

Abstract
Nitric oxide (NO) functions as a diffusible transmitter in most tissues of the body and exerts its effects by binding to receptors harboring a guanylyl cyclase transduction domain, resulting in cGMP accumulation in target cells. Despite its widespread importance, very little is known about how this signaling pathway operates at physiological NO concentrations and in real time. To address these deficiencies, we have exploited the properties of a novel cGMP biosensor, named δ-FlincG, expressed in cells containing varying mixtures of NO-activated guanylyl cyclase and cGMP-hydrolyzing phosphodiesterase activity. Responsiveness to NO, signifying a physiologically relevant rise in cGMP to 30 nM or more, was seen at concentrations as low as 1 pM, making cells by far the most sensitive NO detectors yet encountered. Even cells coexpressing phosphodiesterase-5, a cGMP-activated isoform found in many NO target cells, responded to NO in concentrations as low as 10 pM. The dynamics of NO capture and signal transduction was revealed by administering timed puffs of NO from a local pipette. A puff lasting only 100 ms, giving a calculated peak intracellular NO concentration of 23 pM, was detectable. The results could be encapsulated in a quantitative model of cellular NO-cGMP signaling, which recapitulates the NO responsiveness reported previously from crude cGMP measurements on native cells, and which explains how NO is able to exert physiological effects at extremely low concentrations, when only a tiny proportion of its receptors would be occupied.
Keywords: nitrogen monoxide, enzyme-linked receptor, desensitization
Nitric oxide (NO) is an evolutionarily ancient transmitter of fundamental importance to the physiology of the mammalian cardiovascular, nervous, and other systems (1, 2). NO signal transduction takes place through the simplest one-component type of receptor, comprising an NO binding site (a prosthetic heme) coupled to a guanylyl cyclase (GC) transduction domain (3, 4). The elevation in cellular cGMP that follows NO binding is curtailed by one or more phosphodiesterase (PDE) enzymes that convert it to GMP. There have been countless descriptions of changes in the levels of cGMP in association with NO-mediated transmission in different cells and tissues. It has become increasingly evident, however, that the levels of cGMP measurable by traditional methods, such as radioimmunoassay, may not be relevant to physiological NO signaling. For example, smooth muscle relaxation (5) and cGMP-dependent phosphorylation events in platelets (6) are seen at NO concentrations below those giving increases in cGMP measurable with such methods; NO can still relax vascular smooth muscle despite deletion of 94% of the NO-activated GC (7); and engagement of 2% or less of the available GC activity is sufficient to stimulate cGMP-dependent phosphorylation in platelets (8). These findings indicate that cells possess a very large receptor excess and that new approaches are required to understand the elementary properties of NO capture and signal amplification by cells under physiological conditions. A related gap in knowledge is in the dynamics of NO signaling in cells because traditional methods of cGMP measurement generally do not have the necessary temporal resolution (but see ref. 9) yet, as with any signaling mechanism, awareness of the dynamics is key to understanding how NO signals are decoded.
A fluorescent cGMP biosensor, named δ-FlincG, appears to overcome both these deficiencies. The biosensor consists of the dual cGMP-binding domain from cGMP-dependent protein kinase fused to circularly permutated EGFP and its sensitivity to cGMP is in the submicromolar range relevant for engaging protein kinases and other downstream targets (10). The selectivity of δ-FlincG has for cGMP over cAMP is high, and its rapid activation and deactivation kinetics should provide a faithful temporal readout of changes in cellular cGMP concentration. Here we have scrutinized the performance of this sensor and have then used it to analyze how cells containing various mixtures of NO-activated GC and cGMP-hydrolyzing PDEs respond to NO delivered in different ways. The results allow some general principles of physiological NO signal transduction to be formulated.
Results
In the first experiments, we followed the responses of HEK 293T (HEK) cells expressing δ-FlincG and different combinations of GC and PDE to the superfusion of clamped NO concentrations achieved, as usual (11), by adding a slow NO releaser in the presence of a slow NO scavenger, 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (CPTIO; see SI Materials and Methods). The cells expressed different levels of GC activity (referred to as GClow, GCmid, and GChigh) and the PDEs were the native type (here called PDEHEK) with or without phosphodiesterase-5 (PDE5). PDE5 is a cGMP-activated isoform participating widely in NO-cGMP signaling (12) and was stably expressed in the cells at different levels (referred to as PDE5low and PDE5high; ref. 13). Table 1 summarizes values for the maximal GC (GCmax) and PDE (PDEmax) activities derived from the data below.
Table 1.
