

Abstract
The present study was performed to investigate the effect of acidosis on the efflux of ATP from skeletal muscle. Infusion of lactic acid to the perfused hindlimb muscles of anaesthetised rats produced dose-dependent decreases in pH and increases in the interstitial ATP of extensor digitorum longus (EDL) muscle: 10 mm lactic acid reduced the venous pH from 7.22 ± 0.04 to 6.97 ± 0.02 and increased interstitial ATP from 38 ± 8 to 67 ± 11 nm. The increase in interstitial ATP was well-correlated with the decrease in pH (r2 = 0.93; P < 0.05). Blockade of cellular uptake of lactic acid using α-cyano-hydroxycinnamic acid abolished the lactic acid-induced ATP release, whilst infusion of sodium lactate failed to depress pH or increase interstitial ATP, suggesting that intracellular pH depression, rather than lactate, stimulated the ATP efflux. Incubation of cultured skeletal myoblasts with 10 mm lactic acid significantly increased the accumulation of ATP in the bathing medium from 0.46 ± 0.06 to 0.76 ± 0.08 μm, confirming the skeletal muscle cells as the source of the released ATP. Acidosis-induced ATP efflux from the perfused muscle was abolished by CFTRinh-172, a specific inhibitor of the cystic fibrosis transmembrane conductance regulator (CFTR), or glibenclamide, an inhibitor of both KATP channels and CFTR, but it was not affected by atractyloside, an inhibitor of the mitochondrial ATP transporter. Silencing of the CFTR gene using an siRNA abolished the acidosis-induced increase in ATP release from cultured myoblasts. CFTR expression on skeletal muscle cells was confirmed using immunostaining in the intact muscle and Western blotting in the cultured cells. These data suggest that depression of the intracellular pH of skeletal muscle cells stimulates ATP efflux, and that CFTR plays an important role in the release mechanism.
Introduction
It is known that the interstitial ATP concentration increases during muscle contractions (Hellsten et al. 1998; Mo & Ballard, 2001; Li et al. 2003), and that ATP is converted extracellularly to adenosine (Cheng et al. 2000), which contributes a large part to the exercise hyperaemia (Kille & Klabunde, 1984). The mechanism by which the muscle contractions could give rise to an increase in interstitial ATP remains unknown; however, it is well-known that muscle contractions decrease the muscle pH (Bangsbo et al. 1993; Street et al. 2001), and our previous studies showed that depression of muscle pH led to the appearance of both adenosine (Mo & Ballard, 2001) and AMP (Cheng et al. 2000) in the venous effluent. The present experiments were performed to test directly whether a localised decrease in muscle pH would stimulate the efflux of ATP into the interstitial space. Therefore, we measured the interstitial ATP concentration of extensor digitorum longus (EDL) muscle before, during and after an acidosis challenge, produced by infusion of lactic acid in vivo, as well as the appearance of ATP in the bathing medium of cultured skeletal myoblasts in the absence or presence of lactic acid.
The second purpose of our experiments was to investigate the mechanism by which ATP left the skeletal muscle cells during pH depression. The extracellular concentration of ATP normally remains very low because of the high ectonucleotidase activity on skeletal muscle cells (Gordon, 1986). The membrane permeability to ATP is also very low, due to its large molecular size and charge (Gordon, 1986), suggesting that a specific transport mechanism must be involved in order to allow ATP to enter the interstitial space quickly enough to elevate its concentration there. The predominant mechanism(s) that permit ATP to cross the skeletal muscle cell membrane have not been investigated previously.
The three main possibilities for the mechanism by which ATP crosses the cell membrane are an ATP transporter, exocytosis or efflux through an ATP channel or pore. The only known ATP transporters are those responsible for the translocation of ATP between intracellular compartments, such as the mitochondrial ATP transporter (Fiermonte et al. 2004) or the antiporter which conducts ATP into the endoplasmic reticulum and Golgi apparatus (Hirschberg et al. 1998). There have been no previous reports that these transporters are expressed on cell membranes, or that they could mediate the translocation of ATP across surface membranes. On the other hand, it is theoretically possible that the mitochondrial ATP transporter could contribute to a localised high concentration of ATP near the surface membrane of the muscle cell, which could drive the efflux of ATP from the cell. We therefore tested the effects of atractyloside, the inhibitor of the mitochondrial ATP transporter, on the acidosis-induced ATP efflux from the muscle cells.
Exocytosis is responsible for the release of ATP as a cotransmitter from nerves (Burnstock, 2006) or as a component of secretory granules (Hutton, 1989); vesicular exocytosis is also proposed to account for the shear-stress-induced ATP release from vascular endothelial cells (Bodin & Burnstock, 2001). We are not aware of any reports of ATP-containing vesicles or secretory granules in skeletal muscle cells, and we have not investigated this possibility in the current study.
There have been several reports over the last 10–15 years that ABC-proteins, such as P-glycoprotein or the cystic fibrosis transmembrane conductance regulator (CFTR), can function as pores that permit the efflux of ATP from cells (Abraham et al. 1993; Reisin et al. 1994; Prat et al. 1996; Cantiello et al. 1998; Schweibert, 1999). Similarly, connexins have been reported to function as ATP pores in cells such as astrocytes (Stout et al. 2002), and ATP release through these gap junction proteins is thought to be involved in the propogation of intercellular Ca2+ waves (Cotrina et al. 1998). Pannexins, another group of gap-junction proteins, are reported to form ATP release channels in erythrocytes and other cells (Lucovei et al. 2006; Dubyak, 2009), whilst, under certain experimental conditions, either volume-sensitive outwardly rectifying Cl− channels (VSORs) or maxi-anion channels can be demonstrated to conduct ATP (Bell et al. 2003; Sabirov & Okada, 2005). ABC proteins, and particularly CFTR, have also been reported to regulate the permeability of separate channels in the cell membrane (Guggino, 2004). It has been proposed that CFTR regulates the function of a separate (as yet unidentified) ATP channel (Braunstein et al. 2001). We investigated the contribution of CFTR to the acidosis-induced release of ATP from skeletal muscle using both specific and non-specific pharmacological inhibitors; since there had been no previous reports of the expression of CFTR on skeletal muscle, we also determined the expression of CFTR on intact skeletal muscle using immunohistochemistry and on cultured skeletal myoblasts using Western blot. Finally, we showed that the acidosis-induced release of ATP from skeletal muscle was abolished after RNA interference had been employed to silence the CFTR gene.
Methods
Surgical preparation
All procedures used in this study were approved by the University of Hong Kong Committee on the Use of Live Animals in Teaching and Research. Sprague–Dawley male rats (weight, 300 to 350 g) were anaesthetised by an intraperitoneal injection of pentobarbital sodium (60–70 mg kg−1, Sagatal, RMB, Animal Health Ltd, Dagenham, UK). A Y-shaped cannula was inserted in the trachea through a midline incision to the neck, in order to maintain the airways patent, and the animals were allowed to breathe spontaneously. A catheter was inserted into the external jugular vein for maintenance of the anaesthesia by bolus injections of pentobarbital sodium: supplementary doses of anaesthetic were administered whenever a withdrawal reflex to toe-pinch could be elicited. Body temperature was maintained between 37.5 and 38.5°C by a heating pad and an external heat lamp. The abdominal aorta was cannulated in order to perfuse the arterial supply of the rat hindlimb muscles with buffer. The inferior vena cava was also cannulated to allow venous drainage and for the venous pH measurement. Hindlimb muscles were perfused at a flow rate of 1.5 ml min−1 for 90 min prior to the experiments using a modified Krebs–Henseleit (K-H) bicarbonate buffer containing 2 g l−1 glucose, 59 g l−1 dextrans (MW 60,000–90,000), 1 g l−1 albumin and 100 μl l−1 antifoam A (Sigma, St Louis, MO, USA) equilibrated against 95% O2–5% CO2 to give a pH of 7.4. When all experimental procedures had been completed, animals were killed by intravenous injection of an overdose of sodium pentobarbital.
Microdialysis
One of the hindlimbs was skinned down to the ankle so that the extensor digitorum longus (EDL) muscle was exposed. A microdialysis probe (LM10, Bioanalytical Systems, West Lafayette, IN, USA) was inserted longitudinally into the main body of the EDL muscle using a fine surgical needle, and was perfused at 2 μl min−1 with a fluid of similar composition to the interstitial fluid (NaCl 142 mm, KCl 4 mm, CaCl2 2.4 mm, MgSO4 0.5 mm, NaH2PO4 1.2 mm, glucose 5.6 mm). The exposed muscle was wrapped in plastic film in order to prevent dehydration. Probes were perfused for 90 min prior to sample collection. The dialysate was collected in ice-cooled vials for HPLC analysis. The probe dialysate was protein free, and purification was not required before HPLC analysis. Samples of dialysate were analysed by HPLC, using 15–30 μl dialysate mixed with 105–90 μl ultrapure water. Measured concentrations were corrected for the dilution factor.
