

Abstract
Efavirenz is a second-generation nonnucleoside reverse transcriptase inhibitor (NNRTI) and a common component of clinically approved anti-AIDS regimens. NNRTIs are noncompetitive inhibitors that bind in a hydrophobic pocket in the p66 subunit of reverse transcriptase (RT) ∼10 Å from the polymerase active site. Hydrogen exchange mass spectrometry (HXMS) shows that efavirenz binding reduces molecular flexibility in multiple regions of RT heterodimer in addition to the NNRTI binding site. Of the 47 peptic fragments monitored by HXMS, 15 showed significantly altered H/D exchange rates in the presence of efavirenz. The slow cooperative unfolding of a β-sheet in the NNRTI binding pocket, which was previously observed in unliganded RT, is dramatically suppressed by efavirenz. HXMS also defines an extensive network of allosterically coupled sites, including four distinct regions of allosteric stabilization, and one region of allosteric destabilization. The effects of efavirenz binding extend >60 Å from the NNRTI binding pocket. Allosteric changes to the structural dynamics propagate to the thumb and connection subdomains and RNase H domain of the p66 subunit as well as the thumb and palm subdomains of the p51 subunit. These allosteric regions may represent potential new drug targets.
Introduction
HIV-1 reverse transcriptase (RT) is an essential enzyme in the HIV lifecycle and a major drug target. RT is a heterodimer of 66 and 51 kDa subunits (Fig. 1 A). The p66 subunit consists of a polymerase domain, containing fingers, palm, thumb, and connection subdomains, and an RNase H domain. In addition to the polymerase and RNase H active sites (1), the p66 subunit contains most of the residues that contact the primer/template (P/T) substrate. The polymerase and RNase H primer grips in p66 are important structural motifs that position the nucleic acid substrate at the enzyme active sites (1–3). The p51 subunit is identical in sequence to the p66 polymerase domain, but lacks the C-terminal RNase H domain. Despite a common amino-acid sequence, p51 and p66 adopt different tertiary folds in crystal structures of RT heterodimer.
Figure 1.
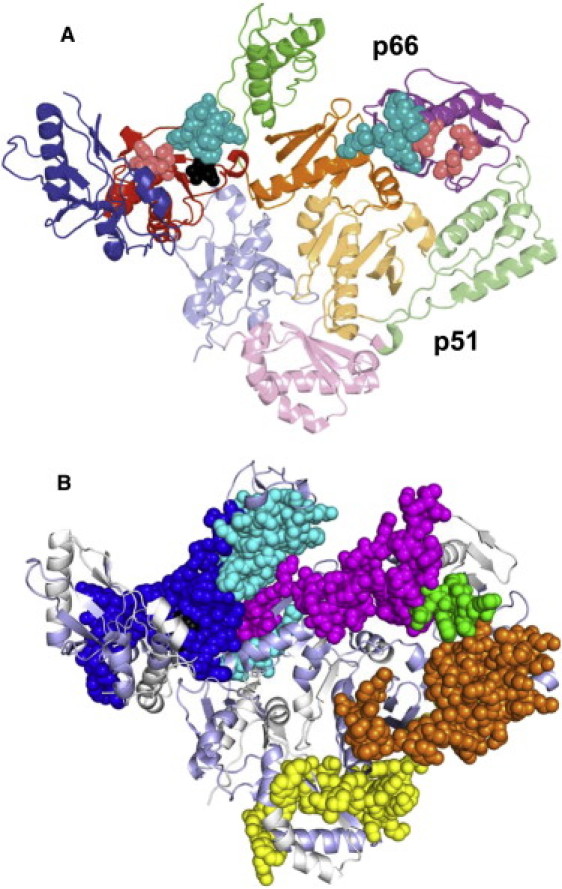
(A) Crystal structure of RT-EFV complex (1IKW). Four subdomains of the polymerase domain in p66 and p51 subunits: (blue) fingers, (red) palm, (green) thumb, and (orange) connection. Faded colors indicate subdomains of p51 subunit. The RNase H domain of p66 subunit is shown in magenta. Balls show: (black) efavirenz; (pink) polymerase and RNase H active sites; and (cyan) polymerase and RNase H primer grips. (B) Allosteric network in RT. Balls show: (dark blue) efavirenz binding region; (cyan) region 1; (magenta) region 2; (green) region 3; (orange) region 4; (yellow) region 5. (pale blue) Peptides with the same H/D exchange in presence and absence of efavirenz and (white) missing coverage. Figure made using PyMOL (52).
Nonnucleoside RT inhibitors (NNRTIs) are an important component of antiretroviral therapy. NNRTIs and protease inhibitors are more potent inhibitors of viral replication than nucleoside RT inhibitors (NRTIs) and integrase inhibitors (4). Using a mass-action-based model, Shen et al. (4) found that the steep dose-response curves of NNRTIs and protease inhibitors in single-round infectivity assays could be explained by the fact that these drugs target the enzymes themselves rather than specific enzyme-substrate complexes. For example, NNRTIs can bind to and inhibit multiple forms of RT, unlike NRTIs that target a single RT-P/T complex in the process of catalyzing nucleotide addition. This is supported by the observation that the NNRTI nevirapine interferes with proper positioning of RT on P/T substrate in the absence of dNTP (5,6). Accumulating evidence indicates that NNRTIs interfere with multiple RT functions in addition to nucleotide addition, and some NNRTIs may even disrupt later steps in the viral life cycle, such as Gag-Pol processing (7).
