

Abstract
To gain a better understanding of the light-induced reduction of protochlorophyllide (PChlide) to chlorophyllide as a key regulatory step in chlorophyll synthesis, we performed transient infrared absorption measurements on PChlide in d4-methanol. Excitation in the Q-band at 630 nm initiates dynamics characterized by three time constants: τ1 = 3.6 ± 0.2, τ2 = 38 ± 2, and τ3 = 215 ± 8 ps. As indicated by the C13′=O carbonyl stretching mode in the electronic ground state at 1686 cm−1, showing partial ground-state recovery, and in the excited electronic state at 1625 cm−1, showing excited-state decay, τ2 describes the formation of a state with a strong change in electronic structure, and τ3 represents the partial recovery of the PChlide electronic ground state. Furthermore, τ1 corresponds with vibrational energy relaxation. The observed kinetics strongly suggest a branched reaction scheme with a branching ratio of 0.5 for the path leading to the PChlide ground state on the 200 ps timescale and the path leading to a long-lived state (>>700 ps). The results clearly support a branched reaction scheme, as proposed previously, featuring the formation of an intramolecular charge transfer state with ∼25 ps, its decay into the PChlide ground state with 200 ps, and a parallel reaction path to the long-lived PChlide triplet state.
Introduction
Protochlorophyllide (PChlide) represents an important metabolite in the biosynthesis of chlorophyll, the ubiquitous pigment of photosynthesis in plants, green algae, and cyanobacteria (1–4). It is synthesized from 5-aminolevulinic acid, the initial precursor of tetrapyrrole biosynthesis, and is reduced to chlorophyllide (Chlide) by the enzyme NADPH:PChlide oxidoreductase (POR) in one of the last steps of the biosynthetic pathway. In chlorophyll-containing organisms, the POR enzyme requires light for activity, making the photoreduction of PChlide into Chlide a key regulatory step in the synthesis of chlorophyll and the subsequent assembly of the photosynthetic apparatus (5). Thus, the light dependency of the enzymatic reaction both enables the adaptation of chlorophyll biosynthesis to external light conditions and prevents photooxidative damage of cellular and subcellular structures due to the accumulation of phototoxic tetrapyrrole intermediates (6–8). In addition, the enzyme POR exhibits another interesting feature: it is one of only two enzymes found in nature in which catalytic activity is switched on by the absorption of light. Light absorption by the substrate PChlide initiates the catalytic process, and together with NADPH as coenzyme, PChlide is reduced to Chlide (Fig. 1) by trans addition of hydrogen across the C17/C18 double bond (5,9,10).
Figure 1.
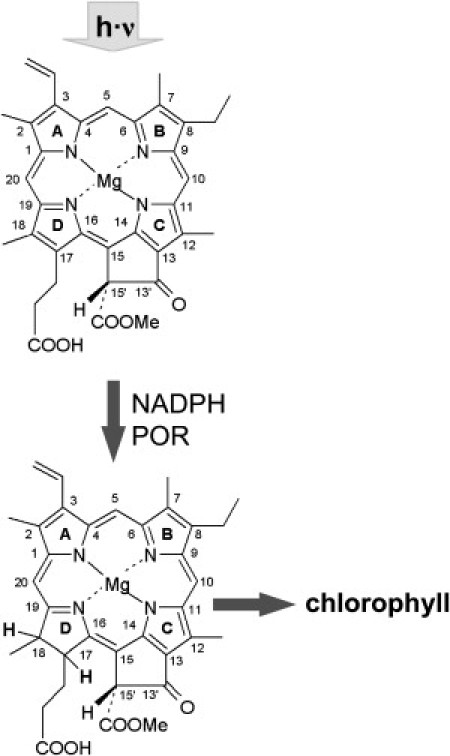
Molecular structure of PChlide and the light-induced reduction to Chlide by the enzyme POR.
Because the enzyme-substrate complex can be assembled in the dark, the use of light to trigger a catalytic reaction gives us an opportunity to study the real-time dynamics of enzyme catalysis by ultrafast, time-resolved, laser spectroscopic methods. In this regard, it was recently shown that a conformational change at the active site of the POR enzyme is necessary to activate catalysis. This first reaction step is then followed by the actual photochemical reaction, which results in the formation of a reaction intermediate (I675∗) with rate constants of 3 and 100 ns−1 (11–14).
We have initiated a comprehensive study on the excited-state dynamics of PChlide alone in solution, i.e., outside the cavity of the POR apoprotein (15–21). The objective of this work is to separate the influence of the POR apoenzyme from the intrinsic excited-state relaxation dynamics of the substrate, and thus elucidate the more complex light-induced processes in the POR enzyme. Furthermore, we expect to gather information on the role of the enzyme's active sites in modulating the photochemistry of the bound substrate for maximum reaction efficiency. Following this strategy, we examined the excited-state dynamics of PChlide in solvents of different physical properties, assuming that the solvent conditions could mimic specific environmental conditions in the enzyme-substrate complex (16,17,19). This resulted in a detailed kinetic model of the excited-state dynamics in PChlide (18,19). The model implies the formation of an intermediate photoproduct that is created via decay along a reactive channel with a time constant of ∼25 ps (see below). Given that the population kinetics is strongly controlled by the polarity but hardly affected by the protic/aprotic properties of the solvent (16,19), the photointermediate is suggested to represent an intramolecular charge transfer state (SICT). The charge transfer character most probably results from the electron-withdrawing effect of the carbonyl group attached at the isocyclic fifth ring in direct conjugation with the π-electron path of the porphyrin macrocycle (Fig. 1). In line with this assignment are previous findings obtained in magnesium octaethylporphyrin (MgOEP), which is structurally related to the porphyrin skeleton in PChlide but lacks the isocyclic fifth ring and thus the electrophilic substituent in conjugation with the π-electron path. Obviously, due to this structural deviation, the intermediate SICT state is not generated (22). Furthermore, the occurrence of an intramolecular charge transfer state on the excited-state hypersurface is a phenomenon that is well described in the literature. For example, it has been reported to be a common feature in the excited-state dynamics of carotenoids when they are substituted with electron-withdrawing groups in conjugation with the π-electron path of the polyene chain (23–25). In the case of peridinin, the most extensively studied carbonyl carotenoid, the charge transfer state has also been characterized by ultrafast infrared (IR) experiments indicating the molecular modes involved in the population of that state (26, 27).