ATP analysis
ATP in the samples was analysed by ion-pair reverse-phase HPLC as previously described (Mo & Ballard, 2001). Samples were chromatographed on a 25 cm × 4.6 mm column, packed with 3 μm particles of LC-18T (Supelco, Bellefonte, PA, USA). The mobile phase consisted of solution A (0.1 m KH2PO4, 0.004 m tetrabutyl ammonium chloride, pH 6.0) and solution B (20% methanol in solution A, pH 5.5). Adenine nucleotides were eluted with a solvent B gradient from 7 to 65%, and the peak of ATP appeared in the phase of 65% solvent B, run from 32 to 45 min. The solvent flow rate was 1 ml min−1. The peak of ATP was identified by comparison of its retention time to the retention times of the standard and the ‘spiked’ sample, in which a certain ratio of the standard and the dialysate sample were mixed, and also by comparison of its absorption spectrum to that of the standard. ATP concentration was quantified using the area under the peak.
Culture of rat skeletal muscle myoblasts
Rat L6 stable skeletal muscle myoblasts were bought from ATCC (Manassas, VA, USA). Cells were cultured in a complete culture medium (ATCC-formulated Dulbecco's modified Eagle's medium supplemented with fetal bovine serum to a final concentration of 10% and penicillin to a final concentration of 1%) at 37°C in a humidified 5% CO2 in air atmosphere.
Experimental procedures
Effect of lactic acid on interstitial ATP of perfused rat muscle
In the first group of 15 rats, after all the surgical procedures had been completed, the muscles were perfused with pH 7.4 buffer for a 90 min stabilization period. Then, a localised decrease in the muscle pH was induced by infusion of graded doses of lactic acid (0, 2.5, 5 or 10 mm) into the arterial supply of the muscle: lactic acid readily permeates the cells, and thus depresses both the intracellular and extracellular pH. Each lactic acid concentration was infused for 20 min, during which two 10 min samples of interstitial microdialysate were collected for ATP analysis, and samples of the venous effluent were collected for pH measurement at 5 and 15 min of perfusion. In a second group of seven rats, this experiment was repeated in the presence of 2 mmα-cyano-4-hydroxycinnamic acid (CHCA), an inhibitor of the cellular uptake of lactic acid.
Effect of sodium lactate on interstitial ATP of perfused rat muscle
In order to distinguish whether ATP output was stimulated by pH depression, or by a specific effect of the infused lactate, the responses to infusion of 10 mm lactic acid or 10 mm sodium lactate were compared in a separate group of eight rats. The muscles were perfused with pH 7.4 buffer for a 20 min control period, then lactic acid (10 mm) was infused for 20 min. A 20 min recovery period was allowed, then sodium lactate (10 mm) was infused for a further 20 min, followed by another 20 min control period. During each 20 min perfusion, two 10 min samples of interstitial microdialysate were collected for ATP determination, and venous effluent samples were collected for pH measurement at 5 and 15 min.
Effect of inhibitors on lactic acid-induced ATP efflux from perfused rat muscle
The lactic acid infusion protocol was performed twice in each of three separate groups of six to eight rats, first in the absence and then in the presence of one of the following inhibitors: atractyloside (20 μm), an inhibitor of the mitochondrial ATP transporter (Wilson & Asimakis, 1987; Roux et al. 1996); glibenclamide (200 μm), a KATP channel blocker which also acts as a non-specific blocker of CFTR (Sheppard & Robinson, 1997; Yamazaki & Hume, 1997); CFTRinh-172 (20 μm), a specific inhibitor of CFTR (Ma et al. 2002). Two 10 min samples of interstitial microdialysate were collected for ATP analysis during the control period and during each lactic acid infusion.
Immunohistochemical determination of CFTR expression on rat skeletal muscle cells
Preparation of skeletal muscle tissue sections
EDL muscles were removed from three rats. The tissues were cut to a size no larger than 3 mm thick, and fixed in 4% paraformaldehyde (Sigma) for 24 h. The tissues were then processed through gradient alcohol followed by xylene and finally embedded in paraffin. The paraffin blocks were sectioned to a thickness of 5 μm on a microtome, and the sections floated on a 40°C water bath containing distilled water. Finally, the sections were transferred onto a SuperfrostPlus microscope slide at room temperature to dry.
Detection of CFTR by immunostaining
Immunostaining of the CFTR was carried out using methods similar to those described by Ajonuma et al. (2005). The sections dried onto Superfrost microscope slides were deparaffinized in xylene, rehydrated in graded ethanol and rinsed twice in phosphate-buffered saline (PBS). The following sequence of treatments was applied, with two 5 min washes in PBS between each treatment: H2O2 (3% in PBS) for 15 min (to block the endogenous peroxidase activity); sodium citrate buffer (pH 6.0) for 30 min (for antigen retrieval); normal goat serum for 20 min (to block non-specific sites), followed by rabbit anti-human polyclonal antibody to residues 55–63 of CFTR (1: 400 v/v in buffer) (Cell Signaling Technology, Inc., Danvers, MA, USA) at 2.5 μg ml−1 for 60 min; biotinylated secondary anti-rabbit antibody (goat-on-rabbit kit, Vector Laboratories, Inc., Burlingame, CA, USA) for 30 min; avidin–biotin complex reagents for 30 min; and finally, 3,3′-diaminobenzendine (DAB) reagent (Vector Laboratories) was added onto the sections. After checking the slides for a brown positive signal, the reactions were stopped by washing slides in tap water. Sections were counterstained with Harris haematoxylin for cell nucleolus staining, dehydrated, and mounted in Depex. The experiment was repeated three times in tissues from three rats. Negative controls were performed for all tissue sections by using normal goat serum (1:400 v/v in buffer) and omitting the primary antibody. The expression of CFTR in sections from rat intestine was also determined using the same method, as a positive control.
Effect of lactic acid on CFTR expression by, and ATP release from, cultured rat skeletal myoblasts
L6 cells (1.5–6 × 105 cells per well) were seeded in a six-well plate in 1 ml of culture medium containing serum and antibiotics; a further 2 ml per well of complete growth medium was added, and the plates were incubated for 48 h in order to allow the cells to grow to confluence. The cell layer was then rinsed briefly with PBS to remove all traces of serum, and 1 ml fresh medium without serum was added to each well of the plate. Lactic acid, at a final concentration of 10 mm, was included in the medium added to half of the wells, and the cells were incubated at 37°C in a 5% CO2 in air atmosphere for 3 h. Then, 200 μl bathing medium was collected into a pre-cooled vial and the cells were collected and blotted for CFTR protein expression using the Western-blotting technique. The samples of bathing medium were stored at −70°C until analysis; after thawing, samples were centrifuged at 4750 g for 5 min, and the supernatants transferred to new pre-cooled vials on ice. The adenosine and ATP concentrations in the supernatants were then analysed by HPLC.
Effect of silencing the CFTR gene using RNA interference on lactic acid-induced ATP release from cultured rat skeletal myoblasts
siRNA protocol for silencing the CFTR gene
Shortly before transfection, (1.5–6) × 105 L6 cells per well were seeded in a six-well plate in 1 ml of culture medium containing serum and antibiotics, and another 2 ml per well of complete growth medium was added.
Two separate siRNAs, Abcc5 and Abcc8 (which target the sequences of the Cftr gene AAATTCTAAAGTGAGAGGAGC and AACCACTCGGCCGCCTACCGG, respectively), were used to suppress the CFTR expression, in order to guard against non-specific (false-positive) results. The transfection reagents (3 μl HiPerFect Transfection Reagent (Qiagen, Valencia, CA, USA), 82 μl culture medium without serum and antibiotics, and 15 μl of 5 μm siRNA (Rn_Abcc5_1 siRNA, or Rn_Abcc8_5 siRNA, Qiagen)) were mixed together and incubated for 5–10 min at room temperature to allow the formation of transfection complexes, and then the complexes were added drop-wise onto the cells in the six-well plates, giving a final siRNA concentration of 5 nm. The plates were gently swirled to ensure uniform distribution of the transfection complexes, and the cells were incubated with the transfection complexes for 48 h under normal growth conditions without changing the medium.
As a control, two separate groups of L6 cells were transfected with either Rn_Mapk1, an siRNA which is known to efficiently knock down rat MAPK1, or a nonsense siRNA. These cells were then subjected to identical treatments to those transfected with the CFTR siRNA.
Effect of lactic acid on ATP release from cultured myoblasts following siRNA transfection
The cells had grown to confluence by the end of the 48 h incubation period. Full growth medium was replaced by serum-free medium with or without 10 mm lactic acid, and the transfected cells were maintained at 37°C in a 5% CO2 in air atmosphere for 3 h. At the end of the incubation period, 200 μl of incubation medium was removed for HPLC determination of ATP and adenosine as described above, and the cells were collected and blotted for CFTR protein expression using the Western-blotting technique.
