

Abstract
The existence of a local renin angiotensin system (RAS) of the kidney has been established. Angiotensinogen (AGT), renin, angiotensin-converting enzyme (ACE), angiotensin receptors, and high concentrations of luminal angiotensin II have been found in the proximal tubule. Although functional data have documented the relevance of a local RAS, the dualism between biosynthesis and endocytotic uptake of its components and their cellular processing has been incompletely understood. To resolve this, we have selectively analyzed their distribution, endocytosis, transcytosis, and biosynthesis in the proximal tubule. The presence of immunoreactive AGT, restricted to the early proximal tubule, was due to its retrieval from the ultrafiltrate and storage in endosomal and lysosomal compartments. Cellular uptake was demonstrated by autoradiography of radiolabeled AGT and depended on intact endocytosis. AGT was identified as a ligand of the multiple ligand-binding repeats of megalin. AGT biosynthesis was restricted to the proximal straight tubule, revealing substantial AGT mRNA expression. Transgenic AGT overexpression under the control of an endogenous promoter was also restricted to the late proximal tubule. Proximal handling of renin largely followed the patterns of AGT, whereas its local biosynthesis was not significant. Transcytotic transport of AGT in a proximal cell line revealed a 5% recovery rate after 1 h. ACE was expressed along late proximal brush-border membrane, whereas ACE2 was present along the entire segment. Surface expression of ACE and ACE2 differed as a function of endocytosis. Our data on the localization and cellular processing of RAS components provide new aspects of the functional concept of a “self-contained” renal RAS.
Keywords: Endocytosis, Endosomes, Epithelium, Kidney, Lysosomes, Glomerular Filtration, Nephron, Transport
Introduction
The renin angiotensin system (RAS)3 serves to maintain extracellular volume homeostasis, a task that is primarily achieved by the kidney. Based on the notion that significant amounts of angiotensin (Ang) II can be formed locally, elements of an intrarenal, “self-contained” RAS have been investigated. These have been assigned chiefly to the proximal tubule where angiotensinogen (AGT), renin, angiotensin-converting enzyme (ACE), basolateral and brush-border membrane angiotensin receptors, and high concentrations of Ang II have been localized (for a review, see Refs. 1–4). Inappropriate activation of these components may lead to the development and maintenance of hypertension (5) and in the long run may cause renal injury (2). The role of Ang II as a sodium- and water-retaining hormone in proximal tubule has been shown by a variety of techniques including single nephron microperfusion and whole animal studies in normal and transgenic organisms. Its effects on proximal volume, salt, and bicarbonate reabsorption as well as on short term activation of sodium-coupled transporters have thus been convincingly demonstrated (6–8), and a focus of its action has been assigned to the early proximal tubule (9). Substantial intratubular Ang II generation has further stimulated the study of its origin in the proximal tubule (10, 11). Here, AGT, the only known substrate for renin, has been localized at the mRNA level, and immunoreactive AGT has been found near or in the brush-border, albeit without clear segmental assignment (12–18). It has been suggested that in contrast to renin (36–40 kDa), AGT is not filtered through the glomerulus because of its large size (61–65 kDa) (15, 16, 19) but is rather secreted into the tubular lumen where substantial concentrations have been measured (300 pmol/ml; 20). In line with this, significant AGT urinary excretion rates have been found at steady state and were enhanced under stimulation by Ang II (21).
Renin has also been localized to the proximal tubule where it was suggested to be endocytosed from the filtrate and stored in subapical vesicles (22, 23), and low but measurable renin concentrations have been determined in the tubule fluid (24). Nonetheless, substantial intratubular levels of Ang I (10-fold greater than in plasma) appear to be formed locally (for review, see Refs. 1, 2). Renin mRNA in the proximal tubule can only be detected by application of highly sensitive PCR techniques (25) so that its local de novo synthesis is likely of limited importance.
ACE, which is abundantly expressed in proximal brush-border membrane, may provide the proximity of enzyme-substrate interaction necessary for generating intratubular Ang II, although alternate, ACE-independent pathways appear to exist (1). Whatever the source of conversion, proximal tubule Ang II levels were approximately 100-fold higher than those in plasma, confirming that the proximal tubule produces and secretes Ang II at levels greater than can be accounted for by glomerular filtration (1, 11, 20). The role of ACE2, an ACE homologue that is highly expressed in the proximal tubule, has received recent interest because ACE2 may counterbalance classical RAS effects in part by metabolizing Ang II to the heptapeptide, Ang-(1–7), which activates the receptor Mas, thus antagonizing the effects of local Ang II (26, 27).
Despite abundant evidence for the expression and function of RAS components in the proximal nephron, we are still left with an incomplete picture of their precise localization with regard to the dualism between local biosynthesis and endocytotic uptake/intracellular processing. In the present study we therefore focused on renin-substrate handling and revisited the presence of other RAS components in proximal tubule. To understand the role of endocytosis herein, we have studied the principal scavenger receptor megalin, which is central to the uptake of a wide spectrum of ligands from proximal tubular fluid by means of its large extracellular ligand-binding domains (28, 29). We could demonstrate (i) that substantial amounts of AGT, as shown earlier for renin, reach the primary ultrafiltrate and that AGT is endocytosed via megalin; (ii) that renin distribution follows the pattern of AGT; (iii) that uptake of these components is restricted to the early proximal tubule, whereas the late proximal tubule only shows AGT mRNA transcription but no cellular storage; (iv) that ACE expression is also concentrated in late proximal tubule, whereas ACE2 is encountered along the entire proximal nephron; and (v) that ACE and ACE2 display regulatory differences with respect to their apical representation. These data allowed us to reconsider the contributions of individual RAS components along the proximal nephron.
EXPERIMENTAL PROCEDURES
Animals and Cells
Experiments were performed on male Hannover Sprague-Dawley (SD) rats, transgenic rats overexpressing human AGT (TGR[hAGT]/line 1623) (30), and megalin conditional knock-out mice (megalin lox/lox; apoE Cre) (31) termed Cre(+) and control mice (megalin lox/lox) termed Cre(−). The rats weighed 250–300 g, and the mice weighed 20–30 g. Remnant megalin expression in Cre(+) was verified by sampling of urinary vitamin D-binding protein levels (31). The mice (n = 24) were divided into one group for perfusion-fixation and immunohistochemical evaluation and another for biochemical analysis. The animals were allowed free access to standard chow and tap water. The experiments were conducted in accordance to the German Law for the protection of animals. Opossum kidney (OK) cells were provided by J. Biber (Zurich, Switzerland). The cells were cultured at 37 °C in 95% air, 5% CO2 in high glucose (450 mg/dl) DMEM supplemented with 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml). For transport studies, OK cells were seeded on membranes (0.4-μm pore size; PET, Falcon). Before seeding the cells, each filter was pretreated with Matrigel (BD Biosciences). The formation of polarized monolayers was assessed by measuring transepithelial resistance. Monolayers were considered optimal for transport analyses when the trans-epithelial resistance exceeded 180 Ω·cm2.
Autoradiographic Tracing of AGT
Purified AGT from rat plasma (32) was iodinated with Na125I; the 125I-labeled AGT was then separated from the free iodine by high performance liquid chromatography (33). SD rats (n = 4) were ether-anesthetized and received a 125I-AGT bolus of 3 μCi/10 g of body weight, dissolved in 0.9% NaCl, or vehicle via the jugular vein. 20 and 60 min after bolus injection, the rats were perfusion-fixed, and the kidneys were removed, frozen, and sectioned in a cryostat. The sections were mounted on glass slides, kept at −20 °C for 3 weeks to decrease radioactivity, dipped in photoemulsion (Ilford), developed after 1 week of exposure, counterstained with hematoxylin and eosin, and coverslipped.