It is tempting to speculate that a charge transfer complex might also play an important role in the light-driven reduction of PChlide by the enzyme POR, insofar as it allows the selective activation of certain regions of the PChlide molecule. Therefore, to further support this hypothesis on the basis of studies with the isolated substrate, it is essential to correlate the excited-state dynamics in PChlide with the changes in its structure in a next step. In this respect, time-resolved vibrational spectroscopy provides direct experimental access to the evolution of nuclear dynamics during the lifetime of excited states (28–33). Although only complex spectra with overlapping bands of poor fine structure have been obtained in the visible region probed so far, individual vibrational bands related to the characteristic signature of chemical constitution can be resolved in the mid-IR region. This led us to probe the ultrafast dynamics of PChlide in the mid-IR region and to characterize the molecular modes involved, in particular in the relaxation along the reactive path. A recent time-resolved mid-IR study on PChlide (34), which used excitation into the Soret band, reported results similar to those obtained in our previous time-resolved fluorescence and absorption studies. However, the mid-IR data do not support the branched reaction model suggested on the basis of our previous work (15–21).
In the experiments presented below, the vibrational dynamics of PChlide were recorded after excitation into the Q-band, i.e., closer to the ambient excitation conditions of the native protein complex. Thus, only processes that are directly related to the intrinsic reaction dynamics are accessible to observation, which facilitates a more straightforward analysis of the transient IR spectra. The results not only provide insight into the structural changes associated with the initial reaction dynamics in PChlide, they also confirm the reaction model suggested for the excited-state dynamics on the basis of our previous transient absorption and fluorescence measurements (15–17,19–21). In this respect, they reveal the partitioning of the initially excited-state population into reactive and nonreactive pathways.
Materials and Methods
Preparation of PChlide
PChlide a was isolated from 5-day-old, dark-grown oat seedlings (Avena sativa L. cv Tomba) as described previously (17). In brief, the tips of the oat coleoptiles were disrupted by homogenization and PChlide was extracted into ice-cold 10 mM Tricin buffer (pH 7.5) containing 75% (v/v) acetone. After centrifugation, the PChlide was transferred into diethylether and then into a 4:1 mixture of methanol and 0.01 M ammonia. Finally, PChlide was again extracted with a water/methanol mixture and further purified by high-performance liquid chromatography (HPLC) on a reverse-phase RP-18 column in a linear 20–80% acetonitrile gradient. The PChlide was eluted with an m/z peak of 613 g/Mol. The fractions containing highly pure PChlide were lyophilized and stored at 250 K in a refrigerator until use. Much care was taken to use only highly purified PChlide samples because PChlide is known to easily release the central Mg2+ ion, leading to significantly altered absorption and fluorescence spectra (Fig. 2 B).
Figure 2.
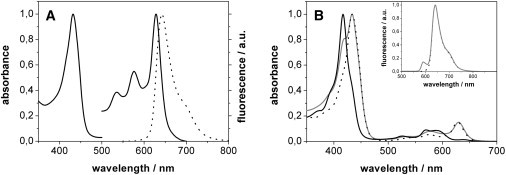
(A) Static UV/vis-spectrum of PChlide in d4-methanol. The absorption (solid line) and fluorescence (dotted line) spectra (λexc = 410 nm) between 500 and 800 nm are given. They are normalized to 1.0, where the absorption of the Q-band is enlarged by the factor of 7 compared with the Soret band. (B) Absorption spectrum of protopheophorbid, separated from PChlide by HPLC (black line), and the absorption and fluorescence spectra (inset, λexc = 410 nm) of PChlide with impurities of protopheophorbid (gray line). For comparison, the static spectra of highly pure PChlide are also given (dotted line).
For the time-resolved measurements, PChlide was diluted in d4-methanol to give an optical density of ∼1.3 at the absorption maximum (λmax = 629 nm, ɛ = 41000 l/mol·cm) in a 0.25 mm optical path length (corresponding to a concentration of ∼1.3 mM). During the measurements, the sample was rotated and moved laterally to the pump and probe beams to excite fresh sample. Before and after each pump-probe experiment, we checked the integrity of the PChlide samples by monitoring the steady-state absorption spectrum in the UV/vis region. All experiments were performed at room temperature.
Pump-probe experiments
A regenerative amplifier laser system (635 Hz, 0.8 mJ/pulse; CPA-2001; CLARK MXR, Dexter, MI) was used to generate short pump and probe pulses. Pump pulses at λpump = 630 nm with an energy of ∼500 nJ were generated by an in-house-built noncollinear parametric amplifier (NOPA). Mid-IR probe pulses were obtained from a two-stage optical parametric amplifier (OPA) and subsequent difference frequency generation (29). The spectral region 1475–1775 cm−1 was probed up to 700 ps after excitation. At the sample position, the focal width of the pump and the probe beam was ∼200 and 150 μm, respectively. The IR pulses were detected by a 32-channel mercury-cadmium-telluride array after dispersion by a polychromator. A pump-probe experiment on a thin silicon sample yielded a system response of ∼350 fs and time zero.