Monitoring of gene silencing at the level of protein expression using Western blotting
Cells were lysed, and proteins denatured using standard methods, then samples were loaded onto the 10% SDS-PAGE separating gel and run at a constant voltage of 130 V for 2.5 h. Transblotting onto PVDF membrane was performed at constant voltage of 30 V overnight at 4°C. The levels of CFTR protein were quantified using rabbit anti-rat CFTR (Cell Signaling), followed by horseradish peroxidase (HRP)-conjugated monkey anti-rabbit IgG. Colour development was achieved by using the ECL Western blotting detection kit (GE Healthcare Biosciences, Piscataway, NJ, USA). Total protein was determined using the Bio-Rad protein assay kit, which is based on the Bradford assay.
Chemicals
All chemicals were purchased from Sigma. Stock standard solutions of ATP (disodium salt), adenosine, methyladenosine, AMP and ADP were dissolved in ultrapure water. Stock solutions of the other pharmacological agents, CHCA, atractyloside, glibenclamide and CFTRinh-172, were prepared in dimethyl sulphoxide (DMSO). The final DMSO concentration did not exceed 1 μl per ml of the perfusion buffer.
Statistical analysis
Data from the in vivo experiments are expressed as means ± s.e.m., and n represents the number of animals used. One-way repeated measures ANOVA was performed in order to compare multiple treatments within the same group of animals. Data from the in vitro experiments are also expressed as means ± s.e.m., and n represents the number of tests. Student's t test was used to compare two treatments, and one-way ANOVA followed by Dunnett's post hoc test was used for multiple comparisons. In all cases significance was established at P < 0.05.
Results
Effect of lactic acid on interstitial ATP of perfused rat muscle
We investigated whether a localised decrease in muscle pH, induced by infusion of lactic acid into the arterial supply of the muscle, would stimulate the efflux of ATP into the interstitial space.
pH depression by infusion of graded doses of lactic acid
Rat EDL muscles were perfused for 20 min with K-H buffer at pH 7.40, during which the venous pH was stable at 7.22 ± 0.04. This was followed by the infusion of lactic acid at concentrations of 2.5, 5 and then 10 mm: 2.5 mm lactic acid slightly reduced buffer pH to 7.11 ± 0.02, but did not significantly reduce venous pH; 5 and 10 mm lactic acid reduced the buffer pH to 6.82 ± 0.01 and 6.41 ± 0.01, respectively, producing significant dose-dependent decreases in the venous pH (Fig. 1A). When the lactic acid infusion was stopped, and the muscle was again perfused with K-H buffer, the venous pH had returned to a value not significantly different from the precontrol after 15 min recovery (Fig. 1A).
Figure 1. The effect of lactic acid (2.5, 5 or 10 mm) on the pH of the venous effluent (A) and the interstitial ATP concentration (B) of rat EDL muscle.

Data are means ± s.e.m., n = 11–15; *P < 0.05 vs. control. The decrease in the venous pH was significantly correlated with the increase in the interstitial ATP concentration (C).
Relationship between pH and interstitial ATP of EDL muscle
The effects of lactic acid infusion on the interstitial ATP concentration are shown in Fig. 1B: interstitial ATP was significantly increased by both 10 and 20 min infusion of lactic acid (5 and 10 mm). Interstitial ATP remained significantly elevated during the first 10 min of post-control, but returned to a value not significantly different from the pre-control between 10 and 20 min after lactic acid infusion was stopped (Fig. 1B). The relationship between venous pH and interstitial ATP output is shown in Fig. 1C: the increase in interstitial ATP was well correlated with the decrease in venous pH (r2 = 0.93; P < 0.05).
Effect of lactic acid in the presence of lactate transporter inhibitor CHCA on interstitial ATP
Infusion of the lactate transporter inhibitor (CHCA, 2 mm) did not significantly alter the interstitial ATP concentration (Fig. 2). However, the presence of CHCA prevented the 5 mm or 10 mm lactic acid infusion-induced increases in interstitial ATP concentrations: the interstitial ATP concentration remained at its pre-control level throughout the infusion of lactic acid at 2.5, 5 or 10 mm, suggesting that cellular uptake of lactic acid was necessary for the stimulation of ATP efflux.
Figure 2. The effect of α-cyano-4-hydroxycinnamic acid (CHCA; 2 mm) on the interstitial ATP concentration of rat EDL muscle before (Control), during or after (Recov) the addition of 2.5, 5 or 10 mm lactic acid to the perfusion medium.

Data are means ± s.e.m., n = 7. None of the ATP concentrations differed significantly from control.
Effect of sodium lactate on interstitial ATP of perfused rat muscle
In order to distinguish whether ATP output was stimulated by pH depression, or by a specific effect of the infused lactate, the responses to infusion of 10 mm lactic acid or 10 mm sodium lactate were compared: lactic acid infusion significantly reduced both the perfusion buffer pH (to 6.41 ± 0.01) and the venous pH (Fig. 3A), whereas sodium lactate infusion did not alter either buffer pH (7.39 ± 0.01) or venous pH (Fig. 3A). The interstitial ATP concentration was significantly increased during lactic acid infusion (Fig. 3B), but it returned to control during the 20 min post-control period. Sodium lactate infusion, on the other hand, failed to significantly alter the interstitial ATP (Fig. 3B).
Figure 3. Comparison of the effects of 10 mm lactic acid and 10 mm sodium lactate on the pH of the venous effluent (A) and the interstitial ATP concentration (B) of rat EDL muscle.

Data are means ± s.e.m., n = 6–8; *P < 0.05 vs. control.
Effect of inhibitors on lactic acid-induced ATP efflux
The mechanism(s) accounting for the increase in interstitial ATP concentration during acidosis challenge were first explored by the application of inhibitors for transporters or ion channels that have been reported to contribute to the efflux of ATP from some cells.
Mitochondrial ATP transporter inhibitor: atractyloside
Infusion of an inhibitor of the mitochondrial ATP transporter, atractyloside (20 μm), did not alter the basal (without lactic acid infusion) ATP concentration (Fig. 4A). Atractyloside failed to block the lactic acid infusion-induced increase in interstitial ATP: in the presence of atractyloside, the interstitial ATP concentration during 5 mm or 10 mm lactic acid infusion was significantly higher than the control level (Fig. 4A).
Figure 4. The effect of atractyloside (20 μm; panel A), glibenclamide (0.2 mm; panel B) or a specific inhibitor of CFTR, CFTRinh-172 (20 μm; C) on the interstitial ATP concentration in rat EDL muscle before (Control), during or after (Recov) the addition of 2.5, 5 or 10 mm lactic acid to the perfusion medium.

Data are means ± s.e.m., n = 7 (atractyloside series), 6 (glibenclamide series) or 8 (CFTRinh-172 series). *P < 0.05 vs. pre-drug control.
KATP channel blocker: glibenclamide
Similarly, administration of glibenclamide (200 μm), a blocker of KATP channels and CFTR, for 20 min did not alter the basal interstitial ATP concentration of EDL muscle (Fig. 4B). However, in the presence of glibenclamide, the interstitial ATP accumulation in response to lactic acid infusion was abolished (Fig. 4B).
Specific inhibitor of CFTR: CFTRinh-172
Glibenclamide has been reported to act as a non-specific blocker of CFTR (Yamazaki & Hume, 1997). Therefore, we used a specific inhibitor of CFTR to determine whether it was involved in lactic acid infusion-induced ATP release. Figure 4C shows that infusion of CFTRinh-172 (20 μm) for 20 min did not alter the basal level of interstitial ATP but it did block the lactic acid infusion-induced increase in interstitial ATP.
Expression of CFTR in rat EDL muscle cells
Immunohistochemistry was performed to confirm the existence of CFTR in the rat skeletal muscle cells: a positive signal was detected on the rat EDL muscle cells (Fig. 5B). A strong positive signal was also detected in the intestinal epithelium (Fig. 5A), which served as a positive control, in accordance with other reports demonstrating the expression of CFTR in the epithelia of various tissues (Simpson et al. 2005). No staining was observed in the negative controls for EDL (Fig. 5D) or intestine (Fig. 5C), which lacked the antibody for CFTR, indicating that the staining for CFTR was specific. CFTR expression in rat muscle was further confirmed using Western blot (Fig. 5E): both intestine and EDL muscle exhibited a heavily stained band at 168 and a lighter stained band at 150 kDa, which have been attributed to a mature fully glycosylated band and an immature core-glycosylated band, respectively (Cheng et al. 1990).
Figure 5. Immunohistochemical localisation of CFTR on rat intestinal epithelium and EDL muscle cells.
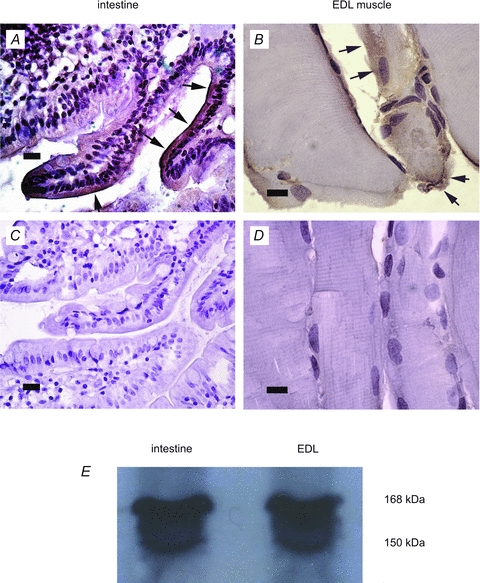
Immunohistochemical localisation of CFTR (brown staining indicated by arrows) on rat intestinal epithelium (A) and EDL muscle cells (B). Cell nucleoli were counterstained with Harris haematoxylin (purple). No brown staining was detectable in the negative controls for intestine (C) or EDL (D), in which the primary antibody was omitted. Scale bar corresponds to 10 μm in EDL sections and 20 μm in intestine samples. E shows CFTR expression in intestine and EDL muscle, measured using Western blotting: mature CFTR is seen at 168 kDa and immature CFTR at 150 kDa.