Fixation and Tissue Processing for Immunohistochemistry
The mice and rats were anesthetized by intraperitoneal injection of sodium pentobarbital (0.06 mg/g of body weight). The kidneys were then perfused retrogradely via the abdominal aorta with 3% paraformaldehyde as described (34). For cryostat sectioning, the tissues were protected from freezing artifacts by subsequent overnight immersion in PBS adjusted to 800 mosmol/kg H2O sucrose, shock frozen, and stored at −80 °C. For the paraffin technique, the tissues were postfixed in 3% paraformaldehyde, dehydrated, and standard paraffin-embedded.
Tissue Processing for Immunoblotting
For isolation of cortical brush-border membrane (BBM), the kidneys were surgically removed and shock frozen, and the cortices were homogenized in isolation buffer containing 300 mm d-mannitol, 5 mm EGTA, 16 mm HEPES, 10 mm Tris base, and protease inhibitor mixture (Complete; Roche Applied Science) as described (35, 36). Purity of the preparation was checked by controlling for the activity of BBM marker enzymes γ-glutamyltransferase and alkaline phosphatase in BBM fractions versus total homogenate using the appropriate kits (Roche Applied Science). Total protein concentration was measured using BCA protein assay reagent kit (Pierce) and controlled by Coomassie staining.
Gel Electrophoresis and SDS-PAGE
After Laemmli's sample buffer was added, the proteins were solubilized at 96 °C for 3 min. SDS gel electrophoresis was performed on 8–10% polyacrylamide gels. After electrophoretic transfer of the proteins to nitrocellulose membranes, equity in protein loading and blotting was verified by membrane staining using 0.1% Ponceau red. The membranes were probed overnight at 4 °C with primary antibodies and then exposed to HRP-conjugated secondary antibodies (DAKO). Immunoreactive bands were detected using an enhanced chemiluminescence kit (Amersham Biosciences) before exposure to x-ray films (Hyperfilm; Amersham Biosciences). For densitometric evaluation of the resulting bands, the films were scanned and analyzed using BIO-PROFIL Bio-1D image software (Vilber Lourmat). All of the parameters were normalized to β-actin abundance.
Antibodies
The following previously characterized or commercially acquired antibodies were used: rabbit anti-rAGT recognizing both hAGT and rAGT (32), goat anti-renin (R & D Systems), guinea pig anti-megalin (37), rabbit anti-ACE (Santa Cruz Biotechnology), rabbit anti-ACE2 (R & D Systems), monoclonal mouse anti-c-Myc (Sigma-Aldrich), rabbit anti-c-Myc (Santa Cruz Biotechnology), monoclonal mouse anti-β-actin (Sigma-Aldrich), rat anti-LAMP-2 (Santa Cruz Biotechnology), mouse anti-Rab11 (BD Transduction), mouse anti-clathrin (Progen), and mouse anti-EEA1 (BD Transduction).
Immunohistochemistry
Immunohistochemical staining was performed on cryostat or paraffin sections of kidneys or on paraformaldehyde-fixed OK cell monolayers. Sections or cells were blocked with 5% skim milk/PBS, incubated with the appropriate antibody and suitable HRP-, cy-2-, or cy-3-coupled secondary antibodies (DAKO, Dianova). For double labeling, the different primary antibodies were administered consecutively. Tissues from at least six rats or mice were evaluated after perfusion fixation. To demonstrate colocalization of different markers by double staining, at least eight cryosections from each tissue were evaluated. Specificity was controlled by parallel incubation of consecutive sections, each incubated only with one single probe. The sections were analyzed using a Leica DMRB microscope or a multilaser confocal scanning microscope (TCS SP-2; Leica Microsystems).
In Situ Hybridization
For nonradioactive in situ hybridization, digoxigenin-labeled sense and antisense riboprobes were generated by in vitro transcription of a 1200-nucleotide rat AGT cDNA fragment from the coding region. In situ hybridization was performed on 5-μm-thick paraffin sections according to an established protocol (34, 38). To more sensitively detect AGT mRNA, radiolabeled in situ hybridization was performed using a 290-nucleotide rat AGT cDNA fragment (PvuII/BamHI) that was subcloned in a pGEM4 vector, linearized with EcoRI, and transcribed in the presence of α-35S-labeled UTP (DuPont) (30). Hybridization was performed on 5-μm-thick cryostat sections, and signal was generated by exposure to photoemulsion (Ilford) covering the sections. The sections were counterstained with hematoxylin and eosin. For control, sense probes were applied in parallel with antisense probes throughout.
Single-nephron Preparation of Proximal Tubule Segments
To quantify steady state AGT mRNA in rat proximal tubules, an established protocol was used (39). Briefly, normal male Hannover SD rats (Janvier, France; 175–215 g of body weight) were deeply anesthetized by isoflurane inhalation; after flank incision the left renal artery was exposed, and the left kidney perfused immediately with 1 mg/ml collagenase type II (PAN Biotech) and 1 mg/ml protease XIV (Roche Applied Science) in 0.8 ml of isolation buffer (140 mmol/liter NaCl, 0.4 mmol/liter KH2PO4, 1.6 mmol/liter K2HPO4, 1 mmol/liter MgSO4, 10 mmol/liter sodium acetate, 1 mmol/liter α-ketoglutarate, 1.3 mmol/liter calcium gluconate, 5 mmol/liter glycine, containing 48 mg/liter trypsin inhibitor and 25 mg/liter DNase I, pH 7.4, at 37 °C). The kidney was removed quickly, decapsulated, cut into slices (0.2–0.4 mm), and transferred in fresh isolation buffer to a thermo shaker (Eppendorf; 850 rpm, 10 min, 37 °C). The resulting tubule suspensions were then transferred to ice-cold isolation buffer containing 1 mg/ml albumin. After sedimentation, the tubules were washed again. Sorting of tubules was performed at 4 °C using a dissection microscope (Leica MZ16) within a time frame of 90 min. The tubules were sorted according to their morphology (supplemental Fig. S1); PT S1 segments were identified by their attachment to glomeruli and tight convolutions, PT S1/S2 segments were identified by their greater length and wider convolutions, and PT S3 segments were identified by their straight course, opaque appearance, slight, corkscrew-like windings, and their transitions to the descending thin limbs. Kidneys from eight rats were prepared. From each rat, three batches were used, each containing 30–50 tubules. In parallel, liver samples from three SD rats were analyzed.
Quantitative RT-PCR
Total RNA was isolated from the isolated tubule preparations using an RNeasy-total-RNA-kit (Qiagen). Contaminating DNA was removed after treatment with DNA-free DNase (Qiagen). cDNA was synthesized by reverse transcription of 5 μg of RNA from each preparation using a cDNA synthesis kit (Invitrogen). Gene expression of AGT was analyzed by quantitative RT-PCR using the TaqMan® system based on real time detection of accumulated fluorescence in a 7500 fast RT-PCR system (Applied Biosystems). Fluorescence for each cycle was analyzed quantitatively, and gene expression was normalized relative to the expression of the housekeeping gene GAPDH mRNA. Specific TaqMan® gene expression assays for AGT and GAPDH were generated by Applied Biosystems (catalogue numbers Rn 00593114-m1 and 43523338E). Expression of AGT was presented as the percentage of GAPDH. Sample analysis was performed in triplicate in a blinded fashion.