During the experiments, every second pump pulse was chopped and the pump-induced absorption differences ΔA(t, λpr) were detected as a function of the delay time t between the pump and probe pulse at probe wavelength λpr and were evaluated on a single-shot basis. The measured absorption changes are plotted against wavelength (absorption difference spectra) or delay time (transients). Negative absorption changes indicate the disappearance of IR absorption and thus the depopulation of electronic ground-state vibrations (bleach bands). Positive absorbance changes display the absorption of newly populated states. The data were quantitatively analyzed by a global multiexponential fit (Eq. 1) for delay times t > 400 fs (to exclude artifact signals around the time zero) using
| (1) |
where A0(λpr) is the pump-induced difference absorption spectrum after long delay times (700 ps in our experiments), and Ai(λpr) are the decay-associated amplitude spectra (DAS) of the corresponding time constants τi.
Results
Fig. 2 A shows the ground-state absorption spectrum of PChlide in methanol. The spectrum reveals a strong Soret band at 434 nm and three weaker Q-absorption bands, with the main band at 629 nm and the other two of lower intensity at 576 nm and 534 nm. The spectral features correspond with highly pure PChlide, which is separated from its demetallated analog, protopheophorbid (Fig. 2 B). Due to the acid's labile nature, PChlide loses the central Mg2+ ion relatively easily and forms protopheophorbid, which exhibits a slightly blue-shifted Soret band at 418 nm and Q-bands at 569 and 595 nm, respectively. To ensure the homogeneity of the PChlide sample used in the study described below, and thus the existence of only one type of ground-state species, PChlide was carefully separated from the protopheophorbid derivative by means of reversed-phase HPLC.
Fig. 3 represents the transient IR-difference spectra of PChlide between 1475 and 1775 cm−1 up to 700 ps, obtained after excitation in the Q-band at 630 nm. The negative band, which appears within 0.5 ps at 1686 cm−1 is assigned to the C13′=O carbonyl stretching mode of the isocyclic fifth ring of PChlide (Fig. 1) in its electronic ground state (20,35–37). In general, the spectral position of the C=O stretching mode depends on the solvent properties; here it coincides well with the C13′=O carbonyl frequency (1687 cm−1) previously obtained for methanol (34) with the use of different solvents. Although the ester C=O stretching modes of the carboxyl groups of ring D and of the fifth ring fall into the same spectral region (typically 1730–1750 cm−1), by their constitution they are largely decoupled from the Q-band excitation and thus are expected to contribute much less to the observed photo-induced IR absorbance differences. However, we cannot exclude the possibility that the small dip at 1747 cm−1 is related to either of the ester C=O modes. Within 700 ps, the bleaching signal at 1686 cm−1 decreases to ∼50% of its maximum value, suggesting a corresponding partial recovery of the PChlide ground state.
Figure 3.
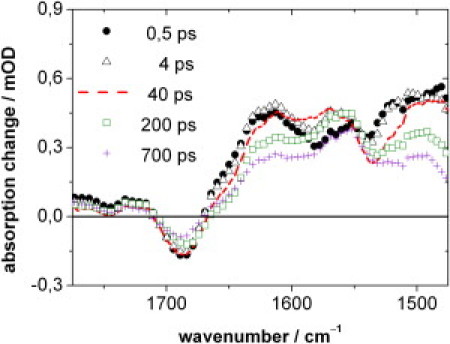
Time-resolved IR-difference spectra of PChlide after excitation at 630 nm at selected delay times.
Apart from this bleaching signal and small signals above 1700 cm−1 with the dip at 1747 cm−1, the difference spectra are dominated by strong positive signals below 1670 cm−1 at all delay times. They show a slight overall decrease with increasing delay time and clearly change shape during the observation period, suggesting the involvement of various molecular structures. In addition to the C=O stretching modes, several delocalized modes with C=C- and C=N-in-plane character are expected in the investigated spectral region (20,35–37). Thus, although a change of frequency or oscillator strength upon electronic excitation is anticipated for these modes, their corresponding negative difference signals are obviously superimposed by positive contributions of vibrational modes in the excited state with larger oscillator strength.
The global fit (Eq. 1) with three exponentials and time constants of τ1 = 3.6 ± 0.2, τ2 = 38 ± 2, and τ3 = 215 ± 8 ps yields the DAS shown in Fig. 4. Fig. 5 displays representative absorbance transients at 1686 and 1546 cm−1 together with the fit. The varying sign of the DAS amplitudes and the course of the absorbance transient at a given wavenumber (e.g., 1546 cm−1) indicate the presence of three processes within the timescale of 700 ps, involving distinct molecular structures. For example, at 1546 cm−1, the initially positive signal decreases quickly and rises again, followed by a much slower partial decay.
Figure 4.
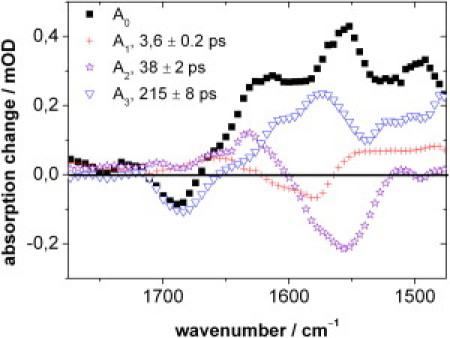
DAS of the kinetic components in the PChlide dynamics as derived from the global fit (Eq. 1) of the transient IR data with time constants τ1 = 3.6 ± 0.2, τ2 = 38 ± 2, and τ3 = 215 ± 8 ps.