Cellular guanylyl cyclase and phosphodiesterase activities
| Cells | GCmax, μM/s | PDEmax, μM/s | GCmax/PDEmax | n |
| GClowPDEHEK | 0.25 ± 0.02 | 0.045 ± 0.003 | 5.66 ± 0.34 | 13 |
| GChighPDEHEK | 20, 20 | 0.035, 0.040 | 571, 500 | 2 |
| GChighPDE5low | 16.6 ± 0.4 | 2.65 ± 0.26 | 6.14 ± 1.4 | 10* |
| GCmidPDE5high | 3.67 ± 0.67 | 16.7 ± 3.53 | 0.27 ± 0.12 | 3 |
*NO was delivered by superfusion in three experiments and was puff-applied in the remainder.
Evaluation of δ-FlincG Signals.
Because of their slow response kinetics, cells containing a low level of NO-activated GC and the native HEK cell PDE (GClowPDEHEK) were advantageous for analyzing the utility of the biosensor. The fluorescence of δ-FlincG rose during the superfusion of NO (0.3–30 nM for 1 min) and declined slowly following washout (Fig. 1 A and B). Maximum signals, implying cGMP concentrations of 1 μM or more (10), were observed at and above 3 nM NO. The responses were inhibited by the NO receptor blocker, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ; ref. 14) and the response decay on washout of NO was greatly slowed by the PDE inhibitor, 3-isobutyl-1-methylxanthine (IBMX, 100 μM; Figs. S1 A and C). IBMX did not significantly affect the rising phase, however, indicating that this phase overwhelmingly reflects NO-stimulated GC activity, the PDE activity being too low to contribute (Fig. S1 A and B). To analyze the GC component, fluorescent responses in Fig. 1B were converted into cGMP concentrations (see SI Materials and Methods), which were seen to rise largely linearly with time, allowing GC activities to be measured. Plotting these activities against NO concentration showed a peak at 10 nM NO followed by a decline, a profile symptomatic of receptor desensitization (Fig. S1 D and E). The EC50 of the rising phase of the curve was 6.9 nM, which is very close to the value found in platelets and cerebellar astrocytes (6, 15). An explicit enzyme-linked receptor mechanism for NO-activated GC has been formulated from detailed analysis of the purified protein (16) and the cellular version of it (Fig. 2A) was previously found to emulate cGMP responses in both these cell types accurately (15). The EC50 determined from the δ-FlincG results was essentially identical to the value (6.0 nM) predicted by this model (Fig. S1E; without desensitization the predicted EC50 is 10 nM). A repeat response to 1 nM NO at the end of the experiment (Fig. 1 A and B) gave a smaller response with a slower rising phase than at the start, signifying desensitization. A similar reduction was seen after a single exposure to 30 nM NO (Fig. S2) and the underlying degree of desensitization derived from the data (37 ± 1% from four experiments of this type) was the same as predicted by the model (38%). Thus, δ-FlincG reports quantitatively coherent changes in cellular cGMP concentration and the GC component derived from δ-FlincG responses appears identical to its counterpart determined by direct measurement in native cells. This agreement extended to the other three cell models tested here because their NO concentration–δ-FlincG response curves all adhered to the predictions of the receptor model (Fig. S1F). Of particular importance to later experiments, the biosensor also reported dynamic (subsecond) changes in cellular cGMP concentration without measurable distortion (Fig. S3). These tests support the use of δ-FlincG for quantitative real-time cGMP imaging.
Fig. 1.

Behavior of GClowPDEHEK cells in response to perfusion (1 min) of different clamped NO concentrations. (A) Responses of a single cell, illustrating how baselining was applied (orange broken line); the mean population responses (seven cells) are shown in B. The red line fits the data to the GC/PDEHEK model (Fig. 2 and SI Materials and Methods) with GCmax = 0.235 μM/s and PDEmax = 0.036 μM/s; for the final response, PDEmax was raised to 0.08 μM/s to fit the faster decay (blue line). The changes in the principal NO receptor species during the course of the experiment, according to the model, are shown in C; with reference to Fig. 2A, Unliganded, GC; Active, NOGC*; and Desensitized, GC*NO. (D) Predicted changes in cGMP concentration giving rise to the fluorescent changes depicted in B, together with the profiles of applied NO concentration (calibrated from the kinetics of fluorescein wash-in and wash-out; see SI Materials and Methods).
Fig. 2.

Model for NO-activated GC (A) and PDE5 (B). The parameters in A had the following values (15): k1 = 3 × 108 M-1 s-1, k-1 = 6 s-1, k2 = k-2 = 28 s-1, k3 = 107 M-1 s-1, k-3 = 1000 s-1, k4 = 2000 s-1, k-4 = 1.8 × 106 M-1 s-1, k5 = 7.34 × 10-4 s-1, k-5 = 4 × 108 M-1 s-1, k6 = 1 s-1, k-6 = 10-3 s-1. The PDE5 parameters are given in Table 2 and the text. Data analysis (see SI Materials and Methods) generated indistinguishable Kp and PDEmax values for the two active PDE5 species (tPDE5* and pPDE5*), and so they were designated equal.