Generation of Fusion Proteins and Constructs of Megalin Domains
Receptor-associated protein (RAP) and AGT were used as GST fusion proteins. To generate rat AGT expression constructs, nucleotide sequences corresponding to amino acids 25–477 were cloned into pGEX-4T-1. The production of GST, RAP-GST, and AGT-GST was performed according to the manufacturer's protocol (Amersham Bioscience). The megalin domains, ligand-binding repeats (LBRs) 1–4 and the C terminus including the transmembrane domain, were cloned into pCEP-Pu containing N-terminal His6 and Myc tags. The constructs were transfected into HEK cells. The resulting proteins were used for pulldown assays.
Pulldown Assay
Fusion proteins RAP-GST, AGT-GST, and GST alone were immobilized on glutathione-Sepharose beads and incubated either with BBM fractions from mouse kidney cortices or with lysates from HEK cells containing megalin receptor LBRs 1–4 or C terminus, dissolved in radioimmune precipitation assay buffer containing 25 mm Tris-HCl, pH 7.6, 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, and 5 mm CaCl2 supplemented with protease inhibitor mixture (Roche Applied Science). After extensive washing with radioimmune precipitation assay buffer, the eluates were loaded on SDS-PAGE, and silver staining or Western blot analysis using anti-megalin or anti-Myc antibody was performed.
Transcytosis
OK cells were cultured at 37 °C on inserts of a PET transfilter system (0.4-μm pore size; Falcon) placed in 6-well plates. The cells were confluent and revealed polar organization and formation of tight junctions, thus allowing us to trace the passage of molecules across the cell layer. The tightness of the cell layers was assessed by measuring transepithelial resistance and paracellular transport (see below). Human AGT and Ang II were purchased (Sigma-Aldrich). 24 h prior to the experiment, the cells were incubated with FCS-free medium. To study transcytosis, the cells were incubated either with Ang II or AGT alone (each 10 μg/ml) or combined with 200 μg/ml RAP-GST or 10 μm colchicine (Sigma-Aldrich), respectively. The cells were also incubated simultaneously with Ang II and 10 μm valsartan. Samples with medium alone served as controls. Each experiment was performed in triplicate and repeated at least five times. After 1 h, fluids from the lower chamber were collected and analyzed for AGT by ammonium sulfate precipitation followed by Western blot; graded quantities of AGT (0.1, 0.25, 0.5, and 1 μg/ml) serving as a standard were processed alike. Ang II was analyzed by enzyme immunoassay (Phoenix Pharmaceuticals Inc.; no cross-reactivity with Ang-(1–7)).
Paracellular Tracer Flux
FITC-dextran (5 kDa) was used at a final concentration of 10 μg/ml in FCS-free cell culture medium and added to the apical compartment of filters. After 1 h, aliquots from the upper and lower compartments were collected, and the concentration of FITC-dextran was determined by photofluorometry (excitation, 492 nm; emission, 520 nm; Shimadzu RF5000). The results were compared with those obtained in filters without cells.
Presentation of Data and Statistical Analysis
Quantitative data are presented as the means ± S.E. For statistical comparison, the Mann-Whitney U test was employed. p values of less than 0.05 were considered statistically significant.
RESULTS
Localization of AGT mRNA, AGT Protein, and Uptake of Radiolabeled AGT in Rat Kidney
In situ hybridization in normal SD rats using 35S-radiolabeled AGT cRNA revealed proximal tubular signal with restriction to the proximal straight tubules (PST; S2 and S3 segments; Fig. 1A). To corroborate these results, nonradioactive in situ hybridization was performed in transgenic TGR(hAGT1623) rats carrying the entire genomic human AGT gene including its 5′-flanking sequences (30); although physiologically irrelevant because of the inability of endogenous rat renin to cleave the human AGT, the construct targets its overexpression to the regular sites of synthesis including liver and brain with the result of ∼10-fold higher plasma human AGT concentrations compared with endogenous AGT (30). In the kidney of TGR(hAGT1623) rats, strong AGT mRNA signals were as well restricted to the PST, whereas the proximal convoluted tubules (PCT) were devoid of AGT mRNA (Fig. 1B); by contrast, AGT protein was localized to subapical compartments of the PCT (Fig. 1C). In normal rat, AGT protein localization was identical, beginning with the onset of the proximal convoluted tubule at the urinary pole and ending within the S2/S3 transition at the entry to the medullary rays (Fig. 1D). Cellular assignments of AGT mRNA and protein were thus mutually exclusive, which infers that AGT protein expression results from its cellular uptake in PCT, whereas in PST, AGT biosynthesis was not associated with cellular storage, which corresponds to AGT handling in the liver parenchymal cell.
FIGURE 1.

Renal AGT mRNA and AGT protein expression in normal SD and AGT transgenic rats. A, AGT transcript in PST (S2 and S3 segments) of control SD rat kidney by 35S-labeled radioactive in situ hybridization; S1 segments and glomeruli (G) show background signal. B and C, double staining for AGT transcript (nonradioactive in situ hybridization; B) and AGT protein immunoreactivity (IR; fluorescence immunohistochemistry; C) in AGT-transgenic rat showing mutually exclusive distribution of signals. Dashed lines mark the border between medullary rays (signal in B) and cortical labyrinth (signal in C). D, HRP bright field staining of AGT in control SD rat kidney; AGT is localized in the subapical compartments of PCT as exemplified in its initial portion at the urinary pole of a glomerulus. E, 125I-labeled AGT is detected by autoradiography in initial PCT; signal is indicated by arrowheads. Signal distribution was similar in D and E. Scale bar in A, D, and E, 20 μm; scale bar in B and C, 100 μm.
To confirm that renal AGT protein results from endocytotic uptake in rat, tracing experiments were performed using intravenous injection of 125I-labeled AGT. There was significant autoradiographic signal in the early PCT in rats perfusion-fixed after 20 or 60 min upon injection, whereas the downstream PCT and PST portions were devoid of signal (Fig. 1E). These observations further support that in rat PCT, cellular AGT protein accumulation derives from endocytotic uptake from the filtrate.
Quantification of AGT mRNA Expression in Isolated, Single-nephron Preparations from Rat Kidney
To quantitatively determine AGT mRNA abundances in the proximal tubule segments, they were manually isolated after enzymatic digestion, and batches from eight rats were analyzed. Individual mRNA abundances were quantitated and plotted as the percentages of the respective GAPDH levels. S1 segments revealed very low AGT mRNA expression (0.02 ± 0.003%); compared with S1, the expression was low as well in S1/S2 (0.64 ± 0.361%; not significant), whereas S3 segments showed substantial AGT mRNA expression (22.77 ± 2.478%; p < 0.0001; Fig. 2). For comparison, liver AGT mRNA expression was in the 10% range of its GAPDH expression (10.06 ± 2.037%).
FIGURE 2.