Figure 5.
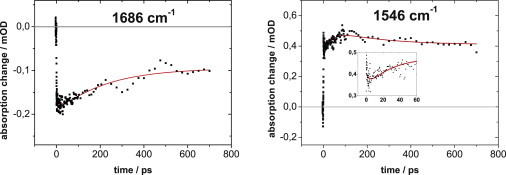
Representative IR absorbance transients of PChlide after excitation at 630 nm. The temporal evolution of the absorption changes is shown at 1686 cm−1 and 1546 cm−1 (symbols) together with the (global) fit of the data with the time constants τ1 = 3.6 ± 0.2, τ2 = 38 ± 2, and τ3 = 215 ± 8 ps (solid line).
As expected, the DAS A0, belonging to the longest-lived component, is almost identical to the difference spectrum at long delay times (700 ps) in Fig. 3. It describes the difference between the molecular states that exist 700 ps after excitation and the PChlide electronic ground state, the latter characterized by the carbonyl stretching mode at 1686 cm−1. Thus, after 700 ps, the PChlide ground state has recovered only partially (cf. Fig. 3) and the positive bands below 1670 cm−1 represent the longer-lived molecular state(s) formed on this timescale.
A2 basically displays a positive band at ∼1625 cm−1 and a negative band around 1565 cm−1 with a shoulder at 1580 cm−1, indicating a relative decrease and increase of absorbance strength in the respective region with τ2 = 38 ps. This is in line with the corresponding absorption features in Fig. 3.
In A3 the positive band around 1565 cm−1 corresponds to the negative band in A2, and the negative band coincides with the PChlide carbonyl stretch at 1686 cm−1. Thus, A3 suggests the decay of a state characterized by the band around 1565 cm−1 and the partial recovery of the PChlide electronic ground state with τ3 = 215 ps.
A1, which describes the fastest process with τ1 = 3.6 ps, exhibits positive and negative signals of small amplitudes around 1660 cm−1 and 1580 cm−1, respectively. Below 1560 cm−1, a broad and unstructured positive feature is observed.
Discussion
To further elucidate the time-resolved mid-IR measurements described above, we briefly summarize the current kinetic model, which accounts for the excited-state dynamics in PChlide on the basis of previous transient absorption and fluorescence studies (15–19,21), in Fig. 6. The key features of this model are as follows: photoexcitation of PChlide into the Franck-Condon state region initiates relaxation along two parallel pathways, one reactive and one nonreactive, with quantum yields of ∼60 and 40%, respectively. Relaxation along the reactive path leads to the population of a secondary excited state (SX), from which an intermediate photoproduct (SICT) with charge transfer character is formed in ∼25 ps. The SICT state converts with its lifetime of ∼200 ps to the initial ground state. On the other hand, in the nonreactive path a subset of the initially excited Franck-Condon state population undergoes relaxation toward the lowest excited singlet state (S1) via vibrational cooling with a time constant of ∼3.5 ps. Decay of the thermally equilibrated S1 state population then occurs on the nanosecond timescale by intersystem crossing into the triplet state.
Figure 6.
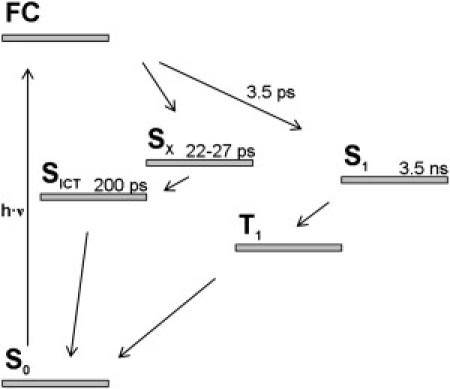
Kinetic model of the excited-state dynamics in PChlide as derived from ultrafast transient absorption and fluorescence studies (15–19,21). FC denotes the Franck-Condon excited state; for other abbreviations, see text.
The analysis of the transient IR data presented in this work yields three time constants of τ1 = 3.6, τ2 = 38, and τ3 = 215 ps, which are in good agreement with the time constants of 3.5, 22–27, and 200 ps estimated from the population dynamics of PChlide in previous spectroscopy studies (cf. Fig. 6) (15–19,21). Thus, the electronic-state kinetics derived from transient absorption and fluorescence studies is reflected by the transient IR spectra. In particular, the C13′=O carbonyl stretching mode serves as a marker band for the involved electronic states. Frequencies between 1650 and 1710 cm−1 have been reported for corresponding bands in the electronic ground state of chlorophyll and related species (37,38), with a downshift in the excited electronic state (e.g., from 1683 to 1666 cm−1 in bacteriochlorophyll (39)). The downshift can be even larger in species with altered electronic structure, or when hydrogen-bonding interactions weaken the C=O bond. For example, the carbonyl stretch in the excited electronic state of 2-(2′-hydroxyphenyl)benzothiazole is found at 1530 cm−1, due to hydrogen bonding of the carbonyl group in the keto tautomer and integration of this group in the conjugated C=C bond system (40,41). Furthermore, a dramatic downshift of the C=O stretching mode in the S1 state of ∼190 cm−1 was revealed by density functional theory calculations on fluorenone (42,43). Thus, the bleaching band at 1686 cm−1 observed in this work is assigned to the C13′=O carbonyl stretch of the PChlide electronic ground state, and the positive band around 1625 cm−1 in A2, which decays with τ2 = 38 ps, is assigned to its counterpart in the excited electronic state S1625. The decay of the excited state S1625 goes along with the concomitant formation of a new electronic state, S1565, in which the electronic structure has changed. This new state is characterized by the negative band in A2 at 1565 cm−1, including a weak shoulder at 1580 cm−1. The dominant contributions to the band around 1565/1580 cm−1 can be assigned to more or less delocalized porphyrin skeleton modes according to density functional theory calculations (20). However, in S1565 the C13′=O stretch must also be located at some frequency, i.e., a negative counterpart to the positive band at 1625 cm−1 in A2 is definitely to be expected. Thus, considering previous results regarding the frequencies of C=O stretching modes (39–43), it is not implausible to (heuristically) assign parts of the negative bands in A2 to the C13′=O stretch in S1565. On the basis of these considerations, the following conclusions can be drawn:
-
1.