With GClowPDEHEK cells, most of the response decay takes place after washout of NO, and so just reflects PDE activity (Fig. S1A). After the repeat application of 1 nM NO at the end of the experiment in Fig. 1B, the decay rate was faster than at the start. As with desensitization, this speeding up could be observed after a single 30 nM NO application (Fig. S2). Based on pharmacological evidence, PDEHEK appeared most similar to the PDE1 subtype (Fig. S4). Accordingly, the results from the GClowPDEHEK cells (Fig. 1B and Figs. S1, S2, and S4) could be simulated (red lines) by the receptor model incorporating a mean GCmax of 0.25 μM/s, together with a Michaelis–Menten-type PDE having a Km of 4 μM and a mean maximal activity of 44.6 nM/s, increasing on average by 75% following exposure to 30 nM NO. The results in Fig. 1 A and B can be interpreted, therefore, as an NO concentration-dependent rise in cGMP (into the low micromolar range) that becomes progressively curtailed because of slowly reversing NO-dependent desensitization of the GC limb of the pathway, with the decay of cGMP back to baseline following NO washout taking several minutes because of the low PDEHEK activity (Fig. 1 C and D).
GChighPDEHEK Cells.
Cells having more GC but no additional PDE (13) exhibited unprecedented sensitivity to NO in that a concentration as low as 1 pM generated a measurable signal (Fig. S1F). In an example (Fig. 3A), 3 and 10 pM NO gave responses of increasing amplitude with, as before, decay taking several minutes. With this high level of sensitivity, we anticipated that the cells would be able to detect environmentally derived NO, which is expected to be 10–100 pM depending on conditions (16). As a test, the NO scavenger (CPTIO) routinely present in the superfusion solution was removed, whereupon δ-FlincG fluorescence rose to the plateau previously seen on applying 100 pM NO. This response to CPTIO removal was inhibited by blocking NO receptors with ODQ and was restored with 8-bromo-cGMP (Fig. 3A). The NO-evoked changes could be replicated (red line, Fig. 3A) assuming a GCmax about 100-fold higher than in the GClow cells (20 μM/s) together with the native PDEHEK activity (Table 1). The ambient NO concentration accounting for the response to CPTIO removal was 12 pM which, from the simulation, raised cGMP into the low micromolar range (Fig. 3B).
Fig. 3.

Behavior of GChighPDEHEK cells (A, 5 cells), GChighPDE5low cells (C, 17 cells), and GCmidPDE5high cells (D, 22 cells) on perfusion of clamped NO concentrations. A also shows the effect of removal of the NO scavenger, CPTIO (100 μM), followed by perfusion of the NO antagonist, ODQ (3 μM), and restoration of the fluorescence with 8-bromo-cGMP (8-Br-cGMP, 1 mM) at the end. The red lines are fits to the GC/PDEHEK model (A, GCmax = 20 μM/s, PDEmax = 0.035 μM/s; C, GCmax = 5 μM/s, PDEmax = 2.5 μM/s; D, GCmax = 3 μM/s, PDEmax = 18 μM/s). (B) Changes in NO and cGMP concentrations for the simulation in A. The Inset in C shows the response of a cell in a different experiment to a 95-s exposure to 30 pM NO (points, Left) simulated with the model (red line), and the predicted changes in cGMP concentration, GC, and PDE activities (Right; scale bar for GC and PDE activities, 0.05 μM/s; GCmax = 21.5 μM/s, PDEmax = 3.2 μM/s). The Inset in D is an expansion of the response to 10 nM NO (Left) together with the predicted corresponding change in cGMP and the NO concentration profile (right). Note in C and D the small rapid undershoots and slow rebound increases in fluorescence following exposure to the high NO concentrations. Their origin has not been investigated because of their small size and poor reproducibility between experiments.
GChighPDE5low and GCmidPDE5high Cells.
When supplemented with low or high levels of PDE5, the maximum PDE activity was raised 50- or 350-fold (Table 1). Nevertheless, the GChigh cells still showed remarkable NO sensitivity, with concentrations down to 10 pM being detectable by the PDE5low cells (Fig. 3C and Fig. S1F). The GCmidPDE5high cells were an order of magnitude less sensitive (Fig. 3D and Fig. S1F). The most obvious difference with PDE5 present was a fade of the response during NO applications ( Fig. 3 C and D, Insets), similar to the fade in cGMP levels observed on exposure of these cells to S-nitrosoglutathione (13), and a much more rapid recovery on removal of NO. Interpretation of these records requires knowledge of the kinetics of PDE5, which was found using a different method of NO delivery in the following experiments.
Exposing the PDE5 Kinetics Using NO Puffs.
