (9), and ethyl acetoacetate (10) in THF led to the desired product 11a in 64% yield and 66% ee after 3 days at room temperature (entry 1). The absolute configuration of the major enantiomer was determined to be R by comparing the measured optical rotation with those reported data.[5] The demethylated catalyst 2 yielded this product in a similar yield after a reaction of 5 days at room temperature, and the ee value obtained was slightly lower (entry 2). The quinidine-derived amine 3, which is a pseudo-enantiomer of 1, gave the opposite enantiomer as the major product in 57% ee (entry 3). When a similarly demethylated catalyst 4 was used, the reaction became very sluggish and the ee value obtained was much lower (40%, entry 4). These results indicate that both the reaction rate and enantioselectivity are highly sensitive towards the subtle changes in the catalyst structure. While these catalysts generate only mediocre ee values of the product, 1,2-cyclohexanediamine (5) and its thiourea derivative 6 and sulfonamide derivative 7 proved to be even poorer catalysts for this reaction, because low yields and/or poor ee values of the product were obtained (entries 5–7).
This screening identified the quinine-derived amine 1 as the best catalyst for this reaction. Then we studied the solvent effects on this reaction by using 1 as the catalyst. As revealed in Table 1, a slightly lower ee values of 61% was obtained with 1,4-dioxane (entry 8). When chloroform and dichloromethane were used as the solvent, the ee values obtained were also inferior (59% and 50%, respectively, entries 9 and 10). The reaction is also slower in these three solvents (entries 8–10). Other common organic solvents, such as toluene (entry 11), 2,2,2-trifluoroethanol (entry 12), CH3CN (entry 13), DMSO (entry 14), and acetone (entry 15), all led to poorer ee values of the product. Next different acid cocatalysts were evaluated. Weak acids, such as benzoic acid (entry 16) and 2-nitrobenzoic acid (entry 17), are not effective in promoting the reactivity and the enantioselectivity of this reaction at all. In contrast, good yields of the product were obtained with a stronger acid, such as TFA (entry 18), p-toluenesulfonic acid (entry 19), and trifluoromethanesulfonic acid (entry 20), albeit the ee values obtained were slightly lower with these acid cocatalysts. Thus, THF was identified as the best solvent and HCl was identified as the best acid cocatalyst for this reaction. In order to obtain better yield and enantioselectivity in this reaction, the catalyst loading and ratio of the three substrates were further studied. Dropping the catalyst loading to 5 mol % led to lower yields and ee value of the product (entry 21). On the other hand, increasing the catalyst loading to 20 mol % only resulted in slightly better product yield (entry 22). Nevertheless, we found that much better product yield could be achieved if an excess amount of urea and acetoacetate was used together with a 20 mol % loading of the catalyst. For example, the yield improved to 91% when the molar ratio of 8a/9/10 was changed to 1:1.5:3 (entry 23). The yield was further improved to 97% if this ratio was 1:2:5 (entry 24). Moreover, the ee values obtained for the product remained almost steady in these two cases. Finally, the temperature effects were studied, and it was found that lowering the reaction temperature to 0 ° C can improve the ee value to 73% (entry 25). Further dropping the reaction temperature is impractical since the reaction becomes very sluggish. Although 10 mol % of catalyst 1 has to be used to achieve a good yield of the product, it is possible to recover the catalyst in high yields (93 to 98% yield, entries 25 and 26) during the chromatographic purification of the product. Furthermore, the recycled catalyst shows almost the same reactivity and selectivity as the original catalyst (entry 26).
The scope of this reaction was then evaluated using compound 1 as the catalyst and HCl as the cocatalyst under the optimized conditions (Table 1, entry 25). The results are collected in Table 2. As shown in Table 2, besides benzaldehyde (entry 1), substituted benzaldehydes may also be applied as the substrates in this reaction. The electronic nature of the substituent on the benzene ring was found to have influences on both the reactivity and the enantioselectivity of this reaction. Electron-donating groups, such as methyl or methoxy groups, diminish slightly the reaction yield and the ee values of this reaction as compared to the unsubstituted phenyl group (entries 2–4 vs. 1). In contrast, electron-withdrawing groups on the phenyl ring have no influence on the enantioselectivities (entries 5–10). For example, similar ee values of 72% and 74% were obtained for the strong electron-withdrawing para- and meta-nitro-substituted benzaldehydes, respectively (entries 9 and 10 vs. 1). Nonetheless, while weak electron-withdrawing groups only reduce the reaction yields slightly (entries 5–7), strong electron-withdrawing groups, such as cyano and nitro groups, diminish the yield dramatically (entries 8–10). Although aromatic aldehydes are often studied in the asymmetric synthesis of DHPMs,[5] aliphatic aldehydes are seldom used as the substrates.[5b,c] To our knowledge, aliphatic aldehydes have never been studied in an enamine-mediated asymmetric Biginelli reaction. To test the scope our catalyst, we studied heptanal in our reaction, and found the reaction gave comparable enantioselectivity of the product (72% ee) as aromatic substrate, although the yield of the product is much lower (43%, entry 11).