The negative band at 1686 cm−1 in A3 clearly indicates the partial recovery of the PChlide electronic ground state with τ3 = 215 ps. This ground-state repopulation is in accordance with the 200-ps recovery kinetics observed in the transient absorption experiments (Fig. 6) (16,19,21).
-
2.
For the extent of the partial recovery, an estimate of 50% is derived from the decrease in the amplitude at this wavenumber relative to that at early delay times when bleaching reached the maximum value.
-
3.
The direct link between A3 (describing the recovery of the electronic ground state via decay of an excited electronic state) and A2 is given by the coinciding positive and negative bands around 1565/1580 cm−1 in the two spectra. Together with the positive band at 1625 cm−1 in A2, they suggest that an excited electronic state, S1625, characterized by a C13′=O stretch at 1625 cm−1, converts with τ2 = 38 ps into another excited state, S1565, with a characteristic band at ∼1565/1580 cm−1. S1565 then decays with τ3 = 215 ps into the PChlide ground state. This scenario clearly supports the kinetic scheme summarized in Fig. 6, thereby identifying S1625 and S1565 with Sx and SICT, respectively.
-
4.
The decay of the C13′=O stretch at 1625 cm−1 in A2 with τ2 = 38 ps and the simultaneous appearance of the band at ∼1565/1580 cm−1 indicates a significant change in the electronic structure upon the S1625 → S1565 transition. The 38-ps time constant is in accordance with the 22–27 ps rise time of the intermediate photoproduct, which was seen in previous transient absorption studies and ascribed to an intramolecular charge transfer state (SICT) (18,19,21). The increase in electron density at the C13′=O bond resulting from an intramolecular charge transfer directly affects the C13′=O bond order and reduces the double-bond character. Furthermore, the formation of the SICT state in a hydrogen-bonding solvent is most likely assisted by dynamic hydrogen-bonding interactions between the C13′=O site and the solvent (34,44). This could give rise to another increase in the C13′=O bond length. As a result of these effects, a red shift of the C13′=O stretch is to be expected in the SICT state. It is conceivable that this red shift is at least partially reflected in the appearance of the band at 1565/1580 cm−1. As argued above, such a remarkable downshift of the C=O stretching frequency in an excited state is not unusual. Thus, it is tempting to speculate that the transient changes in the C13′=O stretching mode are related to the molecular changes that occur upon formation of the SICT state.
Finally, A0 describes the difference between the S1-excited state and the PChlide electronic ground state (Fig. 6). Besides the carbonyl stretching mode of the latter at 1686 cm−1, it is dominated (below 1670 cm−1) by strongly overlapping modes of S1, mostly with C=C- and C=N-in-plane character. Due to the long lifetime of 3.5 ns for the S1 state, this difference spectrum appears static on the timescale of the experiment. Accordingly, triplet formation and subsequent decay into the PChlide ground state is not observed.
As discussed above, A2 and A3 strongly suggest consecutive processes, with S1625 decaying into S1565, and S1565 returning to the PChlide ground state. In addition, there is a fast 3.6 ps dynamics represented by A1. Several different variants can be used to implement a third process into this sequential reaction scheme. However, the fact that the amplitude of A1 is of the same order of magnitude as A2 and A3, together with the time ordering τ1 << τ2 << τ3, excludes the insertion of an additional state with the lifetime τ1 after S1565 or between S1565 and S1625. Instead, the process associated with τ1 could feed S1625. Consequently, to explain the nonzero A0, this scenario requires the existence of a further reaction branch, leading to a state with a lifetime much longer than 700 ps. This indeed is an essential feature of the scheme in Fig. 6. Hence, the 3.6 ps process could also feed the long-lived state, corresponding to the S1-excited state. The time regime of A1 is consistent with that of vibrational energy relaxation and/or cooling. These relaxational processes are expected in both channels, i.e., during population of the S1 and Sx states (Fig. 6), albeit to different extents depending on how much excess energy is released in either channel. In this case, A1 would reflect the relatively small frequency shift (few wavenumbers) of each vibrational mode that is involved in vibrational relaxation or cooling. Such a DAS would then be composed of pairs of adjacent positive and negative signals (difference bands), with the positive one related to the decaying (unrelaxed, hot) vibrational species and the negative one related to the correspondingly arising (relaxed, cold) vibrational species. Thus, all affected vibrational modes of either S1 or Sx or both (i.e., those modes with positive amplitude in A0 and A2) could contribute to A1 in this way. However, the rather structureless shape of A1 and the shape of A0 and A2 do not allow such decomposition. In addition, considering excitation at 630 nm (i.e., at λmax of the PChlide Q-absorption band) and the small energy gap of ΔE (FC, SX) = 255 cm−1 and ΔE (FC, S1) = 424 cm−1 (estimated from fluorescence data (17)), a rather small effect is anticipated. This is in line with previous results from Franck-Condon simulations of vibrational relaxation effects in chlorophyll a (45), in which the Franck-Condon active modes exhibited rather small Huang-Rhys factors resulting in only small spectral changes of the transient absorption spectra. Therefore, although vibrational cooling in S1 or Sx or both is not in contradiction to the data presented here, a more specific assignment based on the transient IR absorption data is not reasonable at this point. On the other hand, the time-resolved fluorescence measurements (17) clearly indicate a 3.5-ps rise time of the long-lived S1 fluorescence in contrast to the short-lived fluorescence of the Sx state that appears instantaneously. Thus, the 3.6-ps process reflected by the amplitude spectrum A1 can be attributed to vibrational energy relaxation in the S1-excited state.