Effect of lactic acid on CFTR expression by cultured rat skeletal myoblasts
To further confirm that CFTR was expressed on skeletal muscle cells, and that skeletal muscle cells were the source of the ATP released into the interstitial space of intact muscle, we performed further experiments using cultured skeletal myoblasts. The total protein contents of control cells and cells incubated in culture medium supplemented with 10 mm lactic acid were similar at the end of the 3 h incubation period. The expression of CFTR protein, determined by Western blotting, was significantly increased in the cells incubated in lactic acid (Fig. 6A): the CFTR/β-actin ratio was increased by 64% following 3 h exposure to 10 mm lactic acid (Fig. 6B). However, the expression of MAPK, which was used as a reference protein for the control, was not affected by lactic acid incubation: the MAPK/β-actin ratio was 0.39 ± 0.08 in cells kept at pH 7.4 and 0.33 ± 0.05 in cells incubated with lactic acid.
Figure 6. The influence of silencing the CFTR gene in cultured rat skeletal myoblasts on CFTR expression (A and B) and the appearance of ATP (C) and adenosine (D) in the bathing medium of the cells.
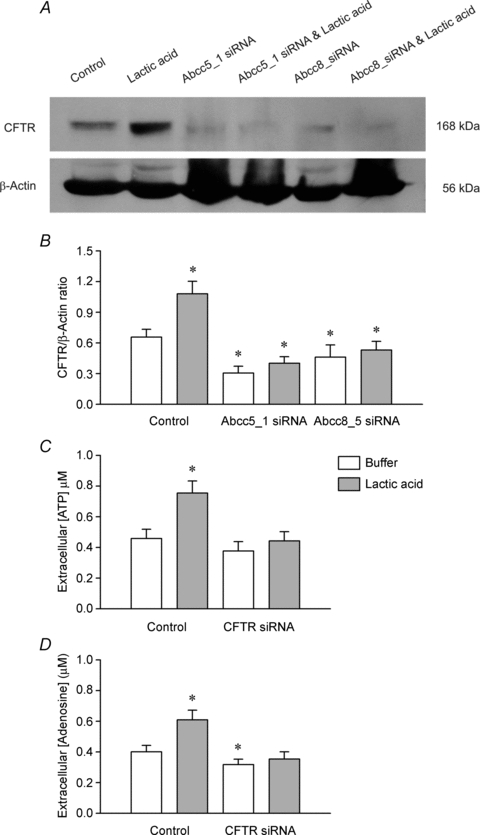
Two separate short interfering RNAs (siRNAs) were used: CFTR expression was measured using Western blotting (A) and the 168 kDa band was normalised to the expression of β-actin in the same tissue (B). Data are means ± s.e.m., n = 6; *P < 0.05 vs. the control (non-transfected) group. The appearance of ATP and adenosine in the bathing medium was measured in non-transfected cells (Control) and cells that had been transfected with the Abcc5_1 siRNA (siRNA group) in the absence or presence of 10 mm lactic acid. Data are means ± s.e.m., n = 9; *P < 0.05 vs. the control (non-transfected) group.
Effect of lactic acid on ATP release from cultured rat skeletal myoblasts
The ATP concentration in the bathing medium of the L6 cells incubated at pH 7.4 for 3 h averaged 0.46 ± 0.06 μm (Fig. 6C). Addition of 10 mm lactic acid to the bathing medium significantly elevated the extracellular ATP concentration to 0.76 ± 0.08 μm (Fig. 6C). The adenosine concentration in the bathing medium was also significantly increased by incubation with lactic acid, from 0.40 ± 0.04 to 0.61 ± 0.06 μm (Fig. 6D).
Effect of silencing the CFTR gene using RNA interference on lactic acid-induced ATP release from cultured rat skeletal myoblasts
The total protein contents of control cells and of cells treated with lactic acid and/or transfected with an siRNA were similar at the end of the incubation period. Forty-eight hours after transfection with CFTR-siRNA, the expression of CFTR protein was significantly decreased from control in the transfected cells (Fig. 6A and B), but the expression of MAPK1 was unchanged (data not shown). Treatment with 10 mm lactic acid failed to stimulate an increase in CFTR protein expression in the CFTR-siRNA-transfected cells (Fig. 6B). In the MAPK-siRNA-transfected cells, MAPK expression was significantly decreased (Fig. 7A), but CFTR expression was unchanged (data not shown), demonstrating that the gene silencing was specific.
Figure 7. The influence of control transfections using either a nonsense siRNA or MAPK siRNA on the appearance of ATP in the bathing medium of cultured rat skeletal myoblasts.

Western blot of non-transfected and MAPK siRNA-transfected cells is shown in A. Treatment of either group of transfected cells with 10 mm lactic acid significantly increased the appearance of ATP in the bathing medium (B). Data are means ± s.e.m., n = 8–11; *P < 0.01 with vs. without lactic acid.
The concentration of ATP in the bathing medium of CFTR-siRNA-transfected cells kept at pH 7.4 was not significantly different from that of the control cells (Fig. 6C), although, the adenosine concentration was slightly decreased (Fig. 6D). However, incubation of CFTR-siRNA-transfected cells with lactic acid failed to elevate either the ATP (Fig. 6C) or adenosine (Fig. 6D) concentrations.
The concentration of ATP in the bathing medium of MAPK-siRNA-transfected cells or nonsense-siRNA transfected cells kept at pH 7.4 was also not significantly different from that of the control cells (Fig. 7B). However, transfection with MAPK-siRNA or nonsense-siRNA failed to block the lactic acid-induced release of ATP into the bathing medium (Fig. 7B): the increase in ATP concentration in the bathing medium of MAPK-siRNA-transfected cells was not significantly different from that of the control cells following 3 h incubation with 10 mm lactic acid.
Discussion
The main findings of this study were that depression of the pH with lactic acid caused a dose-dependent increase in the interstitial ATP of skeletal muscle (Fig. 1), which could be blocked (1) by inhibition of the entry of lactate to cells using CHCA (Fig. 2), (2) by the non-specific inhibitor of CFTR, glibenclamide (Fig. 4B), (3) by the specific inhibitor of CFTR, CFTRinh-172 (Fig. 4C), and (4) by silencing the CFTR gene using an siRNA (Fig. 6). Sodium lactate, which failed to depress the pH, also failed to produce an increase in interstitial ATP. We have also confirmed, both by immunostaining and by Western blotting, the expression of CFTR on the skeletal muscle cells (Figs 5 and 6). This is the first time that the lactic acid-induced increase in interstitial ATP has been reported: there was a significant correlation between the decrease in pH produced by the lactic acid and the increase in interstitial ATP that resulted. These data suggest that CFTR plays an essential role in the lactic acid-induced efflux of ATP into the interstitial space of skeletal muscle.
It is known that the interstitial ATP concentration increases during muscle contractions (Hellsten et al. 1998). Extracellular ATP can act through P2Y1 receptors of muscle cells to inhibit chloride channels (Voss, 2009), which is suggested to help prevent muscle fatigue during high-frequency stimulation. ATP is also converted extracellularly to adenosine (Hellsten, 1999; Cheng et al. 2000), accounting for a large part of the active hyperaemia. However, it is not known how the muscle contractions give rise to the increase in interstitial ATP. Previous work has found a strong correlation between the decrease in venous pH and the adenosine release from skeletal muscle during either muscle contractions (Achike & Ballard, 1993) or pH depression by lactic acid infusion (Ballard, 1991) or pH depression by systemic hypercapnia (Mo & Ballard, 1994). Similarly, a reduction of pH to 6.8 by CO2 has been reported to cause an increase in adenosine release from Langendorff-perfused guinea-pig hearts (Mustafa & Mansour, 1984). Collectively, these findings suggest that the adenosine release resulted from the pH change rather than from some specific effect of lactic acid.
The first aim of our current study was to determine whether pH depression stimulated the output of ATP from skeletal muscle: we have clearly shown that lactic acid infusion increased the interstitial ATP of perfused skeletal muscle in vivo; using the cultured skeletal myoblasts, we have also confirmed the skeletal muscle cells as the source of the released ATP. Given the strong correlation between pH depression and adenosine output in the previous studies, and the strong correlation between pH depression and the increase in ATP observed in the current study (Fig. 1C), it is likely that an increase in interstitial ATP preceded the increase in adenosine observed in all of the above studies. This would imply that the ATP release in the present study also resulted from the pH depression, rather than from a specific effect of lactic acid. This conclusion is supported by the observation that 10 mm sodium lactate, which did not significantly decrease the pH, failed to increase interstitial ATP (Fig. 3), whereas 10 mm lactic acid did decrease the pH and did increase the interstitial ATP.