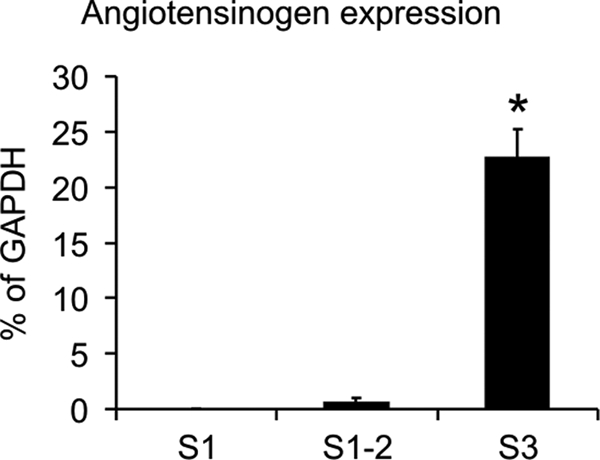
Quantitative RT-PCR analysis of AGT mRNA expression from isolated single-nephron preparations. The percentages of AGT mRNA relative to the respective GAPDH mRNA abundances are shown for proximal S1, S1/S2, and S3 segments. *, p < 0.001.
Proximal Tubular AGT Accumulation in Conditional Megalin-deficient Mice
The observed AGT uptake suggests mediation by the proximal tubular scavenger receptor, megalin. We therefore studied conditional megalin knock-out mice (Cre+) with mosaic deficiency of megalin in the proximal tubule (supplemental Fig. S2A). In these mice, immunoreactive AGT was localized in the subapical endosomal/lysosomal compartment of megalin-positive cells, whereas megalin-deficient cells were AGT-negative (Fig. 3A). Similarly, Western blot analysis showed significantly decreased AGT abundance in BBM fractions from Cre(+) compared with Cre(−) mice (−42.7 ± 2.1%; p < 0.01; Fig. 3B). By contrast, plasma AGT levels were not different between strains (Cre(−) 100.0 ± 24.9% compared with Cre(+) 121.0 ± 29.8%; Fig. 3C). Substantial urinary AGT excretion was found in Cre(+) but was nearly undetectable in Cre(−) mice as revealed by Western blot (Fig. 3D). These data suggest that intracellular AGT accumulation in PCT is megalin- and endocytosis-dependent, corroborating the evidence that AGT is reabsorbed from the ultrafiltrate.
FIGURE 3.

Renal AGT protein expression of megalin conditional knock-out mice. A, double immunofluorescence staining of megalin (red) and AGT (green) in Cre(+) kidney revealing mosaic distribution of megalin signal in a single PCT profile. Megalin-positive cells show subapical AGT distribution below the BBM, whereas megalin-deficient cells are devoid of AGT staining. Western blot for AGT from BBM fractions (B), plasma (C), and urine (D) of Cre(−) and Cre(+) mice. Note that in Cre(+), AGT is reduced in BBM (B), whereas urinary AGT excretion is strongly increased (D). Arrows mark the respective, specific bands for AGT. The values are the means ± S.E. (n = 5). *, p < 0.05; bar, 10 μm.
Megalin Binds AGT
Megalin is a type I transmembrane protein containing an extracellular region, a single transmembrane domain, and a C-terminal cytoplasmic tail (29). The extracellular domain, responsible for ligand binding, contains four clusters of cysteine-rich LDL receptor LBRs, growth factor repeats, an EGF repeat, and spacer regions. RAP is a ligand for all members of the LDL family including megalin and may be used as an inhibitor (40). To assess whether there is specific interaction between megalin and AGT, pulldown assays were performed using GST and the fusion proteins GST-RAP and GST-AGT. Only GST-RAP and GST-AGT precipitated megalin from BBM fractions of rat kidney cortex as shown by SDS-PAGE and silver staining or Western blot analysis (Fig. 4A). For GST, no specific interaction was observed. To map the AGT-binding sites on the diverse megalin domains, lysates from HEK cells transfected with LBR 1–4 and the C terminus were incubated with the fusion proteins. RAP and AGT precipitated all four LBRs, whereas the C-terminal domain did not bind to the fusion proteins (Fig. 4B).
FIGURE 4.

Megalin binds AGT. A, silver staining and Western blot of megalin pulldown assay using GST, GST-RAP, and GST-AGT, showing that megalin binds to RAP and AGT. B, pulldown assay using Myc-tagged LBR 1–4 and the Myc-tagged C terminus (C-term) of megalin. Protein fragments of Myc-tagged LBR and C terminus (left, B). As opposed to the C terminus, all four LBR bind to GST-RAP (middle, B) and GST-AGT (right, B).
Intracellular Routes and Transcytosis of AGT
To identify the intracellular route of AGT, its localization in cellular compartments was detected using anti-clathrin for clathrin-coated pits/vesicles, anti-EEA1 for early endosomes, anti-Rab11 for recycling endosomes, and anti-LAMP-2 for lysosomes. AGT was regularly found in partial colocalization with clathrin and early endosomal markers (Fig. 5, A and B). Partial overlap was also encountered with Rab11, and a significant merge signal with LAMP-2 suggested storage of AGT in lysosomes (Fig. 5, C and D).
FIGURE 5.

Intracellular localization of AGT in the proximal tubule. A–D, double immunofluorescence staining of AGT (green) with anti-clathrin antibody staining clathrin-coated pits or vesicles (red, A), anti-EEA1 antibody staining early endosomes (red, B), anti-Rab11 antibody staining recycling endosomes (red, C), and anti-LAMP-2 antibody staining lysosomes (red, D). In the merge pictures, sites of double staining are indicated by arrowheads. The dotted lines mark the position of the apical plasma membrane beneath the brush-border membrane and the basal lamina, respectively, as indicated. Bar, 5 μm.
To analyze whether AGT is subject to apical-to-basolateral transcytosis, OK cells cultured on permeable filters in dual chambered devices were used. OK cells expressed megalin in high concentrations as shown by immunohistochemistry and Western blot analysis (supplemental Fig. S2, B and C). Under the applied culture conditions, OK cell monolayers are polarized. To investigate the presence of tight junctions, trans-epithelial resistance was measured; trans-epithelial resistance increased continuously until usage of the cells at 180 Ω·cm2 (supplemental Fig. S2D). Paracellular flux was estimated by adding low molecular mass (4-kDa) FITC-dextran to the upper chamber; transepithelial passage of FITC-dextran is an established means to estimate paracellular permeability, producing results comparable with those obtained with [3H]mannitol (41). Photofluorometric measurements performed after 1 h revealed little apical-basal diffusion of FITC-dextran; only 0.167 ± 0.02% of total FITC-dextran added to the apical chamber was recovered from the lower chamber (p < 0.001), suggesting only minor paracellular flux in the OK cell monolayers.
Transcytosis of AGT and for comparison also Ang II was measured either alone or upon addition of RAP or colchicine. The upper chamber was incubated with 10 μg/ml AGT. After 1 h, 548 ± 30 ng/ml AGT was measured in the lower chamber, corresponding to a 5% recovery rate. Apical addition of RAP reduced the amount of transcytosed AGT significantly (−68.4 ± 13.6%; p < 0.05), and colchicine had an even stronger effect (−83.3 ± 4.3%; p < 0.05) (Fig. 6A). These results are in agreement with transcytotic transport of AGT.
FIGURE 6.

Proximal tubular processing of AGT and Ang II. Opossum kidney cells cultured in dual chamber devices were incubated apically with 10 μg of AGT or 10 μg of Ang II for 1 h. Samples from the lower chambers were analyzed. A, in comparison with control, the amount of transcytosed AGT is significantly reduced upon RAP or colchicine administration (Western blot evaluation). B, transcytosed Ang II is significantly lower upon RAP, valsartan, or colchicine administration (competitive ELISA). The values are the means ± S.E. (n = 6). *, p < 0.05.