In comparison with the mid-IR absorption dynamics observed upon 400-nm excitation of PChlide in methanol (34), it is most striking that the ground-state recovery with τ3 = 215 ps clearly indicated in our experiments by the negative band at 1686 cm−1 in A3 is not detected. However, the initial downshift of the C13′=O stretching mode from 1686 cm−1 in the ground state to 1625 cm−1 in the excited state (S1625) is also seen (1687 → 1625 cm−1), whereas the corresponding PChlide excited state is ascribed to a solvated S1/ICT complex. Because of the absence of any ground-state recovery up to 3 ns, the reaction dynamics is approached by a sequential mechanism whereby multiphasic decay with time constants of 60 ps and 3 ns occurs via solvation of the PChlide excited state and the subsequent formation of the PChlide triplet state. On the other hand, the dynamic features uncovered at 630 nm excitation in this study (i.e., the repopulation of the depleted ground state with τ3 = 215 ps along with the growing-in of the band at 1565/1580 cm−1 with τ2 = 38 ps) are consistent with our previous absorption and fluorescence studies (15–17,19–21) and support the branching of the excited-state population into two reaction channels. It therefore appears that excitation into the Soret and Q-band leads to differences in the initial reaction dynamics and most likely affects the routing of the excitation energy into distinct relaxation channels.
In conclusion, the transient IR difference spectra presented here for PChlide in solution allow a detailed insight into the primary reaction dynamics. In particular, the transient changes in the C13′=O stretching mode of the isocyclic fifth ring within the PChlide structure provide an important structural probe of this dynamics. Thus, the first changes in nuclear coordinates after excitation are reflected by the decay of the C13′=O stretching mode at 1625 cm−1 with a time constant of 38 ps and the concomitant formation of a new electronic state, S1565, characterized by a band at 1565/1580 cm−1. These data suggest a significant structural change in the C13′=O mode (most likely along with other delocalized C=C and C=N modes of the porphyrin skeleton) upon formation of S1565. It seems reasonable to suppose that the structural changes along the C13′=O coordinate are related to the formation of an intramolecular charge transfer state on the excited-state hypersurface, as seen in the VIS spectral region in our previous studies (18,19,21).
An ultrafast reactive motion that involves the generation of a charge transfer state may also be a crucial process in the photoreduction of PChlide by the enzyme PChlide reductase. It is quite conceivable that the enzymatic reaction is initiated by a charge transfer mechanism that creates an electron-deficient site at the porphyrin skeleton and thus facilitates the actual reduction of the C17/C18 double bond in a subsequent reaction step. Furthermore, the kinetics of the key vibrational modes recorded in this time-resolved IR absorption study is in full agreement with the kinetic model suggested on the basis of results from previous absorption and fluorescence studies (15–19,21). In particular, the results corroborate the main branching of the initially excited-state population into reactive and nonreactive paths, with 50% of the population decaying via the reactive route to the charge transfer state, and the other 50% undergoing relaxation toward the vibrationally relaxed S1-excited state via the nonreactive route.
Overall, the results of this study contribute to a broader understanding of the early relaxation dynamics in PChlide at the molecular level. The molecular structural details detected in the excited-state dynamics of isolated PChlide may be helpful in elucidating the catalytic mechanism of the PChlide reduction by the enzyme POR.
Acknowledgments
This study was supported by grants from the Deutsche Forschungsgemeinschaft (Graduiertenkolleg 792 to R.D., M.C., and R.G.; and HE 2657 and PO 563 to S.S, B.D, M.S., J.P., and G.H.).