The control levels of interstitial ATP varied between 25 and 60 nm in the different series of experiments (Figs 1–4), although the great majority of values fell into the 40–50 nm range. To some extent, this must represent biological variation; it is also possible that some ATP release from the muscle could have been induced by tissue damage caused by the insertion of the microdialysis probe: this would affect the background level of ATP, but it could not account for either the increase in ATP during lactic acid infusion or the return to control levels when the infusion was stopped. Finally, the recovery from individual microdialysis probes might have varied between experiments: this could contribute to the variability in control levels, but within any given experiment, it would affect both the control and the treatment samples equally.
We selected lactic acid infusion as the method to lower pH because it is a highly physiological stimulus, and because lactic acid is readily transported into the cell, depressing both the intra- and extracellular pH. The extracellular lactate concentration is about 1–2 mm at rest (Karlsson & Saltin, 1970; Bangsbo et al. 1993), rising to around 5–10 mm in moderate exercise (Tesch & Wright, 1983), or 12–14 mm following exercise to exhaustion (Karlsson & Saltin, 1970; Thomas et al. 2005). Intracellular lactate can increase above 30 mm during intense exercise (Bangsbo et al. 1993). Thus, the range of lactic acid doses that we used (2.5–10 mm) lay within the normal physiological range. During the infusion of lactic acid, a gradient for H+ would exist from the blood to the interstitial space: thus the interstitial pH would be similar to, or higher than, venous pH. The maximum decrease in venous pH in our study was about 0.2 units (to around 7.0), which is also within the physiological range: interstitial pH drops to around 7.0 during moderate exercise (Street et al. 2001), whereas intracellular pH may fall as low as 6.5 during exhausting exercise (Juel, 2008).
Lactic acid is transported into cells by the monocarboxylate transporters (MCTs): 14 MCTs are currently recognised (Halestrap & Meredith, 2004), of which, only two (MCT1 and MCT4) are known to transport lactic acid into skeletal muscle cells (Halestrap & Meredith, 2004). MCT1 is mainly expressed in oxidative muscles, whereas fast-twitch glycolytic muscles, such as the EDL in the current study, mainly express MCT4 (Bonen, 2000). Both of these MCTs transport lactate and H+ at a 1:1 ratio (Juel & Halestrap, 1999), and the transport rate is influenced by the concentration gradients for both substrates, (Juel, 1996). The Km for MCT4 was reported as 10 mm in intact muscle (Mason & Thomas, 1988) although later studies with sarcolemmal vesicles have reported values around 15–20 mm (Juel & Halestrap, 1999). Nevertheless, we may be confident that with the range of lactic acid concentrations that we employed, each incremental increase in the lactic acid dose would result in an incremental increase in the transport of lactate and H+ into the cells. Thus, during lactic acid infusion, the depression of intracellular pH is expected to be in proportion to the decrease in extracellular pH. Lactic acid infusion produced a significant and dose-dependent increase in the interstitial ATP: since there are no known mechanisms for extracellular ATP formation, this must result from increased efflux of ATP from the cells.
In order to test whether the ATP efflux was stimulated by the increased intra- or extracellular lactic acid, we used CHCA, a structural analogue of lactate, which acts as a potent competitive inhibitor of the MCTs (Juel, 1988). Around 85% of lactic acid transport into cells is carrier mediated (Roth & Brooks, 1990), and the diffusion of free undissociated lactic acid accounts for the rest: thus, the application of CHCA was expected to greatly reduce the cellular uptake of lactic acid. CHCA has not been reported to affect the cellular metabolism, and a previous study showed that it did not directly change pHi in normoxia or hypoxia (Mo & Ballard, 1997). The present study showed that, in the presence of CHCA, the lactic acid infusion-induced ATP output was abolished (Fig. 2). This strongly suggests that it is increased intracellular lactic acid that stimulates ATP efflux from skeletal muscle.
Our next objective was to determine whether the increased intracellular lactate or the decreased intracellular pH was responsible for the increased ATP efflux. To investigate this, we infused neutral sodium lactate to the muscle. MCTs are much more sensitive to the lactate gradient than the H+ gradient (Juel, 1996): thus, it is expected that the addition of 10 mm lactate should drive the uptake of lactate plus H+ into the cell. However, in the absence of a gradient of H+ concentration into the cell, it is expected that the accompanying protons will be rapidly extruded from the cell, mainly by the Na+/H+ exchanger (NHE), with a smaller contribution from bicarbonate-dependent exchange (Aickin & Thomas, 1977; Juel, 2008). This is similar to the situation in the recovery from exercise, where H+ is extruded from the cell about 50% more rapidly than the accompanying lactate (Bangsbo et al. 1993). Therefore, sodium lactate infusion is expected to significantly elevate the intracellular lactate concentration, but to produce little or no change in intracellular pH. The interstitial ATP concentration clearly followed the expected changes in intracellular H+, and not the change in intracellular lactate in this series of experiments. We interpret this as evidence that it is the depression of intracellular pH, rather than the change in lactate concentration, that stimulates ATP efflux from muscle.
We also found that the lactic acid-induced ATP efflux could be reproduced in cultured skeletal myoblasts (Fig. 6C). This shows that skeletal muscle cells have the capability to release ATP in response to a decrease in pH. We assume, therefore, that the skeletal muscle cells were the major contributor to the ATP efflux in the in vivo experiments. We could not rule out the possibility that other cells facing the interstitial space, such as vascular smooth muscle or nerve, also released ATP during acidosis in the intact muscle, since vascular smooth muscle is reported to express CFTR (Robert et al. 2004), a key player in ATP efflux from muscle (for discussion, see below). However, as skeletal muscle cells make up the bulk of the tissue, they would be likely to account for the majority of the ATP release. Vascular endothelium and erythrocytes also express CFTR (Sprague et al. 1998; Tousson et al. 1998), but would be unlikely to contribute to the ATP in the interstitial space, as we have previously found that ATP cannot cross the vascular wall unless the blood concentration reaches about 10 μm.
Having established that pH depression stimulated ATP efflux from muscle cells, we next investigated the mechanism by which ATP left the cell during acidosis. We showed that the specific inhibitor of CFTR, CFTRinh-172 (Ma et al. 2002; Wang et al. 2004; Servetnyk et al. 2006), abolished the lactic acid-induced ATP efflux from the perfused muscle in vivo (Fig. 4C), and that silencing the CFTR gene abolished the lactic acid-induced ATP efflux from the cultured skeletal myoblasts (Fig. 6C). We further confirmed the expression of CFTR protein on the intact muscle using immunohistochemistry, and in both the cultured cells and the intact muscle using Western blot. These data strongly suggest that CFTR plays an important role in the acidosis-induced ATP efflux from muscle cells. However, CFTR is unlikely to account for the basal (unstimulated) ATP release: silencing the CFTR gene using RNA interference significantly decreased CFTR protein expression in the myoblasts, but did not reduce the accumulation of ATP in the bathing medium at pH 7.4 (Fig. 6B and C).
We first reported CFTR expression in rat soleus muscle in our 2007 conference paper (Tu & Ballard, 2007). Recently, while this paper was under revision, two other groups have corroborated our finding of CFTR expression in skeletal muscle (Divangahi et al. 2009; Lamhonwah et al. 2010). Both groups report that CFTR expression is localised in the sarcoplasmic reticulum and sarcolemma of muscle, and propose that it plays a role in calcium homeostasis: CFTR-deficient muscle exhibited increased calcium mobilisation upon depolarisation (Divangahi et al. 2009) and higher end-exercise pH values (Lamhonwah et al. 2010). Lamhonwah's group propose that CFTR in muscle may regulate chloride-dependent bicarbonate transport, which may help to explain the activation of CFTR at low pH that we observed in the current study.
CFTR is one of a group of ATP-binding cassette (ABC) proteins, which also includes the multidrug resistance transporter and P-glycoprotein (Holland et al. 2003). Many studies have reported the presence of ATP channels and/or ATP release in cells expressing one of the ABC proteins (Abraham et al. 1993; Reisin et al. 1994; Pasyk & Foskett, 1997; Roman et al. 1997; Cantiello et al. 1998; Schweibert, 1999). CFTR is a plasma-membrane cAMP/protein kinase A-activated Cl− channel that is best known for its role in epithelial Cl− secretion (Gadsby et al. 2006). Laboratories using patch-clamp studies have reported that ATP channels are only present in CFTR-containing patches, and that cAMP activates both Cl− and ATP conductances (Reisin et al. 1994; Schweibert et al. 1995; Prat et al. 1996; Pasyk & Foskett, 1997), which led to the proposal that ATP could exit cells through the open CFTR Cl− channel. This was supported by the demonstration that an open CFTR channel would permit the efflux of several large organic anions, including gluconate, pyruvate and isothionate, some of which were larger in size than ATP (Lindsell & Hanrahan, 1998). However, this proposal remains controversial, since other studies have reported that around one-third of CFTR-containing patches did not contain an ATP conductance (Sugita et al. 1998), and that inhibitors of the CFTR Cl− channel had different effects on the Cl− and ATP conductances (Sugita et al. 1998). Furthermore, both purified CFTR reconstituted into phospholipid vesicles (Li et al. 1996) and recombinant CFTR in planar lipid bilayers (Reddy et al. 1996) failed to conduct ATP. An alternative explanation has been proposed that CFTR regulates the conductance of a separate ATP channel or transporter, in a manner analogous to its regulation of the epithelial Na+ channel (Stutts et al. 1995) or outwardly rectifying Cl− channel (Schweibert et al. 1995). Our present study has demonstrated that CFTR plays an obligatory role in the acidosis-induced ATP efflux, but we have not addressed the question of whether or not CFTR itself conducts the ATP. This is the subject of our on-going investigation, and we will report the findings separately.