To comparatively estimate transcytosis of Ang II, the upper chamber was incubated with 10 μg/ml Ang II. After 1 h, a minute recovery of 5.4 ± 0.8 ng/ml Ang II (0.05%) as measured by competitive ELISA was established in the lower chamber. The addition of RAP, valsartan, and colchicine reduced this amount significantly (−59.9 ± 2.8%, −45.1 ± 5.8%, and −56.0 ± 1.2%, respectively; p < 0.05); coadministration of RAP and valsartan had no additive effect (−48.6 ± 5.0%; p < 0.05) (Fig. 6B).
Coexpression of Other RAS Components in Proximal Tubule
Endocytotic uptake of renin has earlier been demonstrated in the proximal convoluted tubule (22, 23). Endocytosed AGT was consistently colocalized with renin in the subapical endosomal/lysosomal compartment of the proximal convolutions, suggesting a possible interaction that may result in the intracellular cleavage of AGT (Fig. 7A). In megalin-deficient portions of the proximal tubule in Cre(+) mice, immunoreactive renin was lacking, whereas in the presence of megalin, immunoreactive renin was consistently present (Fig. 7B). This result suggests that uptake of renin from the proximal tubule is megalin-dependent. Western blot analysis of BBM fractions (Fig. 7C) and plasma (Fig. 7D) exhibited no quantitative differences in renin abundance between strains (BBM, 100.0 ± 5.0% in Cre(−) compared with 88.2 ± 15.2% in Cre(+); plasma, 100.0 ± 24.9% in Cre(−) compared with 120.1 ± 29.8% in Cre(+); not significant). Urinary excretion of renin was, however, substantially higher in Cre(+) than in Cre(−) mice as revealed by Western blot; in Cre(−), urinary renin levels were almost undetectable (Fig. 7E).
FIGURE 7.

Proximal tubular renin expression in megalin conditional knock-out mice. A, double immunofluorescence staining of AGT (green) and renin (red) in Cre(−) kidney shows consistent colocalization of both proteins in intracellular vesicles (arrowheads). B, double staining of megalin (red) and renin (green) in Cre(+) kidney shows that megalin-deficient cells are devoid of renin. C–E, renin abundances from BBM fractions (C), plasma (D), and urine (E) in Cre(−) versus Cre(+) (Western blots). Arrows point to renin (lower, 43-kDa band); the upper band corresponds to the 53–55-kDa prorenin range. Only the urinary renin levels differ significantly. The values are the means ± S.E. (n = 5). *, p < 0.05; bar, 10 μm.
To assess whether the expression of ACE and ACE2 is affected by megalin deficiency, immunohistochemistry and Western blot analysis were performed in kidneys from Cre(−) and Cre(+) mice. ACE was mainly expressed in the BBM of the proximal tubule with increasing abundance from the S1 to the S3 segments as revealed by immunohistochemistry; in the S3 segments it was clearly evident that megalin-deficient cells showed markedly reduced BBM abundance of ACE (Fig. 8, A and B). Western blot analysis of BBM fractions demonstrated significant reduction in ACE expression in Cre(+) compared with Cre(−) (−21.4 ± 2.2%; p < 0.05; Fig. 8C), suggesting that proximal tubular abundance of ACE partially depends on megalin expression.
FIGURE 8.

ACE and ACE2 expression in megalin conditional knock-out mice. A, double immunofluorescence staining of ACE in Cre(−) kidney. The lower dotted lines indicate the border between cortical labyrinth and medullary rays/outer stripe; signal is high in the latter. B, in PST of Cre(+) kidney, megalin-deficient cells exhibit reduced ACE signal in BBM compared with megalin-expressing cells. C, BBM fractions show an overall mild reduction of ACE in Cre(+) compared with Cre(−) by Western blot. D, double immunofluorescence staining of ACE2 in Cre(−) kidney. The lower dotted lines indicate transition from outer stripe to inner stripe of outer medulla. E, in Cre(+) kidney, megalin-deficient cells exhibit increased ACE2 signal in BBM compared with megalin-positive cells. F, BBM fractions show higher ACE2 abundance in Cre(+) compared with Cre(−) by Western blot. The upper dotted lines mark the renal capsule. The values are the means ± S.E. (n = 5). *, p < 0.05. Scale bar in A and D, 20 μm; scale bar in B and E, 10 μm.
By contrast, ACE2 was found in BBM along the entire proximal tubule with no segmental differences. Its expression correlated inversely with megalin because enhanced ACE2 abundance was found in megalin-deficient compared with megalin-positive cells (Fig. 8, D and E). The results were confirmed by Western blot from BBM fractions showing increased expression in Cre(+) compared with Cre(−) (+186.0 ± 33.9%; p < 0.05; Fig. 8F).
DISCUSSION
Angiotensinogen
The main focus of our study was directed to AGT, the substrate from which renin cleaves off the dekapeptide, Ang I. Although its systemic supply is chiefly provided by the liver (14, 42, 43), an intrinsic renal or specifically proximal tubular generation and regulation of AGT has been discussed (2, 16, 18, 25, 44). However, some conclusions drawn from these studies may be questioned because crucial factors such as the endocytotic retrieval from the ultrafiltrate or segmental differences of its biosynthesis along the proximal tubule by AGT have not been considered adequately.
First, our findings have demonstrated rapid and effective proximal tubular uptake of exogenously administered, radiolabeled AGT, which demonstrates that in contrast to published interpretations, AGT is likely to pass through the glomerular filter despite its large molecular size (45). The autoradiographic signal over early proximal tubular epithelia indicates that filtered AGT becomes endocytosed probably via the endosomal-lysosomal pathway as shown for albumin (45). This is supported by our observations on the mosaic pattern of megalin deficiency (36), demonstrating that selectively only the megalin-expressing cells displayed subapical AGT accumulation. We also demonstrated the specificity for megalin-mediated endocytotic uptake of AGT. Earlier studies had suggested that only the second cluster of extracellular ligand-binding repeats (LBR2) of megalin is relevant for receptor-ligand interaction (29, 46); we found, however, that all four LBR were capable of binding AGT. Identical binding properties of RAP, the endogenous inhibitor of megalin (28, 29), supported specificity of our approach. Our data thus provide good evidence that AGT is reabsorbed from the glomerular ultrafiltrate by specific, receptor-mediated endocytosis with megalin acting as the receptor. Further support for the uptake from the filtrate by AGT comes from earlier data reporting that under colchicine-induced inhibition of endocytosis, the kidneys were rapidly depleted from AGT (14). By contrast, basolateral uptake of AGT from the circulation is not supported by available data.
Second, our data clearly show that immunoreactive AGT is only found in PCT, which agrees with a previous report (47); our findings in normal rat and mouse kidneys, in the megalin-positive portions of the Cre(+) mice, and in the AGT-overexpressing hAGT1623 rats with 100-fold elevated systemic levels of human AGT (43) support this evidence. In the latter, both endogenous and transgenic AGT were recognized by the same antibody. Third, AGT mRNA transcript was located predominantly in the late proximal tubule irrespective of its origin either from the endogenous or hAGT1623 transgenic source. This result was obtained by in situ hybridization as well as quantification of AGT mRNA within isolated tubule preparations by quantitative RT-PCR; quantification demonstrated nearly absent signal in S1 and S1/S2 segments but substantial amounts in S3 segments. Our data are at variance with previous data obtained from isolated PCT and PST segments of rat kidney (12) showing similar AGT mRNA expression levels in both parts by PCR evaluation. The reason for this discrepancy is unclear but may be methodologically based, because contemporary real time PCR approaches are more reliable than conventional PCR techniques, which may have obscured the differences between PCT and PST transcripts for AGT in that former study. The rapid conservation of the in vivo condition by perfusion-fixation reliably preserves transcripts cell-specifically so that in situ hybridization should provide reliable information, and because both approaches showed the same results, our data are plausible.