References
- 1.Beale S.I. Enzymes of chlorophyll biosynthesis. Photosynth. Res. 1999;60:43–73. [Google Scholar]
- 2.Bollivar D.W. Recent advances in chlorophyll biosynthesis. Photosynth. Res. 2006;90:173–194. doi: 10.1007/s11120-006-9076-6. [DOI] [PubMed] [Google Scholar]
- 3.Tanaka R., Tanaka A. Tetrapyrrole biosynthesis in higher plants. Annu. Rev. Plant Biol. 2007;58:321–346. doi: 10.1146/annurev.arplant.57.032905.105448. [DOI] [PubMed] [Google Scholar]
- 4.Masuda T., Fujita Y. Regulation and evolution of chlorophyll metabolism. Photochem. Photobiol. Sci. 2008;7:1131–1149. doi: 10.1039/b807210h. [DOI] [PubMed] [Google Scholar]
- 5.Heyes D.J., Hunter C.N. Making light work of enzyme catalysis: protochlorophyllide oxidoreductase. Trends Biochem. Sci. 2005;30:642–649. doi: 10.1016/j.tibs.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Yang J., Cheng Q. Origin and evolution of the light-dependent protochlorophyllide oxidoreductase (LPOR) genes. Plant Biol (Stuttg) 2004;6:537–544. doi: 10.1055/s-2004-821270. [DOI] [PubMed] [Google Scholar]
- 7.Buhr F., El Bakkouri M., Reinbothe C. Photoprotective role of NADPH:protochlorophyllide oxidoreductase A. Proc. Natl. Acad. Sci. USA. 2008;105:12629–12634. doi: 10.1073/pnas.0803950105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schoefs B., Franck F. Protochlorophyllide reduction: mechanisms and evolutions. Photochem. Photobiol. 2003;78:543–557. doi: 10.1562/0031-8655(2003)078<0543:prmae>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 9.Lebedev N., Timko M.P. Protochlorophyllide photoreduction. Photosynth. Res. 1998;58:5–23. [Google Scholar]
- 10.Wilks H.M., Timko M.P. A light-dependent complementation system for analysis of NADPH:protochlorophyllide oxidoreductase: identification and mutagenesis of two conserved residues that are essential for enzyme activity. Proc. Natl. Acad. Sci. USA. 1995;92:724–728. doi: 10.1073/pnas.92.3.724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heyes D.J., Hunter C.N., Groot M.L. Ultrafast enzymatic reaction dynamics in protochlorophyllide oxidoreductase. Nat. Struct. Biol. 2003;10:491–492. doi: 10.1038/nsb929. [DOI] [PubMed] [Google Scholar]
- 12.Sytina O.A., Heyes D.J., Groot M.L. Conformational changes in an ultrafast light-driven enzyme determine catalytic activity. Nature. 2008;456:1001–1004. doi: 10.1038/nature07354. [DOI] [PubMed] [Google Scholar]
- 13.Sytina O.A., Heyes D.J., Groot M.L. Ultrafast catalytic processes and conformational changes in the light-driven enzyme protochlorophyllide oxidoreductase (POR) Biochem. Soc. Trans. 2009;37:387–391. doi: 10.1042/BST0370387. [DOI] [PubMed] [Google Scholar]
- 14.Heyes D.J., Scrutton N.S. Conformational changes in the catalytic cycle of protochlorophyllide oxidoreductase: what lessons can be learnt from dihydrofolate reductase? Biochem. Soc. Trans. 2009;37:354–357. doi: 10.1042/BST0370354. [DOI] [PubMed] [Google Scholar]
- 15.Dietzek B., Maksimenka R., Schmitt M. Excited-state processes in protochlorophyllide a—a femtosecond time-resolved absorption study. Chem. Phys. Lett. 2004;397:110–115. doi: 10.1021/jp0556456. [DOI] [PubMed] [Google Scholar]
- 16.Dietzek B., Kiefer W., Schmitt M. Solvent effects on the excited-state processes of protochlorophyllide: a femtosecond time-resolved absorption study. J. Phys. Chem. B. 2006;110:4399–4406. doi: 10.1021/jp0556456. [DOI] [PubMed] [Google Scholar]
- 17.Dietzek B., Kiefer W., Schmitt M. The excited-state chemistry of protochlorophyllide a: a time-resolved fluorescence study. ChemPhysChem. 2006;7:1727–1733. doi: 10.1002/cphc.200600172. [DOI] [PubMed] [Google Scholar]
- 18.Schmitt M., Dietzek B., Popp J. Femtosecond time-resolved spectroscopy on biological photoreceptor chromophores. Laser Photonics Rev. 2007;1:57–78. [Google Scholar]
- 19.Dietzek B., Tschierlei S., Popp J. Protochlorophyllide a: a comprehensive photophysical picture. ChemPhysChem. 2009;10:144–150. doi: 10.1002/cphc.200800536. [DOI] [PubMed] [Google Scholar]
- 20.Hanf R., Tschierlei S., Popp J. Probing the structure and Franck-Condon region of protochlorophyllide a through analysis of the Raman and resonance Raman spectra. J. Raman Spectrosc. 2009;41:414–423. [Google Scholar]
- 21.Dietzek B., Tschierlei S., Popp J. Dynamics of charge separation in the excited-state chemistry of protochlorophyllide. Chem. Phys. Lett. 2010;492:157–163. [Google Scholar]
- 22.Dietzek B., Maksimenka R., Schmitt M. The excited-state dynamics of magnesium octaethylporphyrin studied by femtosecond time-resolved four-wave-mixing. Chem. Phys. Lett. 2005;415:94–99. [Google Scholar]
- 23.Frank H.A., Bautista J.A., Wasielewski M.R. Effect of the solvent environment on the spectroscopic properties and dynamics of the lowest excited states of carotenoids. J. Phys. Chem. B. 2000;104:4569–4577. [Google Scholar]
- 24.Zigmantas D., Hiller R.G., Polivka T. Dynamics of excited states of the carotenoid peridinin in polar solvents: dependence on excitation wavelength, viscosity, and temperature. J. Phys. Chem. B. 2003;107:5339–5348. [Google Scholar]