NO was applied locally to the GChighPDE5low cells by brief puffs from a pipette containing 1 nM NO (Fig. 4A). The amplitudes and kinetics of the NO exposures were calibrated using Texas red dye (Fig. S5 and Movie S1). With a 1-s puff, the peak NO concentration surrounding the cells of interest reached 0.5 nM. To capture the transient fluorescent signal faithfully, the data had to be acquired at a more rapid rate than before (usually one frame every 0.75 s rather than every 2–5 s). This increased frame rate led to the appearance of a “background” fluorescent signal, attributable to cGMP generation as a result of the well-known phenomenon of light-induced NO release (Fig. S6) which was estimated usually to provide about 3 pM NO (SI Materials and Methods). When this background was exaggerated, a 1-s puff of NO led to a long-term suppression of δ-FlincG fluorescence, signifying that the single NO puff induced an enduring period of enhanced PDE5 activity (Fig. S6). The kinetics of PDE5 activation and recovery was analyzed by delivering a series of four puffs spaced 12–30 s apart, followed by test puffs at various intervals (up to 10 min). With 12-s spacing, the responses to the four puffs initially overlapped but then became progressively discrete, a pattern that was associated with a speeding of the decay phases (Fig. 4B and Inset). With 30-s spacing, the thinning of the response, together with a loss of background cGMP, appeared complete before the second puff and both the background and the response to the puff remained suppressed 4 min after the last of the “induction” puffs (Fig. 4C). As a rough measure of the rate of recovery, the width of the responses at half-height was measured relative to the last induction puff. In all four experiments of this type, recovery was incomplete after 10 min (Fig. 4F).
Fig. 4.

Puff applications of NO to GChighPDE5low cells. (A) Sequence of images showing the transient δ-FlincG fluorescence increase in a field of cells subjected to a 2-s puff of NO from a neighboring pipette. The final frame shows the δ-FlincG response (red line) in the cell outlined in red in the first frame in relation to the delivered NO concentration (black broken line). (B) Four NO puffs were applied 12 s apart; data are means of five runs (six cells) and are fitted by the GC/PDE5 model (GCmax = 15.2 μM/s, PDEmax = 2.34 μM/s). The Inset shows the responses aligned, numbered according to their order. (C) Four NO puffs were given 30 s apart, followed by a test puff 4 min after the last one, showing only partial recovery (three cells). The red line is a fit to the model (GCmax = 35.5 μM/s, PDEmax = 4.17 μM/s). (D) Predicted changes in cGMP concentration from the model in relation to the NO concentration profiles in the experiment in B. (E) The cGMP concentration changes predicted for the experiment in C. (F) Rates of recovery in four experiments (each in a different color) quantified from measurement of the width of the responses at half-height (using the “Peak Analyzer” in OriginPro 8) and expressed relative to the width of the last of the induction puffs (6.94 ± 0.69 s). (G) Changes in the amount of transiently (tPDE5*) and persistently (pPDE5*) active PDE5 species during the experiment shown in C, according to the model.
A minimum model of the PDE5 kinetics comprises a two-step activation by cGMP forming a transiently active species, tPDE5*, and then a cGMP-dependent transition to a persistently active form, pPDE5* (Fig. 2B), most likely representing phosphorylated PDE5 known to be formed in these cells on exposure to NO (13, 15) and which is slow to reverse (13, 17). With such brief puffs, the decay of the response occurs after NO washout and so purely reflects PDE activity. Moreover, the protocols generated information rich enough to yield values of all the kinetic constants governing PDE5 activation and recovery from each experiment (see Table 2 and SI Materials and Methods). On combining the PDE5 and NO receptor models, the results could be recapitulated (red lines, Fig. 4 B and C), allowing the underlying changes in cGMP to be elucidated (Fig. 4 D and E). Thus, the induction phase is seen to comprise rapid activation of GC and the attendant rise in cGMP, and then a relatively slow activation of PDE5 by cGMP, forming first tPDE5* and then pPDE5*, which ultimately becomes the dominant species (Fig. 4G). Only about 5% of the total PDE5 content needs to be in the persistently active state (pPDE5*) to account for the prolonged response suppression, and the half-time for recovery from this state is about 10 min. The model also accurately depicts the behavior of GChighPDE5low and GCmidPDE5high cells superfused with NO (red lines, Fig. 3 C and D), allowing the triphasic shapes of the responses to be understood in terms of the underlying alterations in GC and PDE5 activities (Fig. 3 C and D, Insets). A combination of desensitization (45–46%) and the buildup of pPDE5* (to 20% and 2.5% of total in Fig. 3 C and D, respectively) explains the near-complete suppression of the responses to 100 pM or 1 nM NO seen at the end. On average, GCmax in GCmidPDE5high cells was a quarter of that of the GChighPDE5low cells, whereas PDEmax was fivefold higher (Table 1; see also ref. 13).
Table 2.