NNRTIs are small hydrophobic compounds that bind to a small pocket at the base of the thumb in the p66 palm and allosterically inhibit polymerase activity (8). However, the nature and extent of allostery in RT is not well defined. Unlike regulatory sites in allosteric enzymes, the NNRTI binding pocket does not appear to play a role in RT function. In fact, the binding pocket is not present in crystal structures of RT in the absence of bound NNRTIs (1,9,10). Crystal structures of RT-NNRTI complexes indicate that formation of the binding pocket is accompanied by further structural changes, most notably an extended conformation of the p66 fingers and thumb relative to unliganded RT (11). X-ray crystallography and computer simulations suggest that NNRTIs may function by restricting molecular motions (2,12–15). Various inhibition mechanisms have been proposed, including an arthritic thumb model, a primer grip model, and an active site distortion model.
Recent studies suggest that RT binds NNRTIs by a selected-fit binding mechanism (16–18). According to the population shift model, a protein samples a wide ensemble of states, only a small fraction of which is capable of binding ligand. The presence of a ligand stabilizes the binding competent states, which become the dominant population. The current view of allosteric transitions in proteins posits a population shift in either protein conformation, dynamics, or both (19). In the past, allosteric communication was thought to occur via a discrete pathway. The view of allostery has evolved to encompass multiple pathways and conformational fluctuations. In the former, allosteric communication occurs through changes in structure or dynamics along a series of continuous pathways (20), whereas in the latter, allostery arises by energetic coupling of intramolecular interactions and protein-solvent interactions (21).
Hydrogen/deuterium exchange is a powerful method for probing conformational fluctuations in proteins (22). Amide hydrogens in stable hydrogen bonds or buried in the protein interior are protected from exchange, but structural fluctuations can transiently disrupt local structure, exposing these protected hydrogens for exchange,
where k1 and k−1 are the rate constants for the local unfolding/refolding that exposes protected amides from exchange and kint is the intrinsic, chemical rate of H/D exchange at the given conditions. Interpretation of measured exchange rates depends on the relative magnitudes of kint and k−1. When kint << k−1, exchange occurs by the EX2 mechanism; the observed rate of exchange can be expressed as kobs = (k1/k−1)kint = Kunf kint. Under these conditions, locally or globally unfolded states are visited transiently, and refolding is fast compared to the chemical rate of exchange. Thus unfolding/refolding will typically occur many times before a particular hydrogen exchanges with deuterium and the observed rate of exchange is proportional to an equilibrium constant for local (or global) unfolding, Kunf. When kint >> k−1, then exchange occurs by the EX1 mechanism and the observed rate of exchange is a direct measure of the rate of unfolding; kobs = k1.
H/D exchange measured using mass spectrometry (HXMS) provides exchange rates at the peptide level rather than at the level of individual amide hydrogens (22). Upon quenching exchange by decreasing pH, proteins are digested with the acid protease pepsin, and the overall degree of exchange in each peptic fragment (typically 5–20 residues) is obtained from the mass. In the case of EX1 exchange, all amide hydrogens in a given region exchange when that region unfolds. If unfolding is slow (t1/2 > 1 s), this leads to characteristic bimodal isotopic envelopes in the mass spectra of peptides from that region, with the relative areas of the upper and lower mass/charge ratio (m/z) peaks corresponding to the populations in solution that have sampled the unfolded state (upper m/z peak) or remained folded (lower m/z peak). Under physiological conditions in the absence of denaturants, such double isotopic envelopes are rare but have been observed (23). Because the p51 subunit has the same amino-acid sequence as the polymerase domain of the p66 subunit, the subunits were separated after H/D exchange but before peptic digestion and analyzed in separate experiments by HPLC-MS. As a result, any double isotopic envelopes observed in these experiments are not artifacts due to different H/D exchange behavior in the two subunits.
Previously, we used HXMS to study the structure and dynamics of RT in solution (24). The HXMS studies of RT heterodimer showed that the structure of the palm, thumb, and connection subdomains of both subunits and the RNase H domain of p66 are significantly more flexible than expected from the crystal structure. An especially tantalizing finding was that β-sheet β12-β13-β14, which forms part of the binding pocket in the presence of NNRTI, undergoes EX1 exchange. This behavior is due to slow cooperative unfolding/refolding with t1/2 for unfolding ∼20 s in the absence of NNRTI. The present work employs HXMS to probe the structure and dynamics of RT bound to efavirenz (EFV), a second generation NNRTI in clinical use. The functional implications are discussed.