Table 2.
Three-component reaction of aldehydes, urea, and acetoacetate for the asymmetric synthesis of DHPMs[a]
| Entry | R | Product | Yield [%][b] | ee [%][c] |
|---|---|---|---|---|
| 1 | Ph | 11a | 81 | 73 |
| 2 | 4-MeC6H4 | 11b | 53 | 69 |
| 3 | 4-MeOC6H4 | 11c | 71 | 51 |
| 4 | 2-MeC6H4 | 11d | 53 | 53[d] |
| 5 | 4-FC6H4 | 11e | 61 | 73 |
| 6 | 4-ClC6H4 | 11f | 63 | 76 |
| 7 | 4-BrC6H4 | 11g | 68 | 78 |
| 8 | 4-CNC6H4 | 11h | 20 | 76 |
| 9 | 4-NO2C6H4 | 11i | 14 | 72 |
| 10 | 3-NO2C6H4 | 11j | 21 | 74 |
| 11 | n-C6H13 | 11k | 43 | 72 |
All reactions were conducted with compounds 8 (0.25 mmol), 9 (0.5 mmol), and 10 (1.25 mmol) in the presence of catalyst 1 (0.05 mmol, 20 mol %) and HCl (0.05 mmol, 20 mol %) in THF (1.5 mL) at 0 °C for 6 days.
Yield of the isolated product after column chromatography.
Unless otherwise noted, ee values were determined by the HPLC analysis on a ChiralCel OD-H column.
Determined by the HPLC analysis on a ChiralPak AD-H column.
On the basis of the mechanism proposed for the secondary-amine-catalyzed synthesis of DHPMs,[5e–h] we believe the present reaction also works through a dual activation mechanism realized by the quinine amine catalyst 1. The formation of the R-configured product may be interpreted by the proposed transition state in Scheme 1. As shown in Scheme 1, the imine formed between benzaldehyde and urea is hydrogen-bonded to the protonated quinuclidine backbone of catalyst 1. Such a hydrogen bond not only activates the imine for a nucleophilic attack, but also helps bring the imine closer to the reaction center and limit its possible orientations. The primary amine group on the side chain of the catalyst activates acetoacetate through the formation of an enamine. Attack of this enamine onto the Si face of the imine gives an intermediate, which, after hydrolysis, intramolecular cyclization, and dehydration reaction, yields the observed R-configured product 11a.
Scheme 1.
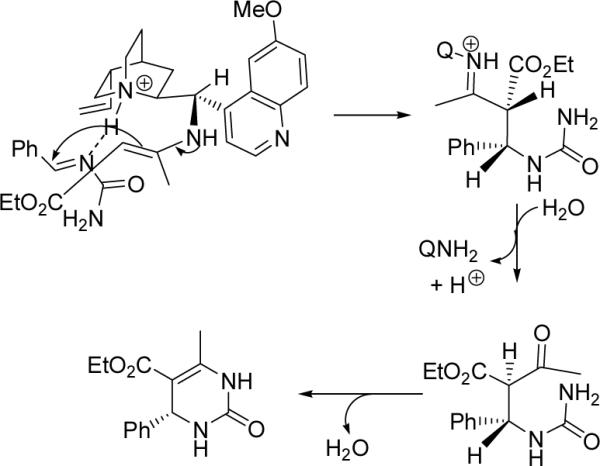
Plausible transition state for the formation of the R-configured enantiomer (QNH2 = catalyst 1).
Conclusions
We have demonstrated that chiral primary amines, such as the amine derived from quinine, may be used the catalyst for the three-component reaction of aldehydes, urea, and acetoacetate in the enantioselective synthesis of 3,4-dihydropyrimidin-2(1H)-ones (DHPMs). Under the optimized conditions, the corresponding DHPMs may be obtained in moderate to good yields and good ee values (up to 78% ee).