Glibenclamide also abolished the acidosis-induced ATP release from the intact muscle: glibenclamide is best-known as a KATP channel blocker, although it also blocked the CFTR chloride channel (Sheppard & Robinson, 1997) and the CFTR-associated ATP channel (Sugita et al. 1998) in patch clamp studies. The most likely explanation for its effects in the present study is that it inhibited CFTR. However, with the existing evidence, we could not altogether rule out the possibility that the KATP channel was involved in the ATP efflux, for example through the formation of a large dynamic signalling complex with CFTR (Seino, 1999; Shibasaki et al. 2004): KATP channels comprise two subunits, of which, one is a K+ channel from the Kir6.0 family, and the other is a sulfonylurea (SUR) receptor (Seino, 1999), which, like CFTR itself, is a member of the ABC protein family; the possible involvement of the KATP channel in the ATP efflux is the subject of further investigation in our laboratory.
Mitochondrial ATP transporters are a family of transporter proteins that are found in the inner membranes of mitochondria (O’Rourke, 2007). They shuttle metabolites, nucleotides, and cofactors through this membrane and thereby connect and/or regulate cytoplasm and matrix functions. There have been no previous reports that this family of proteins was involved in the transport of ATP across the cell membrane. Nevertheless, it is theoretically possible that they might contribute to an increase in the cytoplasmic ATP concentration, which might, in turn, lead to the ATP efflux. However, atractyloside, a specific inhibitor of the mitochondrial ATP transporter (Wilson & Asimakis, 1987; Roux et al. 1996) did not block the lactic acid infusion-induced ATP release (Fig. 4A). The concentration of atractyloside was sufficient to fully inhibit the mitochondrial ATP transporter (Asimakis & Conti, 1984; Kopustinskiene et al. 2003); thus, we conclude that the ATP transporter was unlikely to be involved in the ATP output during acidosis challenge.
The mechanism by which pH depression activates CFTR-dependent ATP efflux from the muscle is currently under investigation in our laboratory. In the present study, incubation of the cultured myoblasts with lactic acid for 3 h increased the expression of CFTR protein (Fig. 6B), and our preliminary findings suggest that expression is also increased at the mRNA level (unpublished observation). However, it is unlikely that a significant increase in gene expression could have occurred within 10 min, yet the ATP efflux was increased in the first 10 min of lactic acid infusion in the in vivo studies (Fig. 3B), suggesting that other signal transduction mechanisms are involved. Other laboratories have reported that the CFTR-associated ATP efflux is activated through the cAMP/protein kinase A pathway (Sugita et al. 1998; Sprague et al. 2001), and our preliminary findings also suggest that a protein kinase A inhibitor can block the acidosis-induced ATP efflux (unpublished observation). However, there have been no previous reports of a link between pH depression and increased cAMP. The chloride conductance of sweat gland CFTR has previously been reported to be regulated by cytosolic pH (Reddy et al. 1998), although in that study the chloride conductance was reduced by pH depression, apparently by inhibition of the phosphorylation activation of the channel. However, a more recent patch-clamp study on recombinant CFTR reported that acidic intracellular pH increased the open probability of CFTR (Chen et al. 2009): the authors concluded that pH changes were sensed directly by the CFTR chloride channel, through modulation of the interaction of MgATP with one of its ATP-binding sites. Further investigation is needed, therefore, to elucidate fully the mechanism by which intracellular pH depression stimulates the ATP efflux from muscle.
In conclusion, we have demonstrated that depression of the intracellular pH using lactic acid caused an efflux of ATP from skeletal muscle cells, and that CFTR played an important role in that process: this is the first report of a role for CFTR in ATP efflux from muscle. These data will contribute significantly to our understanding of the mechanism of ATP release from muscle.
Acknowledgments
We are grateful to Mr M. K. Yip and Mr C. P. Mok for technical assistance. This work was funded by Research Grants Council Direct Allocation Grants nos 10206730 and 10207994 and grant no. 200807176238 from the University of Hong Kong Committee on Research & Conference Grants.
Glossary
Abbreviations
- CFTR
cystic fibrosis transmembrane conductance regulator
- EDL
extensor digitorum longus
- VSOR
volume-sensitive outwardly rectifying Cl− channel
Author contributions
All authors contributed to conception and design, or analysis and interpretation of data, and drafting the article or revising it critically for important intellectual content, and gave final approval of the version to be published.
References
- Abraham EH, Prat AG, Gerweck L, Seneveratne T, Arceci RJ, Kramer R, Guidotti G, Cantiello HF. The multidrug resistance (mdr1) gene product functions as an ATP channel. Proc Natl Acad Sci U S A. 1993;90:312–316. doi: 10.1073/pnas.90.1.312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achike FI, Ballard HJ. Influence of stimulation parameters on the release of adenosine, lactate and CO2 from contracting dog gracilis muscle. J Physiol. 1993;463:107–121. doi: 10.1113/jphysiol.1993.sp019586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aickin CC, Thomas RC. An investigation of the ionic mechanism of intracellular pH regulation in mouse soleus muscle fibres. J Physiol. 1977;273:295–316. doi: 10.1113/jphysiol.1977.sp012095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ajonuma LC, Ng EHY, Chow PH, Hung CY, Tsang LL, Cheung ANY, Brito-Jones C, Lok IH, Haines CJ, Chan HC. Increased cystic fibrosis transmembrane conductance regulator (CFTR) expression in the human hydrosalpinx. Hum Reprod. 2005;20:1228–1234. doi: 10.1093/humrep/deh773. [DOI] [PubMed] [Google Scholar]
- Asimakis GK, Conti VR. Myocardial ischemia: correlation of mitochondrial adenine nucleotide and respiratory function. J Mol Cell Cardiol. 1984;16:439–447. doi: 10.1016/s0022-2828(84)80615-x. [DOI] [PubMed] [Google Scholar]
- Ballard HJ. The influence of lactic acid on adenosine release from skeletal muscle in anaesthetized dogs. J Physiol. 1991;433:95–108. doi: 10.1113/jphysiol.1991.sp018416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangsbo J, Johansen L, Graham T, Saltin B. Lactate and H+ effluxes from human skeletal muscles during intense, dynamic exercise. J Physiol. 1993;462:115–133. doi: 10.1113/jphysiol.1993.sp019546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell PD, Lapointe JY, Sabirov R, Hayashi S, Peti-Peterdi J, Manabe KI, Kovacs G, Okada Y. Macula densa cell signaling involves ATP release through a maxi anion channel. Proc Natl Acad Sci U S A. 2003;100:4322–4327. doi: 10.1073/pnas.0736323100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodin P, Burnstock G. Evidence that release of adenosine triphosphate from endothelial cells during increased shear stress is vesicular. J Cardiovasc Pharmacol. 2001;38:900–908. doi: 10.1097/00005344-200112000-00012. [DOI] [PubMed] [Google Scholar]
- Bonen A. Lactate transporters (MCT proteins) in heart and skeletal muscles. Med Sci Sports Exerc. 2000;32:778–789. doi: 10.1097/00005768-200004000-00010. [DOI] [PubMed] [Google Scholar]
- Braunstein GM, Roman RM, Clancy JP, Kudlow BA, Taylor AL, Shylonsky V, Jovov B, Peter K, Jilling T, Guggino WB, Ismailov II, Collawn JF, Benos DJ, Schweibert LM, Fitz JG, Schweibert EM. CFTR facilitates ATP release by stimulating a separate ATP release channel for autocrine control of cell volume. J Biol Chem. 2001;276:6621–6630. doi: 10.1074/jbc.M005893200. [DOI] [PubMed] [Google Scholar]