PDE5 kinetic parameters
| Parameter | Mean | SEM | Units |
| Kp | 0.351* | 0.052 | μM |
| kp1 | 1.81 × 104† | 0.67 × 104 | M-1 s-1 |
| kp-1 | 0.0868† | 0.0129 | s-1 |
| kp2 | 0.246† | 0.054 | s-1 |
| kp-2 | 0.154† | 0.044 | s-1 |
| kp3 | 1.13 × 105‡ | 0.02 × 105 | M-1 s-1 |
| kp-3 | 1.21 × 10-3‡ | 0.19 × 10-3 | s-1 |
*Mean median value in four experiments (three to four runs per experiment); the overall mean (n = 14) was 0.45 ± 0.14 μM.
†From the averaged induction phases of four experiments.
‡From eight runs in four experiments. Leaving aside the transition to pPDE5* (Fig. 2B), the cGMP agonist affinity for PDE5 (kp-1/kp1) is 4.8 μM and the cGMP EC50 [the affinity divided by (1 + kp2/kp-2)] is 1.85 μM. The computer-derived values of the parameters kp1, kp-1, kp2, and kp-2 in the table are closely comparable to those deduced manually from cGMP measurement in rat platelets, where no persistent PDE5 activity was discernible (15).
Cellular NO Capture.
To evaluate the ability of cells to capture NO, GChighPDE5low cells were exposed to a wide range of puff durations (100 ms–30 s), with 1 nM NO in the puffer pipette. A puff lasting 100 ms was just detectable (Fig. 5A). From the calibrations, the peak applied NO concentration was 56 pM but, with such brief puffs, the intracellular concentration is expected to be lower because diffusion of NO through unstirred layers becomes limiting (see Fig. 5B, Inset and SI Materials and Methods). With the 100-ms puff, the calculated peak intracellular NO concentration is 23 pM. The response amplitude rose with increasing puff durations (Fig. 5A) in accordance with the rise in peak NO concentration (Fig. 5A, Inset), the maximum size being reached with a 3-s puff (peak [NO] = 0.85 nM). As was seen when NO was superfused (Fig. 3C), the response faded during the 30-s puff (peak [NO] = 1 nM) and subsequent 1-s puffs (peak [NO] = 0.5 nM) gave response amplitudes that were much reduced compared with earlier. Also evident was a cumulative speeding of the response decay as the experiment progressed. This experiment provides a rigorous test of the complete model for NO signal transduction developed above because it interrogates all aspects of it, except for receptor desensitization which is negligible at these low NO concentrations (6, 15). The model output (red line) mimicked the experimental data (Fig. 5A), allowing them to be explained by the underlying time-dependent alterations in GC and PDE5 activities (Fig. 5 B and C). Movie S2 depicts a shorter experiment of this type.
Fig. 5.

Effect of varying the NO puff duration. (A) NO puffs lasting 0.1–30 s were delivered to a GChighPDE5low cell, with repeat 1-s puffs at the end. The Inset plots the peak NO concentration and peak δ-FlincG response for the different puff durations. The red line is a fit to the GC/PDE5 model (GCmax = 17.5 μM/s, PDEmax = 3.23 μM/s). (B) Predicted changes in cGMP concentration in relation to the NO concentration profiles. The Inset illustrates the effect of diffusion through unstirred layers on the access of NO to the intracellular compartment at two sample NO puff durations (see SI Materials and Methods). (C) Changes in the amount of transiently (tPDE5*) and persistently (pPDE5*) active PDE5 species during the experiment shown in A, according to the model.
Discussion
The methodology used here has allowed analysis of cellular NO signal transduction at the subnanomolar concentrations likely to operate physiologically, and in real time. From the results, NO-receptive cells are seen to be easily the fastest and most sensitive NO detectors yet encountered. Furthermore, the finding that the primary steps of cellular NO signal transduction can be encapsulated in a quantitative model provides a conceptual understanding of the underlying principles.
NO synthases are complex enzymes that generate NO only slowly, typically at just a few molecules per second (18). Based on their NO synthase content and taking into account NO consumption by circulating red blood cells, it has been calculated that endothelial cells could donate only 20–100 pM NO to the underlying smooth muscle (19). From assumptions about the sensitivity of NO-activated GC, such concentrations were considered unrealistically low, prompting speculation by the authors that additional sources of NO must be present. NO release at brain synapses is similarly predicted to generate, at the extreme, only subnanomolar NO concentrations in closely neighboring structures (20). In showing that such low NO concentrations are biologically active, the results give credence to these theoretical estimates, as does the observation made with an NO detector cell line that 40–100 pM NO exists just outside stimulated endothelial or neuronal cells in vitro (21). At best (GChighPDEHEK cells), an NO concentration as low as 3 pM was seen to generate a signal corresponding to about 0.13 μM cGMP, a concentration well within range of downstream cGMP-dependent protein kinases (22, 23). This extraordinary degree of sensitivity is unlikely to be of merely theoretical interest: Few native cell types have been subjected to the necessary examination but one that stands out is the cerebellar astrocyte, a nonneuronal cell type having a high GC activity and a very low PDE content dominated by PDE5 (24, 25). Reanalysis of published data on cGMP responses to NO in cerebellar astrocytes (15) using the present model suggests maximal GC and PDE5 activities of 95 and 0.5 μM/s, respectively, from which it is predicted that these cells would react to just 1 pM NO with a rise of cGMP into the 0.1-μM range. This level of sensitivity suggests that the astrocytes are tuned to integrate NO signals at a distance from their source(s), presumably in synapses or blood vessels. Even cells with more closely matched GC and PDE activities (GChighPDE5low cells) responded to NO in the low-picomolar range. The triphasic cGMP response shape (a peak followed by a quasi-plateau) and slower kinetics found in these cells on prolonged NO exposure most closely resembles that reported for smooth muscle (5, 26). GCmax and PDEmax in rat platelets are higher and more similar at around 100 μM/s each (6, 15). Reflecting this higher activity, the cGMP responses should be faster and more transient than in GChighPDE5low cells, as has been observed experimentally using traditional cGMP measurements and high (> 1 nM) clamped NO concentrations (6). Nevertheless, the mix in rat platelets should still allow 10 pM NO to generate a peak cGMP concentration of more than 0.1 μM.