Experimental Section
General
1H NMR spectra were obtained on a Varian INOVA 500 MHz or a GE 300 MHz spectrometers using residual solvent as the standard. TLC was performed with silica gel GF254 precoated on aluminum plates, and spots were visualized with UV and/or iodine vapor. Flash column chromatography was performed on silica-gel. HPLC analysis was performed on a Shimadzu instrument with LC-20AT pump and SPD-20AV UV-Vis detector. ChiralCel and ChiralPak HPLC columns were purchased from Daicel Chemical Industry, Ltd. Compounds used in this study were purchased from Aldrich, Alfa-Aesar, Acros, TCI, or Strem, and were used as received. Toluene, CH2Cl2, CHCl3, and CH3CN were distilled from CaH2. THF was freshly distilled from benzophenone and sodium metal. DMSO was dried over molecular sieves. Catalysts 1,[8] 2,[9] 3,[8] 4,[9] 6,[10] and 7[11] were synthesized according to the reported procedures. Unless otherwise specified, all reactions were carried out at ambient temperature in oven-dried glassware.
General procedure for the three component reaction of aldehyde, urea and acetoacetate
To a mixture of the aldehyde (0.25 mmol) and urea (30.0 mg, 0.5 mmol) in THF (1.5 mL) at 0 °C was added ethyl acetoacetate (162.7 mg, 1.25 mmol), catalyst 1 (16.2 mg, 0.05 mmol) and HCl (4.0 M in dioxane, 13 μL, 0.05 mmol). The mixture was further stirred for 6 days at this temperature. Then the reaction mixture was directly transferred to a silica gel column and purified by column chromatography (1:1 hexane/ethyl acetate) to afford the DHPM products. All the products are known compounds and have identical spectroscopic data as those reported.[5b,c]
Catalyst recovering
Once the DHMP product was seperated, the column was flushed with 1:2:4 Et3N/CH3OH/EtOAc as the eluent to isolate catalyst 1 (as its HCl salt, Rf = 0.62). The fractions were combined and the solvents were removed under reduced pressure. The residue was disolved in CH2Cl2 (10 mL), washed with aq. NaHCO3 (5 mL), and dried over Na2SO4. The solvent was removed to afford the catalyst (15.0 mg, 93% recoevery).
(R)-5-Ethoxycarbonyl-4-(n-hexyl)-6-methyl-3,4-dihydropyrimidin- 2(1H)-one (11k)[7,12]
Yield 28.8 mg, 43%, white solid, m.p. 126–128 °C. [α]D23 = +40.0 (c 1.02, MeOH). 1H NMR (500 MHz, CDCl3): δ 8.04 (s, br, 1H), 5.75 (s, br, 1H), 4.29–4.32 (m, 1H), 4.14–4.24 (m, 2H), 2.29 (s, 3H), 1.49–1.60 (m, 2H), 1.23–1.43 (m, 1H), 0.87–0.92 (m, 3H) ppm.
Supplementary Material
Acknowledgments
Generous financial support of this research from the National Institutes of Health-National Institute of General Medical Sciences (Grant no. SC1GM082718) and the Welch Foundation (Grant No. AX-1593) is gratefully appreciated.
Footnotes
Supporting information for this article is available on the WWW under http://www.eurjoc.org/ or from the author.
Supporting Information: 1H NMR data and spectra of the products; HPLC conditions and chromatograms.
References
- [1].For reviews, see: Kappe CO. Eur. J. Med. Chem. 2000;35:1043–1052. doi: 10.1016/s0223-5234(00)01189-2.; Heys, Moore CG, Murphy PJ. Chem. Soc. Rev. 2000;29:57–67.
- [2].a) Biginelli P. Chem. Ber. 1891;24:1317–2962. [Google Scholar]; b) Biginelli P. Chem. Ber. 1893;26:447. [Google Scholar]; c) Biginelli P. Gazz. Chim. Ital. 1893;23:360. [Google Scholar]
- [3].For an example, see: Atwal KS, Swanson BN, Unger SE, Floyd DM, Moreland S, Hedberg A, O'Reilly BC. J. Med. Chem. 1991;34:806–811. doi: 10.1021/jm00106a048.
- [4].For a review, see: Gong L-Z, Chen X-H, Xu X-Y. Chem. Eur. J. 2007;13:8920–8926. doi: 10.1002/chem.200700840.
- [5].For examples, see: Muñoz-Muñiz O, Juaristi E. Arkivoc. 2003;xi:16–26.; Huang Y, Yang F, Zhu C. J. Am. Chem. Soc. 2005;127:16386–16387. doi: 10.1021/ja056092f.; Li N, Chen X-H, Song J, Luo S-W, Fan W, Gong L-Z. J. Am. Chem. Soc. 2009;131:15301–15310. doi: 10.1021/ja905320q.; Chen X-H, Xu X-Y, Liu H, Cun L-F, Gong L-Z. J. Am. Chem. Soc. 2006;128:14802–14803. doi: 10.1021/ja065267y.; Xin J, Chang L, Hou Z, Shang D, Liu X, Feng X. Chem. Eur. J. 2008;14:3177–3181. doi: 10.1002/chem.200701581.; Gonzalez-Olvera R, Demare P, Regla I, Juaristi E. Arkivoc. 2008;vi:61–72.; Wu Y-Y, Chai Z, Liu X-Y, Zhao G, Wang S-W. Eur. J. Org. Chem. 2009:904–911.; Sohn J-H, Choi H-M, Lee S, Joung S, Lee H-Y. Eur. J. Org. Chem. 2009:3858–3862.; Wang Y, Yang H, Yu J, Miao Z, Chen R. Adv. Syn. Catal. 2009;351:3057–3062.