- 25.Polívka T., Sundström V. Ultrafast dynamics of carotenoid excited states—from solution to natural and artificial systems. Chem. Rev. 2004;104:2021–2071. doi: 10.1021/cr020674n. [DOI] [PubMed] [Google Scholar]
- 26.Van Tassle A.J., Prantil M.A., Fleming G.R. Excited state structural dynamics of the charge transfer state of peridinin. Isr. J. Chem. 2007;47:17–24. [Google Scholar]
- 27.Bonetti C., Alexandre M.T.A., Kennis J.T. Identification of excited-state energy transfer and relaxation pathways in the peridinin-chlorophyll complex: an ultrafast mid-infrared study. Phys. Chem. Chem. Phys. 2010;12:9256–9266. doi: 10.1039/b923695c. [DOI] [PubMed] [Google Scholar]
- 28.Herbst J., Heyne K., Diller R. Femtosecond infrared spectroscopy of bacteriorhodopsin chromophore isomerization. Science. 2002;297:822–825. doi: 10.1126/science.1072144. [DOI] [PubMed] [Google Scholar]
- 29.Peters F., Herbst J., Diller R. Primary reaction dynamics of halorhodopsin, observed by sub-picosecond IR-vibrational spectroscopy. Chem. Phys. 2006;323:109–116. [Google Scholar]
- 30.Schumann C., Gross R., Lamparter T. Subpicosecond midinfrared spectroscopy of the Pfr reaction of phytochrome Agp1 from Agrobacterium tumefaciens. Biophys. J. 2008;94:3189–3197. doi: 10.1529/biophysj.107.119297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wolf M.M.N., Schumann C., Diller R. Ultrafast infrared spectroscopy of riboflavin: dynamics, electronic structure, and vibrational mode analysis. J. Phys. Chem. B. 2008;112:13424–13432. doi: 10.1021/jp804231c. [DOI] [PubMed] [Google Scholar]
- 32.Gross R., Wolf M.M.N., Diller R. Primary photoinduced protein response in bacteriorhodopsin and sensory rhodopsin II. J. Am. Chem. Soc. 2009;131:14868–14878. doi: 10.1021/ja904218n. [DOI] [PubMed] [Google Scholar]
- 33.Maksimenka R., Dietzek B., Schmitt M. Population dynamics of vibrational modes in stilbene-3 upon photoexcitation to the first excited state. Chem. Phys. Lett. 2005;408:37–43. [Google Scholar]
- 34.Sytina O.A., van Stokkum I.H.M., Groot M.L. Protochlorophyllide excited-state dynamics in organic solvents studied by time-resolved visible and mid-infrared spectroscopy. J. Phys. Chem. B. 2010;114:4335–4344. doi: 10.1021/jp9089326. [DOI] [PubMed] [Google Scholar]
- 35.Nabedryk E., Leonhard M., Breton J. Fourier transform infrared difference spectroscopy shows no evidence for an enolization of chlorophyll a upon cation formation either in vitro or during P700 photooxidation. Biochemistry. 1990;29:3242–3247. doi: 10.1021/bi00465a015. [DOI] [PubMed] [Google Scholar]
- 36.Hartwich G., Geskes C., Mäntele W. Fourier transform infrared spectroscopy of electrogenerated anions and cations of metal-substituted bacteriochlorophyll a. J. Am. Chem. Soc. 1995;117:7784–7790. [Google Scholar]
- 37.Mäntele W.G., Wollenweber A.M., Breton J. Infrared spectroelectrochemistry of bacteriochlorophylls and bacteriopheophytins: Implications for the binding of the pigments in the reaction center from photosynthetic bacteria. Proc. Natl. Acad. Sci. USA. 1988;85:8468–8472. doi: 10.1073/pnas.85.22.8468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mattioli T.A., Williams J.C., Robert B. Changes in primary donor hydrogen-bonding interactions in mutant reaction centers from Rhodobacter sphaeroides: identification of the vibrational frequencies of all the conjugated carbonyl groups. Biochemistry. 1994;33:1636–1643. doi: 10.1021/bi00173a004. [DOI] [PubMed] [Google Scholar]
- 39.Hamm P., Zurek M., Zinth W. Femtosecond infrared spectroscopy of reaction centers from Rhodobacter sphaeroides between 1000 and 1800 cm−1. Proc. Natl. Acad. Sci. USA. 1995;92:1826–1830. doi: 10.1073/pnas.92.6.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rini M., Dreyer J., Elsaesser T. Ultrafast vibrational relaxation processes induced by intramolecular excited state hydrogen transfer. Chem. Phys. Lett. 2003;374:13–19. [Google Scholar]
- 41.Rini M., Kummrow A., Elsaesser T. Femtosecond mid-infrared spectroscopy of condensed phase hydrogen-bonded systems as a probe of structural dynamics. Faraday Discuss. 2003;122:27–40. doi: 10.1039/b201056a. discussion 79–88. [DOI] [PubMed] [Google Scholar]
- 42.Zhao G.J., Han K.L. Ultrafast hydrogen bond strengthening of the photoexcited fluorenone in alcohols for facilitating the fluorescence quenching. J. Phys. Chem. A. 2007;111:9218–9223. doi: 10.1021/jp0719659. [DOI] [PubMed] [Google Scholar]
- 43.Liu Y.F., Ding J.X., Sun J. Time-dependent density functional theory study on the electronic excited-state geometric structure, infrared spectra, and hydrogen bonding of a doubly hydrogen-bonded complex. J. Comput. Chem. 2009;30:2723–2727. doi: 10.1002/jcc.21297. [DOI] [PubMed] [Google Scholar]
- 44.Zhao G.-J., Han K.-L. Site-specific solvation of the photoexcited protochlorophyllide a in methanol: formation of the hydrogen-bonded intermediate state induced by hydrogen-bond strengthening. Biophys. J. 2008;94:38–46. doi: 10.1529/biophysj.107.113738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Struve W.S. Vibrational equilibration in absorption difference spectra of chlorophyll a. Biophys. J. 1995;69:2739–2744. doi: 10.1016/S0006-3495(95)80145-1. [DOI] [PMC free article] [PubMed] [Google Scholar]