Analyzing cell signaling pathways quantitatively is one of the major current aims in biology but success in this respect for NO has appeared a rather distant prospect. Gratifyingly, the key GC and PDE5 kinetics to emerge from the δ-FlincG recordings in HEK cells cohere well with what had been deduced from crude radioimmunoassay measurements of cGMP accumulation in native cells (6, 9, 15, 24, 27). That the complexities of NO signal transduction in several different cell types are reducible to variations on a common mechanism suggests that there are general principles at work and the elaboration of an in silico model serves to illuminate those principles.
First, the extraordinary sensitivity of cells to NO shown here is primarily the consequence of the properties of its receptor which endows NO with biological activity even at concentrations some 10,000-fold lower than its EC50 (10 nM in cells) or binding affinity (Kd = 20 nM; Fig. 2A), when only about 0.01% of the receptor population would be occupied by agonist. This scenario is very unusual but, for an enzyme-linked receptor, the efficiency of signal transduction (defined as the enzyme output per unit agonist concentration) reaches its highest value under these conditions. Formally, when [NO] ≪ Kd, the steady-state transduction efficiency is simply the ratio of GCmax to the NO binding Kd *, which comes to about 1,000 s-1 for GChigh HEK cells, or 5,000 s-1 for rat platelets and cerebellar astrocytes. Thus, in the latter cell types, 10 pM NO results in 50 nM cGMP being formed each second at steady state. Moreover, the rapidity of the receptor kinetics ensures that 90% of the steady-state output is achieved in about 800 ms, which helps explain the efficaciousness of brief NO puffs (Fig. 5).
The other factor accounting for the high cellular NO sensitivity is related to the PDE activity, either because it is low (GChighPDEHEK cells) or because of the behavior of PDE5. The PDE5 isoform has a low affinity for activation by cGMP (Kd = 5 μM; Table 2) and is switched on slowly (half-time about 20 s at 0.1 μM cGMP), allowing cGMP to accumulate before being hydrolyzed (see, e.g., Fig. 3C). The overall degree of signal amplification (defined as the ratio of the concentration-time integrals of cGMP produced to NO delivered) is thus favored by relatively brief NO exposures. With a 1-s exposure to NO concentrations giving a peak cGMP concentration of about 0.1 μM, the amplification comes to about 20,000 for both the GChighPDE5low cells and rat platelets and about 0.3 million for cerebellar astrocytes (assuming no basal PDE5 activity in each case). Usually, cells utilize multicomponent cascades to produce these high levels of amplification and it is remarkable that they can be achieved with the simplest possible one-component transducer. A problem inherent in any high-amplification device, however, is that it can easily saturate with a strong signal. Short- or longer-term alterations in the PDE5 activity and/or changes in receptor availability (amount of desensitization) combat this problem by adjusting the gain according to the past history of NO exposure.
Such highly efficient NO capture and signal transduction relies on a very unusual arrangement. In conventional transmission, the numbers of agonist molecules released are greatly in excess of the numbers of receptors with which they can combine. In excitatory brain synapses, for example, the ratio is about 30∶1 (28). Based on the maximal activity of highly purified NO-activated GC (taken as 20 μmol·mg-1·min-1) and the molecular mass of the protein (150 kDa), a maximal activity in cells of 100 μM/s (as found in rat platelets and cerebellar astrocytes; see above) implies a cellular NO receptor concentration of 2 μM, which is up to a millionfold higher than the NO concentrations predicted to be active physiologically. This excess means that NO-receptive cells will act as powerful sinks for the NO generated in the vicinity which, in turn, will create a gradient for the diffusion of NO into those cells. This “ligand capture” by target cells helps answer the puzzle of how a transient low-level NO signal that is subject to rapid three-dimensional dispersal is harnessed to biological advantage. It also predicts that there would be little free NO available in and around target cells for other reactions.