- Burnstock G. Historical review: ATP as a neurotransmitter. Trends Pharmacol Sci. 2006;27:166–176. doi: 10.1016/j.tips.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Cantiello HF, Jackson GR, Grosman CF, Prat AG, Borkan SC, Wang Y, Reisin IL, O’Riordan CR, Ausiello DA. Electrodiffusional ATP movement through the cystic fibrosis transmembrane conductance regulator. Am J Physiol Cell Physiol. 1998;274:C799–C809. doi: 10.1152/ajpcell.1998.274.3.C799. [DOI] [PubMed] [Google Scholar]
- Chen JH, Cai ZW, Sheppard DN. Direct sensing of intracellular pH by the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel. J Biol Chem. 2009;284:35495–35506. doi: 10.1074/jbc.M109.072678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SH, Gregory RJ, Marshall J, Paul S, Souza DW, White GA, O’Riordan CR, Smith AE. Defective intracellular transport and processing of CFTR is the molecular basis of most cystic fibrosis. Cell. 1990;63:827–834. doi: 10.1016/0092-8674(90)90148-8. [DOI] [PubMed] [Google Scholar]
- Cheng B, Essackjee HC, Ballard HJ. Evidence for control of adenosine metabolism in rat oxidative skeletal muscle by changes in pH. J Physiol. 2000;522:467–477. doi: 10.1111/j.1469-7793.2000.t01-1-00467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrina ML, Lin JHC, Alves-Rodrigues A, Liu SJ, Li J, Azmi-Ghadimi H, Kang J, Naus CCG, Nedergaard M. Connexins regulate calcium signaling by controlling ATP release. Proc Natl Acad Sci U S A. 1998;95:15735–15740. doi: 10.1073/pnas.95.26.15735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Divangahi M, Balghi H, Danialou G, Comtois AS, Demoule A, Ernest S, Haston C, Robert R, Hanrahan JW, Radzioch D, Petrof BJ. Lack of CFTR in skeletal muscle predisposes to muscle wasting and diaphragm muscle pump failure in cystic fibrosis mice. Plos Genetics. 2009;5:e1000586. doi: 10.1371/journal.pgen.1000586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubyak GR. Both sides now: multiple interactions of ATP with pannexin-1 hemichannels. Am J Physiol Cell Physiol. 2009;296:C235–C241. doi: 10.1152/ajpcell.00639.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiermonte G, De Leonardis F, Todisco S, Palmieri L, Lasorsa FM, Palmieri F. Identification of the mitochondrial ATP-Mg/Pi transporter. Bacterial expression, reconstitution, functional characterization, and tissue distribution. J Biol Chem. 2004;279:30722–30730. doi: 10.1074/jbc.M400445200. [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Vergani P, Csanady L. The ABC protein turned chloride channel whose failure causes cystic fibrosis. Nature. 2006;440:477–483. doi: 10.1038/nature04712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon JL. Extracellular ATP: effects, sources and fate. Biochem J. 1986;233:309–319. doi: 10.1042/bj2330309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guggino W. The cystic fibrosis transmembrane regulator forms macromolecular complexes with PDZ domain scaffold proteins. Proc Am Thorac Soc. 2004;1:28–32. doi: 10.1513/pats.2306011. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Meredith D. The SLC16 gene family – from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447:619–628. doi: 10.1007/s00424-003-1067-2. [DOI] [PubMed] [Google Scholar]
- Hellsten Y. The effect of muscle contraction on the regulation of adenosine formation in rat skeletal muscle cells. J Physiol. 1999;518:761–768. doi: 10.1111/j.1469-7793.1999.0761p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellsten Y, Maclean D, Radegran G, Saltin B, Bangsbo J. Adenosine concentrations in the interstitium of resting and contracting human skeletal muscle. Circulation. 1998;98:6–8. doi: 10.1161/01.cir.98.1.6. [DOI] [PubMed] [Google Scholar]
- Hirschberg CB, Robbins PW, Abeijon C. Transporters of nucleotide sugars, ATP, and nucleotide sulfate in the endoplasmic reticulum and Golgi apparatus. Annu Rev Biochem. 1998;67:49–69. doi: 10.1146/annurev.biochem.67.1.49. [DOI] [PubMed] [Google Scholar]
- Holland IB, Cole SPC, Kuchler K, Higgins CF. ABC Proteins: From Bacteria to Man. London: Academic Press; 2003. [Google Scholar]
- Hutton JC. The insulin secretory granule. Diabetologia. 1989;32:271–281. doi: 10.1007/BF00265542. [DOI] [PubMed] [Google Scholar]
- Juel C, Halestrap AP. Lactate transport in skeletal muscle – role and regulation of the monocarboxylate transporter. J Physiol. 1999;517:633–642. doi: 10.1111/j.1469-7793.1999.0633s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juel C. Intracellular pH recovery and lactate efflux in mouse soleus muscles stimulated in vitro: the involvement of sodium/proton exchange and a lactate carrier. Acta Physiol Scand. 1988;132:363–371. doi: 10.1111/j.1748-1716.1988.tb08340.x. [DOI] [PubMed] [Google Scholar]
- Juel C. Lactate/proton co-transport in skeletal muscle: regulation and importance for pH homeostasis. Acta Physiol Scand. 1996;156:369–374. doi: 10.1046/j.1365-201X.1996.206000.x. [DOI] [PubMed] [Google Scholar]
- Juel C. Regulation of pH in human skeletal muscle: adaptations to physical activity. Acta Physiol Scand. 2008;193:17–24. doi: 10.1111/j.1748-1716.2008.01840.x. [DOI] [PubMed] [Google Scholar]
- Karlsson J, Saltin B. Lactate, ATP, and CP in working muscles during exhaustive exercise in man. J Appl Physiol. 1970;29:598–602. doi: 10.1152/jappl.1970.29.5.598. [DOI] [PubMed] [Google Scholar]
- Kille JM, Klabunde RE. Adenosine as a mediator of postcontraction hyperemia in dog skeletal muscle. Am J Physiol Heart Circ Physiol. 1984;246:H274–H282. doi: 10.1152/ajpheart.1984.246.2.H274. [DOI] [PubMed] [Google Scholar]
- Kopustinskiene DM, Toleikis A, Saris NE. Adenine nucleotide translocase mediates the KATP-channel-openers-induced proton and potassium flux to the mitochondrial matrix. J Bioenerg Biomembr. 2003;35:141–148. doi: 10.1023/a:1023746103401. [DOI] [PubMed] [Google Scholar]
- Lamhonwah AM, Bear CE, Huan LJ, Chiaw PK, Ackerley CA, Tein I. Cystic fibrosis transmembrane conductance regulator in human muscle dysfunction causes abnormal metabolic recovery in exercise. Ann Neurol. 2010;67:802–808. doi: 10.1002/ana.21982. [DOI] [PubMed] [Google Scholar]
- Li C, Ramjeesingh M, Bear CE. Purified cystic fibrosis transmembrane conductance regulator (CFTR) does not function as an ATP channel. J Biol Chem. 1996;271:11623–11626. doi: 10.1074/jbc.271.20.11623. [DOI] [PubMed] [Google Scholar]
- Li J, King NC, Sinoway LI. ATP concentrations and muscle tension increase linearly with muscle contraction. J Appl Physiol. 2003;95:577–583. doi: 10.1152/japplphysiol.00185.2003. [DOI] [PubMed] [Google Scholar]
- Lindsell P, Hanrahan JW. Adenosine triphosphate-dependent asymetry of anion permeation in the cystic fibrosis transmembrane conductance regulator chloride channel. J Gen Physiol. 1998;111:601–614. doi: 10.1085/jgp.111.4.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucovei S, Bao L, Dahl G. Pannexin 1 in erythrocytes: function without a gap. Proc Natl Acad Sci U S A. 2006;103:7655–7659. doi: 10.1073/pnas.0601037103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma TH, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJV, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason MJ, Thomas RC. A microelectrode study of the mechanisms of l-lactate entry into and release from frog sartorius muscle. J Physiol. 1988;400:459–479. doi: 10.1113/jphysiol.1988.sp017132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo FM, Ballard HJ. Adenosine output from dog gracilis muscle during systemic hypercapnia and/or amiloride-SITS infusion. Am J Physiol Heart Circ Physiol. 1994;267:H1243–H1249. doi: 10.1152/ajpheart.1994.267.4.H1243. [DOI] [PubMed] [Google Scholar]
- Mo FM, Ballard HJ. Intracellular lactate controls adenosine output from dog gracilis muscle during moderate systemic hypoxia. Am J Physiol Heart Circ Physiol. 1997;272:H318–H324. doi: 10.1152/ajpheart.1997.272.1.H318. [DOI] [PubMed] [Google Scholar]
- Mo FM, Ballard HJ. The effect of systemic hypoxia on interstitial and blood adenosine, AMP, ADP and ATP in dog skeletal muscle. J Physiol. 2001;536:593–603. doi: 10.1111/j.1469-7793.2001.0593c.xd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustafa SJ, Mansour MM. Effect of perfusate pH on coronary flow and adenosine release in isolated rabbit heart. Proc Soc Exp Biol Med. 1984;176:22–26. doi: 10.3181/00379727-176-41836. [DOI] [PubMed] [Google Scholar]