The detection of AGT transcript in PST is consistent with substantial local AGT biosynthesis and also implies that renal processing of translated AGT lacks its cellular storage like in the liver, where AGT is released into the bloodstream as it is produced (14). In contrast to liver, however, intrarenally generated AGT appears to be released into the urine (15). Here AGT probably mixes with remnant AGT quantities from the filtrate to be available (i) locally in the late proximal tubule and consequently (ii) in the downstream nephron segments and (iii) in the urine. Partial support for this concept comes from previous interpretations based on proximal versus distal (2, 13, 15) or urinary AGT availability (44). Our observation that relative to GAPDH abundance, AGT mRNA abundance in S3 was even higher than in liver supports that significant amounts of AGT may be released from the PST segments.
Changes in proximal tubular AGT content in response to dietary or hormonal stimuli or partial nephrectomy, which have been interpreted in the context of intrarenal AGT generation (16, 44, 47, 48), should thus be reconsidered with appropriate care for segmental specificity. In line with this, transgene-induced hypertensive effects of renal AGT overexpression, as described in mice carrying androgen-regulated AGT gene constructs (49), likely differ from those that are produced under the control of the intrinsic promoter (43); in the former model, AGT overexpression affects the entire proximal tubule, whereas in the latter, as demonstrated here, only the PST is addressed. Local RAS effects may therefore result in more pronounced pressor effects in the former than in the latter model (9).
Renin
Our data agree with earlier localization studies of renin in proximal tubule and further suggest that, like AGT, renin is a ligand for megalin as well; whether this applies also for its precursor, prorenin, remains to be clarified (2, 50). Partial megalin deficiency led to substantial urinary renin excretion, which underlines the role of megalin in its tubular retrieval. In the context of an intrinsic tubular RAS, proximal tubular transcription of renin mRNA has been identified in vivo at steady state or under experimental conditions, although high sensitivity PCR techniques had to be administered for its detection (25, 50), and our own attempts to localize renin transcripts in proximal tubule by means of sensitive in situ hybridization were negative (not shown). Accordingly, the normal synthesis rate within the proximal tubule appears to be quite low (15, 24, 50). On the other hand, active, megalin-dependent renin retrieval from the filtrate likely does not preclude its availability in the early proximal tubular fluid with presumably progressive decreases along S2 and S3 segments.
ACE Homologues
We have localized ACE preferentially in the proximal straight tubule and ACE2 ubiquitously along the proximal nephron, confirming published data (Ref. 51; for a review, see Ref. 26). There were lower ACE but markedly enhanced ACE-2 signals in BBM of megalin-deficient compared with megalin-positive cells; this possibly reflected the divergent functions of the two homologues with ACE metabolizing Ang I to Ang II and ACE2 generating the heptapeptide angiotensin-(1–7) (26). Megalin and an intact endosomal apparatus may be necessary for ACE surface expression, whereas altered retrieval from the membrane may be specifically relevant for ACE2 as demonstrated previously for NaPi-2 (36). Although divergent regulation for ACE and ACE2 has been described (52), their membrane surface expression was considered to be similar, because their spontaneous internalization from the membrane was shown to be rather modest compared with other integral membrane proteins (27). The cause for the obvious difference in BBM representation and likely the concomitant surface expression upon the defects in endocytosis and membrane recycling caused by megalin deficiency in Cre(+) mice is unclear but may be related to distinct interactions of ACE versus ACE2 with cytosolic proteins, which may be differently affected by the drop in megalin-induced signaling, endocytosis of other proteins, or recycling steps (29, 36, 53). Our results also implicate that megalin-mediated endocytosis and/or endosomal recycling may modify shedding of soluble enzyme into the proximal tubular lumen in an ACE homologue-specific manner (27).
Cellular Pathways of Proximal Tubular RAS Components
The detail of AGT intracellular localization suggests that some of the reabsorbed protein is encountered during endocytosis and endosomal accumulation as revealed by double staining for clathrin and EEA1 (Ref. 54; for a review, see Ref. 28). Dissociation of AGT from its receptor, megalin, and subsequent lysosomal storage and degradation can be deduced from its extensive colocalization with LAMP-2 (55). Intracellular storage of AGT was regularly encountered as well in large vesicles within PCT of normal and AGT transgenic rats by immunoperoxidase staining (Fig. 1D); vesicles corresponded to the size and localization of AGT-accumulating lysosomes identified earlier by electron microscopy (56). Recycling of AGT to the luminal surface may occur in parallel to the degradative pathway based on its frequent colocalization with Rab11 (57). Early data from isolated rabbit PCT showing release of significant amounts of AGT over 72 h (13) may indirectly support this view, because the absence of AGT transcript, inferred by our analysis, would agree with recycling and release of the substrate in that study. As a third pathway, we have tested the hypothesis of whether AGT transepithelial transport from the ultrafiltrate to the renal interstitium is a potential mechanism to recycle the intact protein.
With the help of a dual-chambered transfilter system, polarized OK cell layers with established BBM formation, abundant endogenous megalin expression, junctional apparatus, and viable transepithelial electrical resistance were capable of transcytosing intact AGT offered from the apical side with an approximate 5% recovery rate at the basolateral side after 1 h. This duration was chosen in analogy to a pulse-chase series of experiments performed in the intestine (54), predicting that much of the offered AGT should by then have reached the basolateral compartment. Transcytosis was further shown to depend on receptor-mediated endocytosis as revealed by its partial inhibition by the megalin competitor RAP and on intact microtubular function; paracellular leakage of AGT was unlikely to occur because the passage of low molecular mass dextran was nearly absent. Transcytosis of AGT may furthermore require its specific intracellular recognition because other ligands for megalin, such as lysozyme or lactoferrin, cannot be transcytosed across proximal epithelium (58). Confounding effects by endogenous AGT synthesis were unlikely because AGT was undetectable in the control group, and because OK cells display only low grade AGT transcription rate (1% of rat liver) (59), this dimension may be neglected. Megalin-dependent proximal tubular transcytosis has earlier been demonstrated for the vitamin A carrier retinol-binding protein (57, 60), serving as a putative means to prevent its urinary loss, which in analogy may be considered for AGT as well. Our parallel demonstration of the transcytosis of Ang II as an established megalin ligand (61) primarily served as a control; its transcytosis was megalin-dependent as well, because RAP had a major inhibitory effect; this was also observed with an angiotensin receptor antagonist. Both inhibitors were not additive, which agrees with the distinct roles proposed for megalin- and AT1-R-mediated pathways for Ang II (61). Compared with AGT, the weak retrieval of Ang II may result from its partial enzymatic degradation in the BBM.
A role for megalin in the transcytosis of RAS components has thus been demonstrated. The markedly increased urinary excretion of AGT in megalin deficiency supports our view that tubular uptake and transcytosis of AGT may be relevant mechanisms for its return to the circulation, although this is obscured by the unknown proportion of AGT that is intracellularly degraded via the lysosomal pathway like most other megalin ligands (28, 58). Likewise, the question as to which pathway dominates, transcytosis of AGT and release of Ang I in the interstitium or cleavage of AGT within the tubule, cannot be answered at present and requires further study.