Materials and Methods
Full details are given in SI Materials and Methods. Briefly, the HEK 293T cell lines called GChighPDEHEK, GChighPDE5low, and GCmidPDE5high were donated by Doris Koesling (Ruhr-Universität Bochum, Bochum, Germany) and the GClowPDEHEK cell line was generated in-house. The cells were grown on coverslips, infected with a recombinant adenovirus expressing δ-FlincG, and used 1–3 d later. For imaging, which was carried out using an inverted microscope, the coverslips were held in a chamber (0.5 mL volume) that was continuously superfused (1.5 mL/ min) with warm (37 °C) solution containing (millimolar): NaCl (136), KCl (2), MgSO4 (1.2), KH2PO4 (1.2), CaCl2 (1.5), glucose (5.5), Hepes (10), NG-nitro-l-arginine (0.03), superoxide dismutase (100 units/mL), CPTIO (0.1), and urate (0.3), pH 7.4. With CPTIO and urate present, addition of the slow NO releaser 1-hydroxy-2-oxo-3-(N-ethyl-2-aminoethyl)-3-ethyl-1-triazene (half-life = 100 min) produces clamped NO concentrations (11) which were applied either by superfusion or from a nearby puffer pipette; access of NO to the cells was quantified by superfusion of fluorescein or by including Texas red dye in the pipette, respectively. Epifluorescent signals were captured by camera, corrected for background, and displayed as the change in intensity relative to baseline divided by the baseline intensity (ΔF/F0). The model (Fig. 2) was implemented in Mathcad 14 (Parametric Technology Corporation). The “Minerr” routine in this software was used to extract the PDE5 kinetic parameters when NO was delivered by puffer pipette but it could not cope with the much longer superfusion experiments which, as a consequence, were fitted manually by minimizing the sum of the squares of the errors between experimental result and model prediction. Data are presented as means ± SEM.
Supplementary Material
Acknowledgments.
We are grateful to Prof. Doris Koesling (Ruhr-Universität Bochum, Bochum, Germany) for generously supplying HEK cell lines. Supported by The Wellcome Trust Programme Grant 081512 (to J.G.). Additional support was provided by National Institutes of Health Grants HL68891 (to W.R.D.) and T323 HL07944 (to K.F.H.), the Totman Trust for Biomedical Research (W.R.D.), and a bursary from the Jean Shanks Foundation (E.J.H.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1013147107/-/DCSupplemental.
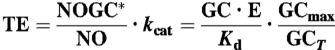 |
[1] |
References
- 1.Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moncada S, Higgs EA. Nitric oxide and the vascular endothelium. Handb Exp Pharmacol. 2006;176(Pt 1):213–254. doi: 10.1007/3-540-32967-6_7. [DOI] [PubMed] [Google Scholar]
- 3.Ignarro LJ. Signal transduction mechanisms involving nitric oxide. Biochem Pharmacol. 1991;41:485–490. doi: 10.1016/0006-2952(91)90618-f. [DOI] [PubMed] [Google Scholar]
- 4.Murad F. The nitric oxide-cyclic GMP signal transduction system for intracellular and intercellular communication. Recent Prog Horm Res. 1994;49:239–248. doi: 10.1016/b978-0-12-571149-4.50016-7. [DOI] [PubMed] [Google Scholar]
- 5.Mullershausen F, Lange A, Mergia E, Friebe A, Koesling D. Desensitization of NO/cGMP signaling in smooth muscle: Blood vessels versus airways. Mol Pharmacol. 2006;69:1969–1974. doi: 10.1124/mol.105.020909. [DOI] [PubMed] [Google Scholar]
- 6.Mo E, Amin H, Bianco IH, Garthwaite J. Kinetics of a cellular nitric oxide/cGMP/phosphodiesterase-5 pathway. J Biol Chem. 2004;279:26149–26158. doi: 10.1074/jbc.M400916200. [DOI] [PubMed] [Google Scholar]
- 7.Mergia E, Friebe A, Dangel O, Russwurm M, Koesling D. Spare guanylyl cyclase NO receptors ensure high NO sensitivity in the vascular system. J Clin Invest. 2006;116:1731–1737. doi: 10.1172/JCI27657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy B, Mo E, Vernon J, Garthwaite J. Probing the presence of the ligand-binding haem in cellular nitric oxide receptors. Br J Pharmacol. 2008;153:1495–1504. doi: 10.1038/sj.bjp.0707687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bellamy TC, Garthwaite J. Sub-second kinetics of the nitric oxide receptor, soluble guanylyl cyclase, in intact cerebellar cells. J Biol Chem. 2001;276:4287–4292. doi: 10.1074/jbc.M006677200. [DOI] [PubMed] [Google Scholar]