- [6].For selected examples of primary amine-catalyzed enantioselective reactions involving an enamine or iminium intermediate, see: Reisinger CM, Wang X, List B. Angew. Chem. 2008;120:8232–8235.;,Angew. Chem. Int. Ed. 2008;47:8112–8115. doi: 10.1002/anie.200803238.; Martin NJA, List B. J. Am. Chem. Soc. 2006;128:13368–13369. doi: 10.1021/ja065708d.; Lu X, Liu Y, Sun B, Cindric B, Deng L. J. Am. Chem. Soc. 2008;130:8134–8135. doi: 10.1021/ja802982h.; Wang X, Reisinger CM, List B. J. Am. Chem. Soc. 2008;130:6070–6071. doi: 10.1021/ja801181u.; McCooey SH, Connon SJ. Org. Lett. 2007;9:599–602. doi: 10.1021/ol0628006.; Liu T-Y, Cui H-L, Zhang Y, Jiang K, Du W, He Z-Q, Chen Y-C. Org. Lett. 2007;9:3671–3674. doi: 10.1021/ol701648x.; Zheng B-L, Liu Q-Z, Guo C-S, Wang X-L, He L. Org. Biomol. Chem. 2007;5:2913–2915. doi: 10.1039/b711164a.; Zhou J, Wakchaure V, Kraft P, List B. Angew. Chem. 2008;120:7768–7771. doi: 10.1002/anie.200802497.; Angew. Chem. Int. Ed. 2008;47:7656–7658. doi: 10.1002/anie.200802497.; Bartoli G, Melchiorre P. Synlett. 2008:1759–1772.; Chen Y-C. Synlett. 2008:1919–1930.; Gogoi S, Zhao C-G, Ding D. Org. Lett. 2009;11:2249–2252. doi: 10.1021/ol900538q.; Mandal T, Zhao C-G. Angew. Chem. 2008;120:7828–7831.; Angew. Chem. Int. Ed. 2008;47:7714–7717. doi: 10.1002/anie.200803236.; Xie J-W, Chen W, Li R, Zeng M, Du W, Yue L, Chen Y-C, Wu Y, Zhu J, Deng J-G. Angew. Chem. 2007;119:393–396. doi: 10.1002/anie.200603612.; Angew. Chem. Int. Ed. 2007;46:389–392.; Huang H, Jacobsen EN. J. Am. Chem. Soc. 2006;128:7170–7171. doi: 10.1021/ja0620890. For a review on primary amine catalysis, see: Peng F, Shao Z. J. Mol. Catal. A. 2008;285:1–13.
- [7].a) Ranu BC, Hajra A, Jana U. J. Org. Chem. 2000;65:6270–6272. doi: 10.1021/jo000711f. [DOI] [PubMed] [Google Scholar]; b) Folkers K, Harwood HJ, Johnson TB. J. Am. Chem. Soc. 1932;54:3751–3758. [Google Scholar]
- [8].a) Brunner H, Bügler J, Nuber B. Tetrahedron: Asymmetry. 1995;6:1699. [Google Scholar]; b) Brunner H, Schmidt P. Eur. J. Org. Chem. 2000:2119. [Google Scholar]
- [9].Chen W, Du W, Duan Y-Z, Wu Y, Yang S-Y, Chen Y-C. Angew. Chem. 2007;119:7811–7814. [Google Scholar]; Angew. Chem. Int. Ed. 2007;46:7667–7670. doi: 10.1002/anie.200702618. [DOI] [PubMed] [Google Scholar]
- [10].Procuranti B, Connon SJ. Chem. Commun. 2007:1421. doi: 10.1039/b618792g. [DOI] [PubMed] [Google Scholar]
- [11].Ng K, Somanathan R, Walsh PJ. Tetrahedron: Asymmetry. 2001;12:1719–1722. [Google Scholar]
- [12].Yadav JS, Reddy BVS, Reddy KB, Raj KS, Prasad AR. J. Chem. Soc., Perkin Trans. 2001;1:1939–1941. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.