Outlook on the Functional Potentials of an Intrarenal, Proximal Tubular RAS
Proximal tubular RAS components are of dual origin, because renin, AGT, Ang I, and Ang II may reach the tubule via the ultrafiltrate and in parallel can also be generated locally by heterogenous mechanisms. Depending on the site of action within the cell, at the luminal or basolateral membrane, or within the interstitial space, RAS effector peptides may therefore be of mixed origin (2, 10, 20). Here, our data permit an improved understanding because the proximal segmental handling of local RAS components has been illustrated. The early PCT displays very effective, megalin-dependent endocytotic uptake and intracellular storage of renin and AGT from the ultrafiltrate, whereas their local biosynthesis apparently can be neglected. In this scenario, transcytotic retrieval, luminal recycling, or intracellular activation of RAS components prevail and may be functionally relevant for endogenous, local Ang I generation. However, because in the proximal convolutions local ACE abundance is minute, local generation of Ang II possibly relies on the activity of other proteases, which could explain the measured, ∼100-fold elevated intratubular Ang II levels (52, 62). Contrastingly, high ACE2 expression levels agree with intensive Ang-(1–7) generation in PCT. On the other hand, the late proximal tubule (PST) displayed substantial AGT transcript, absence of renin or AGT protein accumulation, and strong abundance for ACE and ACE2 in BBM. Hence, local generation of RAS effector peptides may require a sustained base-line release of AGT, which is subject to regulation (2), and possibly mixes with AGT and renin released from the upstream proximal convolutions, to produce luminal Ang I; the abundant expression of ACE homologues may subsequently serve to generate Ang II, Ang-(1–7), or Ang-(1–4) for effects located in PST or further downstream segments. To what extent transcytosis of RAS components occurs in PST cannot be judged here.
In summary, our data support the significance of an intrarenal RAS with its components concentrated in the proximal tubule. Previously unrecognized aspects regarding their localization and cellular processing have been disclosed, providing extended insight into the functional concept of a self-contained RAS in the kidney.
Supplementary Material
Acknowledgments
We thank Thomas Willnow for providing us with the megalin-deficient mice, Chistoph Klett for support in perfusing rats with radiolabeled AGT, and Kerstin Riskowsky and Frauke Grams for expert technical assistance.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1 and S2.
- RAS
- renin angiotensin system
- AGT
- angiotensinogen
- ACE
- angiotensin-converting enzyme
- Ang
- angiotensin
- SD
- Sprague-Dawley
- OK
- opossum kidney
- BBM
- brush-border membrane
- RAP
- receptor-associated protein
- LBR
- ligand-binding repeat
- PST
- proximal straight tubule(s)
- PCT
- proximal convoluted tubule(s).
REFERENCES
- 1.Quan A., Baum M. (2000) Nephron 84, 103–110 [DOI] [PubMed] [Google Scholar]
- 2.Kobori H., Nangaku M., Navar L. G., Nishiyama A. (2007) Pharmacol. Rev. 59, 251–287 [DOI] [PubMed] [Google Scholar]
- 3.Braam B., Mitchell K. D., Fox J., Navar L. G. (1993) Am. J. Physiol. 264, F891–F898 [DOI] [PubMed] [Google Scholar]
- 4.Le T. H., Coffman T. M. (2008) Curr. Opin. Nephrol. Hypertens. 17, 57–63 [DOI] [PubMed] [Google Scholar]
- 5.Guyton A. C. (1991) Science 252, 1813–1816 [DOI] [PubMed] [Google Scholar]
- 6.Baum M., Quigley R., Quan A. (1997) Am. J. Physiol. 273, F595–F600 [DOI] [PubMed] [Google Scholar]
- 7.Geibel J., Giebisch G., Boron W. F. (1990) Proc. Natl. Acad. Sci. U.S.A. 87, 7917–7920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riquier-Brison A. D., Leong P. K., Pihakaski-Maunsbach K., McDonough A. A. (2010) Am. J. Physiol. Renal Physiol. 298, F177–F186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cogan M. G. (1990) Hypertension 15, 451–458 [DOI] [PubMed] [Google Scholar]
- 10.Navar L. G., Lewis L., Hymel A., Braam B., Mitchell K. D. (1994) J. Am. Soc. Nephrol. 5, 1153–1158 [DOI] [PubMed] [Google Scholar]
- 11.Seikaly M. G., Arant B. S., Jr., Seney F. D., Jr. (1990) J. Clin. Invest. 86, 1352–1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terada Y., Tomita K., Nonoguchi H., Marumo F. (1993) Kidney Int. 43, 1251–1259 [DOI] [PubMed] [Google Scholar]
- 13.Yanagawa N., Capparelli A. W., Jo O. D., Friedal A., Barrett J. D., Eggena P. (1991) Kidney Int. 39, 938–941 [DOI] [PubMed] [Google Scholar]
- 14.Richoux J. P., Cordonnier J. L., Bouhnik J., Clauser E., Corvol P., Menard J., Grignon G. (1983) Cell Tissue Res. 233, 439–451 [DOI] [PubMed] [Google Scholar]
- 15.Rohrwasser A., Morgan T., Dillon H. F., Zhao L., Callaway C. W., Hillas E., Zhang S., Cheng T., Inagami T., Ward K., Terreros D. A., Lalouel J. M. (1999) Hypertension 34, 1265–1274 [DOI] [PubMed] [Google Scholar]
- 16.Gociman B., Rohrwasser A., Lantelme P., Cheng T., Hunter G., Monson S., Hunter J., Hillas E., Lott P., Ishigami T., Lalouel J. M. (2004) Kidney Int. 65, 2153–2160 [DOI] [PubMed] [Google Scholar]
- 17.Yang G., Merrill D. C., Thompson M. W., Robillard J. E., Sigmund C. D. (1994) J. Biol. Chem. 269, 32497–32502 [PubMed] [Google Scholar]
- 18.Everett A. D., Scott J., Wilfong N., Marino B., Rosenkranz R. P., Inagami T., Gomez R. A. (1992) Hypertension 19, 70–78 [DOI] [PubMed] [Google Scholar]
- 19.Kobori H., Nishiyama A., Harrison-Bernard L. M., Navar L. G. (2003) Hypertension 41, 42–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Navar L. G., Harrison-Bernard L. M., Nishiyama A., Kobori H. (2002) Hypertension 39, 316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Satou R., Gonzalez-Villalobos R. A., Miyata K., Ohashi N., Katsurada A., Navar L. G., Kobori H. (2008) Am. J. Physiol. Renal Physiol. 295, F283–F239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taugner R., Hackenthal E., Rix E., Nobiling R., Poulsen K. (1982) Kidney Int. Suppl. 12, S33–S43 [PubMed] [Google Scholar]
- 23.Taugner R., Hackenthal E., Inagami T., Nobiling R., Poulsen K. (1982) Histochemistry 75, 473–484 [DOI] [PubMed] [Google Scholar]