- 10.Nausch LW, Ledoux J, Bonev AD, Nelson MT, Dostmann WR. Differential patterning of cGMP in vascular smooth muscle cells revealed by single GFP-linked biosensors. Proc Natl Acad Sci USA. 2008;105:365–370. doi: 10.1073/pnas.0710387105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Griffiths C, Wykes V, Bellamy TC, Garthwaite J. A new and simple method for delivering clamped nitric oxide concentrations in the physiological range: Application to activation of guanylyl cyclase-coupled nitric oxide receptors. Mol Pharmacol. 2003;64:1349–1356. doi: 10.1124/mol.64.6.1349. [DOI] [PubMed] [Google Scholar]
- 12.Bender AT, Beavo JA. Cyclic nucleotide phosphodiesterases: molecular regulation to clinical use. Pharmacol Rev. 2006;58:488–520. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 13.Mullershausen F, Russwurm M, Koesling D, Friebe A. In vivo reconstitution of the negative feedback in nitric oxide/cGMP signaling: role of phosphodiesterase type 5 phosphorylation. Mol Biol Cell. 2004;15:4023–4030. doi: 10.1091/mbc.E03-12-0890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Garthwaite J, et al. Potent and selective inhibition of nitric oxide-sensitive guanylyl cyclase by 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one. Mol Pharmacol. 1995;48:184–188. [PubMed] [Google Scholar]
- 15.Halvey EJ, Vernon J, Roy B, Garthwaite J. Mechanisms of activity-dependent plasticity in cellular nitric oxide-cGMP signaling. J Biol Chem. 2009;284:25630–25641. doi: 10.1074/jbc.M109.030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roy B, Halvey EJ, Garthwaite J. An enzyme-linked receptor mechanism for nitric oxide-activated guanylyl cyclase. J Biol Chem. 2008;283:18841–18851. doi: 10.1074/jbc.M801712200. [DOI] [PubMed] [Google Scholar]
- 17.Mullershausen F, et al. Direct activation of PDE5 by cGMP: Long-term effects within NO/cGMP signaling. J Cell Biol. 2003;160:719–727. doi: 10.1083/jcb.200211041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stuehr DJ, Santolini J, Wang ZQ, Wei CC, Adak S. Update on mechanism and catalytic regulation in the NO synthases. J Biol Chem. 2004;279:36167–36170. doi: 10.1074/jbc.R400017200. [DOI] [PubMed] [Google Scholar]
- 19.Chen K, Popel AS. Theoretical analysis of biochemical pathways of nitric oxide release from vascular endothelial cells. Free Radical Biol Med. 2006;41:668–680. doi: 10.1016/j.freeradbiomed.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 20.Hall CN, Garthwaite J. What is the real physiological NO concentration in vivo? Nitric Oxide. 2009;21:92–103. doi: 10.1016/j.niox.2009.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sato M, Nakajima T, Goto M, Umezawa Y. Cell-based indicator to visualize picomolar dynamics of nitric oxide release from living cells. Anal Chem. 2006;78:8175–8182. doi: 10.1021/ac061791b. [DOI] [PubMed] [Google Scholar]
- 22.Francis SH, Corbin JD. Structure and function of cyclic nucleotide-dependent protein kinases. Annu Rev Physiol. 1994;56:237–272. doi: 10.1146/annurev.ph.56.030194.001321. [DOI] [PubMed] [Google Scholar]
- 23.Vaandrager AB, Hogema BM, de Jonge HR. Molecular properties and biological functions of cGMP-dependent protein kinase II. Front Biosci. 2005;10:2150–2164. doi: 10.2741/1687. [DOI] [PubMed] [Google Scholar]
- 24.Bellamy TC, Wood J, Goodwin DA, Garthwaite J. Rapid desensitization of the nitric oxide receptor, soluble guanylyl cyclase, underlies diversity of cellular cGMP responses. Proc Natl Acad Sci USA. 2000;97:2928–2933. doi: 10.1073/pnas.97.6.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bellamy TC, Garthwaite J. “cAMP-specific” phosphodiesterase contributes to cGMP degradation in cerebellar cells exposed to nitric oxide. Mol Pharmacol. 2001;59:54–61. doi: 10.1124/mol.59.1.54. [DOI] [PubMed] [Google Scholar]
- 26.Mullershausen F, et al. Rapid nitric oxide-induced desensitization of the cGMP response is caused by increased activity of phosphodiesterase type 5 paralleled by phosphorylation of the enzyme. J Cell Biol. 2001;155:271–278. doi: 10.1083/jcb.200107001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roy B, Garthwaite J. Nitric oxide activation of guanylyl cyclase in cells revisited. Proc Natl Acad Sci USA. 2006;103:12185–12190. doi: 10.1073/pnas.0602544103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Franks KM, Bartol TM, Jr, Sejnowski TJ. A Monte Carlo model reveals independent signaling at central glutamatergic synapses. Biophys J. 2002;83:2333–2348. doi: 10.1016/S0006-3495(02)75248-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.