- O’Rourke B. Mitochondrial ion channels. Annu Rev Physiol. 2007;69:19–49. doi: 10.1146/annurev.physiol.69.031905.163804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasyk EA, Foskett JK. Cystic fibrosis transmembrane conductance regulator-associated ATP and adenosine 3′-phosphate 5′-phosphosulfate channels in endoplasmic reticulum and plasma membranes. J Biol Chem. 1997;272:7746–7751. doi: 10.1074/jbc.272.12.7746. [DOI] [PubMed] [Google Scholar]
- Prat AG, Reisin IL, Ausiello DA, Cantiello HF. Cellular ATP release by the cystic fibrosis transmembrane conductance regulator. Am J Physiol Cell Physiol. 1996;270:C538–C545. doi: 10.1152/ajpcell.1996.270.2.C538. [DOI] [PubMed] [Google Scholar]
- Reddy MM, Kopito RR, Quinton PM. Cytosolic pH regulates GCl through control of phosphorylation states of CFTR. Am J Physiol Cell Physiol. 1998;275:C1040–C1047. doi: 10.1152/ajpcell.1998.275.4.C1040. [DOI] [PubMed] [Google Scholar]
- Reddy MM, Quinton PM, Haws C, Wine JJ, Grygorczyk R, Tabcharani JA, Hanrahan JW, Gunderson KL, Kopito RR. Failure of the cystic fibrosis transmembrane conductance regulator to conduct ATP. Science. 1996;271:1876–1878. doi: 10.1126/science.271.5257.1876. [DOI] [PubMed] [Google Scholar]
- Reisin IL, Prat AG, Abraham EH, Amara JF, Gregory RJ, Ausiello DA, Cantiello HF. The cystic fibrosis transmembrane conductance regulator is a dual ATP and chloride channel. J Biol Chem. 1994;269:20584–20591. [PubMed] [Google Scholar]
- Robert R, Thoreau V, Norez C, Cantereau A, Kitzis A, Mettey Y, Rogier C, Becq F. Regulation of the cystic fibrosis transmembrane conductance regulator channel by beta-adrenergic agonists and vasoactive intestinal peptide in rat smooth muscle cells and its role in vasorelaxation. J Biol Chem. 2004;279:21160–21168. doi: 10.1074/jbc.M312199200. [DOI] [PubMed] [Google Scholar]
- Roman RM, Wang Y, Lidofsky SD, Feranchak AP, Lomri N, Scharschmidt BF, Fitz JG. Hepatocellular ATP-binding cassette protein expression enhances ATP release and autocrine regulation of cell volume. J Biol Chem. 1997;272:21970–21976. doi: 10.1074/jbc.272.35.21970. [DOI] [PubMed] [Google Scholar]
- Roth DA, Brooks GA. Lactate and pyruvate transport is dominated by a pH gradient-sensitive carrier in rat skeletal muscle sarcolemmal vesicles. Arch Biochem Biophys. 1990;279:386–394. doi: 10.1016/0003-9861(90)90506-t. [DOI] [PubMed] [Google Scholar]
- Roux P, Le Saux A, Fiore C, Schwimmer C, Dianoux AC, Trezeguet V, Vignais PV, Lauquin GJ, Brandolin G. Fluorometric titration of the mitochondrial ADP/ATP carrier protein in muscle homogenate with atractyloside derivatives. Anal Biochem. 1996;234:31–37. doi: 10.1006/abio.1996.0046. [DOI] [PubMed] [Google Scholar]
- Sabirov RZ, Okada Y. ATP release via anion channels. Pur Signal. 2005;1:311–328. doi: 10.1007/s11302-005-1557-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweibert EM. ABC transporter-facilitated ATP conductive transport. Am J Physiol Cell Physiol. 1999;276:C1–C8. doi: 10.1152/ajpcell.1999.276.1.C1. [DOI] [PubMed] [Google Scholar]
- Schweibert EM, Egan ME, Hwang T-H, Fulmer SB, Allen SS, Cutting GR, Guggino WB. CFTR regulates outwardly rectifying chloride channels through an autocrine mechanism involving ATP. Cell. 1995;81:1063–1073. doi: 10.1016/s0092-8674(05)80011-x. [DOI] [PubMed] [Google Scholar]
- Seino S. ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annu Rev Physiol. 1999;61:337–362. doi: 10.1146/annurev.physiol.61.1.337. [DOI] [PubMed] [Google Scholar]
- Servetnyk Z, Krjukova J, Gaston B, Zaman K, Hjelte L, Roomans GM, Dragomir A. Activation of chloride transport in CF airway epithelial cell lines and primary CF nasal epithelial cells by S-nitrosoglutathione. Respir Res. 2006;7:124. doi: 10.1186/1465-9921-7-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Robinson KA. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a murine cell line. J Physiol. 1997;503:333–346. doi: 10.1111/j.1469-7793.1997.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibasaki T, Sunaga Y, Seino S. Integration of ATP, cAMP, and Ca2+ signals in insulin granule exocytosis. Diabetes. 2004;53(Suppl 3):S59–S62. doi: 10.2337/diabetes.53.suppl_3.s59. [DOI] [PubMed] [Google Scholar]
- Simpson JE, Gawenis LR, Walker NM, Boyle KT, Clarke LL. Chloride conductance of CFTR facilitates basal Cl−/HCO3− exchange in the villous epithelium of intact murine duodenum. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1241–G1251. doi: 10.1152/ajpgi.00493.2004. [DOI] [PubMed] [Google Scholar]
- Sprague RS, Ellsworth ML, Stephenson AH, Kleinhenz ME, Lonigro AJ. Deformation-induced ATP release from red blood cells requires CFTR activity. Am J Physiol Heart Circ Physiol. 1998;275:H1726–H1732. doi: 10.1152/ajpheart.1998.275.5.H1726. [DOI] [PubMed] [Google Scholar]
- Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. Participation of cAMP in a signal transduction pathway relating erythrocyte deformation to ATP release. Am J Physiol Cell Physiol. 2001;281:C1158–C1164. doi: 10.1152/ajpcell.2001.281.4.C1158. [DOI] [PubMed] [Google Scholar]
- Stout CE, Constantin JL, Naus CCG, Charles AC. Intercellular calcium signalling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- Street D, Bangsbo J, Juel C. Interstitial pH in human skeletal muscle during and after dynamic graded exercise. J Physiol. 2001;537:993–998. doi: 10.1111/j.1469-7793.2001.00993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stutts MJ, Canessa CM, Olsen JC, Hamrick M, Cohn JA, Rossier BC, Boucher RC. CFTR as a cAMP-dependent regulator of sodium channels. Science. 1995;269:847–850. doi: 10.1126/science.7543698. [DOI] [PubMed] [Google Scholar]
- Sugita M, Yue Y, Foskett JK. CFTR Cl− channel and CFTR-associated ATP channel: distinct pores regulated by common gates. EMBO J. 1998;17:898–908. doi: 10.1093/emboj/17.4.898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesch PA, Wright JE. Recovery from short term intense exercise: its relation to capillary supply and blood lactate concentration. Eur J Appl Physiol. 1983;52:98–103. doi: 10.1007/BF00429033. [DOI] [PubMed] [Google Scholar]
- Thomas C, Perrey S, Lambert K, Hugon G, Mornet D, Mercier J. Monocarboxylate transporters, blood lactate removal after supramaximal exercise, and fatigue indexes in humans. J Appl Physiol. 2005;98:804–809. doi: 10.1152/japplphysiol.01057.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tousson A, Van Tine BA, Naren AP, Shaw GM, Schwiebert LM. Characterization of CFTR expression and chloride channel activity in human endothelia. Am J Physiol Cell Physiol. 1998;275:C1555–C1564. doi: 10.1152/ajpcell.1998.275.6.C1555. [DOI] [PubMed] [Google Scholar]
- Tu J, Ballard HJ. Lactic-acid-induced ATP release from rat soleus muscle cells through adenylyl cyclase-cAMP-PKA-regulated CFTR. J HK Coll Cardiol. 2007;15:83. [Google Scholar]
- Voss AA. Extracellular ATP inhibits chloride channels in mature mammalian skeletal muscle by activating P2Y1 receptors. J Physiol. 2009;587:5739–5752. doi: 10.1113/jphysiol.2009.179275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XF, Reddy MM, Quinton PM. Effects of a new cystic fibrosis transmembrane conductance regulator inhibitor on Cl− conductance in human sweat ducts. Exp Physiol. 2004;89:417–425. doi: 10.1113/expphysiol.2003.027003. [DOI] [PubMed] [Google Scholar]
- Wilson DE, Asimakis GK. Phosphate-induced efflux of adenine nucleotides from rat-heart mitochondria: evaluation of the roles of the phosphate/hydroxyl exchanger and the dicarboxylate carrier. Biochim Biophys Acta. 1987;893:470–479. doi: 10.1016/0005-2728(87)90098-3. [DOI] [PubMed] [Google Scholar]
- Yamazaki J, Hume JR. Inhibitory effects of glibenclamide on cystic fibrosis transmembrane conductance regulator, swelling-activated, and Ca2+-activated Cl− channels in mammalian cardiac myocytes. Circ Res. 1997;81:101–109. doi: 10.1161/01.res.81.1.101. [DOI] [PubMed] [Google Scholar]