- 24.Leyssac P. P. (1986) Kidney Int. 30, 332–339 [DOI] [PubMed] [Google Scholar]
- 25.Singh I., Grams M., Wang W. H., Yang T., Killen P., Smart A., Schnermann J., Briggs J. P. (1996) Am. J. Physiol. 270, F1027–F1037 [DOI] [PubMed] [Google Scholar]
- 26.Burns K. D. (2007) Curr. Opin. Nephrol. Hypertens. 16, 116–121 [DOI] [PubMed] [Google Scholar]
- 27.Warner F. J., Lew R. A., Smith A. I., Lambert D. W., Hooper N. M., Turner A. J. (2005) J. Biol. Chem. 280, 39353–39362 [DOI] [PubMed] [Google Scholar]
- 28.Christensen E. I., Verroust P. J., Nielsen R. (2009) Pflügers Arch. 458, 1039–1048 [DOI] [PubMed] [Google Scholar]
- 29.Saito A., Sato H., Iino N., Takeda T. (2010) J. Biomed. Biotechnol. 2010, 1–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ganten D., Wagner J., Zeh K., Bader M., Michel J. B., Paul M., Zimmermann F., Ruf P., Hilgenfeldt U., Ganten U., Kaling M., Bachmann S., Mullins J. J., Murakami K. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 7806–7810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leheste J. R., Melsen F., Wellner M., Jansen P., Schlichting U., Renner-Müller I., Andreassen T. T., Wolf E., Bachmann S., Nykjaer A., Willnow T. E. (2003) FASEB J. 17, 247–249 [DOI] [PubMed] [Google Scholar]
- 32.Hilgenfeldt U., Hackenthal E. (1982) Biochim. Biophys. Acta 708, 335–342 [DOI] [PubMed] [Google Scholar]
- 33.Iwao H., Nakamura N., Ikemoto F., Yamamoto K. (1983) J. Histochem. Cytochem. 31, 776–782 [DOI] [PubMed] [Google Scholar]
- 34.Theilig F., Bostanjoglo M., Pavenstädt H., Grupp C., Holland G., Slosarek I., Gressner A. M., Russwurm M., Koesling D., Bachmann S. (2001) J. Am. Soc. Nephrol. 12, 2209–2220 [DOI] [PubMed] [Google Scholar]
- 35.Biber J., Stieger B., Haase W., Murer H. (1981) Biochim. Biophys. Acta 647, 169–176 [DOI] [PubMed] [Google Scholar]
- 36.Bachmann S., Schlichting U., Geist B., Mutig K., Petsch T., Bacic D., Wagner C. A., Kaissling B., Biber J., Murer H., Willnow T. E. (2004) J. Am. Soc. Nephrol. 15, 892–900 [DOI] [PubMed] [Google Scholar]
- 37.Theilig F., Kriz W., Jerichow T., Schrade P., Hähnel B., Willnow T., Le Hir M., Bachmann S. (2007) J. Am. Soc. Nephrol. 18, 1824–1834 [DOI] [PubMed] [Google Scholar]
- 38.Bachmann S., Peters J., Engler E., Ganten D., Mullins J. (1992) Kidney Int. 41, 24–36 [DOI] [PubMed] [Google Scholar]
- 39.Willmann J. K., Bleich M., Rizzo M., Schmidt-Hieber M., Ullrich K. J., Greger R. (1997) Pflugers Arch. 434, 173–178 [DOI] [PubMed] [Google Scholar]
- 40.Willnow T. E., Rohlmann A., Horton J., Otani H., Braun J. R., Hammer R. E., Herz J. (1996) EMBO J. 15, 2632–2639 [PMC free article] [PubMed] [Google Scholar]
- 41.Balda M. S., Whitney J. A., Flores C., González S., Cereijido M., Matter K. (1996) J. Cell Biol. 134, 1031–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nasjletti A., Matsunaga M., Masson G. M. (1971) Can. J. Physiol. Pharmacol. 49, 292–301 [DOI] [PubMed] [Google Scholar]
- 43.Bohlender J., Ménard J., Wagner J., Luft F. C., Ganten D. (1996) Hypertension 27, 535–540 [DOI] [PubMed] [Google Scholar]
- 44.Kobori H., Harrison-Bernard L. M., Navar L. G. (2002) Kidney Int. 61, 579–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Christensen E. I., Birn H., Rippe B., Maunsbach A. B. (2007) Kidney Int. 72, 1192–1194 [DOI] [PubMed] [Google Scholar]
- 46.Orlando R. A., Exner M., Czekay R. P., Yamazaki H., Saito A., Ullrich R., Kerjaschki D., Farquhar M. G. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 2368–2373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kobori H., Harrison-Bernard L. M., Navar L. G. (2001) J. Am. Soc. Nephrol. 12, 431–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lantelme P., Rohrwasser A., Gociman B., Hillas E., Cheng T., Petty G., Thomas J., Xiao S., Ishigami T., Herrmann T., Terreros D. A., Ward K., Lalouel J. M. (2002) Hypertension 39, 1007–1014 [DOI] [PubMed] [Google Scholar]
- 49.Sachetelli S., Liu Q., Zhang S. L., Liu F., Hsieh T. J., Brezniceanu M. L., Guo D. F., Filep J. G., Ingelfinger J. R., Sigmund C. D., Hamet P., Chan J. S. (2006) Kidney Int. 69, 1016–1023 [DOI] [PubMed] [Google Scholar]
- 50.Moe O. W., Ujiie K., Star R. A., Miller R. T., Widell J., Alpern R. J., Henrich W. L. (1993) J. Clin. Invest. 91, 774–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ikemoto F., Song G. B., Tominaga M., Kanayama Y., Yamamoto K. (1990) Nephron 55, 3–9 [DOI] [PubMed] [Google Scholar]
- 52.Park S., Bivona B. J., Kobori H., Seth D. M., Chappell M. C., Lazartigues E., Harrison-Bernard L. M. (2010) Am. J. Physiol. Renal Physiol. 298, F37–F48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Pollock C. A., Poronnik P. (2007) Curr. Opin. Nephrol. Hypertens. 16, 359–364 [DOI] [PubMed] [Google Scholar]
- 54.He W., Ladinsky M. S., Huey-Tubman K. E., Jensen G. J., McIntosh J. R., Björkman P. J. (2008) Nature 455, 542–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saftig P., Beertsen W., Eskelinen E. L. (2008) Autophagy 16, 510–512 [DOI] [PubMed] [Google Scholar]
- 56.Darby I. A., Congiu M., Fernley R. T., Sernia C., Coghlan J. P. (1994) Kidney Int. 46, 1557–1560 [DOI] [PubMed] [Google Scholar]
- 57.Akhter S., Kovbasnjuk O., Li X., Cavet M., Noel J., Arpin M., Hubbard A. L., Donowitz M. (2002) Am. J. Physiol. Cell Physiol. 283, C927–C940 [DOI] [PubMed] [Google Scholar]
- 58.Marinó M., Andrews D., Brown D., McCluskey R. T. (2001) J. Am. Soc. Nephrol. 12, 637–648 [DOI] [PubMed] [Google Scholar]
- 59.Chan J. S., Chan A. H., Nie Z. R., Sikstrom R., Lachance S., Hashimoto S., Carrière S. (1992) J. Am. Soc. Nephrol. 2, 1360–1367 [DOI] [PubMed] [Google Scholar]
- 60.Christensen E. I., Moskaug J. O., Vorum H., Jacobsen C., Gundersen T. E., Nykjaer A., Blomhoff R., Willnow T. E., Moestrup S. K. (1999) J. Am. Soc. Nephrol. 10, 685–695 [DOI] [PubMed] [Google Scholar]
- 61.Gonzalez-Villalobos R., Klassen R. B., Allen P. L., Navar L. G., Hammond T. G. (2005) Am. J. Physiol. Renal Physiol. 288, F420–F427 [DOI] [PubMed] [Google Scholar]
- 62.Quan A., Baum M. (1996) J. Clin. Invest. 97, 2878–2882 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.