Materials and Methods
All biological reagents and chemicals were acquired from Roche Applied Science (Indianapolis, IN) and Sigma Chemicals (St. Louis, MO) unless otherwise specified. Efavirenz was obtained from the National Institutes of Health AIDS Research and Reference Reagent Program (Germantown, MD). D2O was purchased from Cambridge Isotope Laboratories (Andover, MA). RT buffer D is 0.05 M Tris (RNase, DNase-free, pH 7.0), 25 mM NaCl, 1 mM ethylenediaminetetraacetic acid, and 10% (v/v) glycerol (molecular biology grade redistilled).
The p66 and p51 proteins with N-terminal hexahistidine extensions were prepared separately as described (24,25). Proteins were expressed in Escherichia coli and purified by column chromatography; protein concentrations (monomer units) were determined by absorbance at 280 nm (25). The N-terminus of each monomer was labeled with biotin (24). The p66/p51 heterodimer was formed by equilibrating one unlabeled monomer with the other labeled monomer at 1:1 or 1:1.5 molar ratio in RT buffer D containing 50% (v/v) glycerol for one week at 4°C. The equilibrated protein was dialyzed into 3 × 0.5–1 L of RT buffer D containing 25 μM efavirenz for three days at 4°C. The concentration of RT-EFV complex is calculated using the Kd values for homo- and heterodimerization in the absence and presence of efavirenz and the Kd values for efavirenz binding to monomers, homodimers, and heterodimer (16,25). Solutions of 20–30 μM RT-EFV complex (1:1 p66:p51, [EFV] = 25 μM) contain 84% p66/p51-EFV complex plus 6% p66/p66-, 5% p51/p51-, 2% p66-, and 3% p51-EFV complexes. In 20–30 μM solutions of RT-EFV complex with 1.5-fold excess of labeled subunit, the unlabeled subunit is driven to 97% p66/p51-EFV complex with <3% homodimer- and monomer-EFV complexes.
HXMS
Peptide-mapping experiments were conducted on a LTQ-Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer (ThermoElectron, Waltham, MA) to confirm peptide identification by exact mass (24).
HXMS experiments were performed as described previously (24,26). A 20–30 μM solution of RT-EFV complex was diluted 10-fold into RT buffer D-D2O containing 5% glycerol (v/v) and incubated for 10 different times ranging from 5 s to 8 h at 25°C. Exchange was quenched by sevenfold dilution into 100 mM NaH2PO4 (pH 2.4) containing 100 μL of Ultralink immobilized neutraavidin protein beads (Pierce, Rockford, IL) at 5°C. The beads were previously washed with the quench buffer. The biotin-labeled subunit is bound to the beads and removed by centrifugation, while the unlabeled subunit remains in the supernatant. Use of 100 μL of beads ensured complete separation of the two subunits.
Deuterium-labeled protein in the supernatant was digested on ice with 5 μL of 1 mg/mL porcine pepsin in H2O for 5 min and analyzed by HPLC-MS as described (24,27). Data for deuterium uptake were collected using a LTQ-XL linear ion trap mass spectrometer. Additional data were collected using a LTQ-FT-ICR mass spectrometer.
Peptide centroid masses were calculated using the software HXExpress (28). Deuterium levels corrected for back exchange were calculated for each peptide using the equation
| (1) |
where D is the number of amide hydrogens exchanged with deuterium, m is the centroid mass of the peptide at a given time point, m0% is the mass of the undeuterated peptide, m100% is the mass of the fully deuterated peptide, and N is the number of amide hydrogens in the peptide. For peptides with double isotopic envelopes, the centroid mass was calculated for the entire range including peaks at both high and low m/z. The number of deuteria exchanged in unliganded RT (24) was subtracted from the number exchanged in the RT-EFV complex. Significant differences in deuteria exchanged were determined by repeating the 5 s time point 10 times. The average standard deviation for all peptides was 0.4 deuterium. Differences ≥1.2 deuteria exchanged (3 SD) were considered significant. Protein solutions containing 84% and 97% p66/p51-EFV complex gave identical results within error. Analysis of four repetitions of the complete H/D exchange time course for each subunit indicates reproducibility of the results.
A control experiment for nonspecific effects of efavirenz on the H/D exchange behavior of RT was performed. A concentrated solution of efavirenz in dimethylformamide (DMF) was added to RT heterodimer to a final concentration of 50 μM efavirenz and 1.6% DMF. HXMS was measured immediately after addition of efavirenz using an incubation time of 5 s in D2O. Formation of the specific RT-EFV complex is slow (16). The t1/2 for efavirenz binding is ∼15 min at the concentrations used here. At 5 s, no differences were observed between RT solutions containing 1.6% DMF with and without efavirenz.
Results
HXMS was performed and the measured deuterium uptake of RT-EFV complex was compared with the previously reported deuterium uptake of unliganded RT (24). Excepting the presence of efavirenz, experimental conditions were identical. RT-EFV complexes were diluted into D2O, samples were removed at various times, and the exchange was quenched. Peptic fragments provided ∼75% coverage for both subunits. The HXMS data for the two subunits of RT-EFV complex are consistent with the overall secondary structure in the crystal structures (29,30) with the exception of peptide 417–425 in p51 (Fig. 2).
Figure 2.
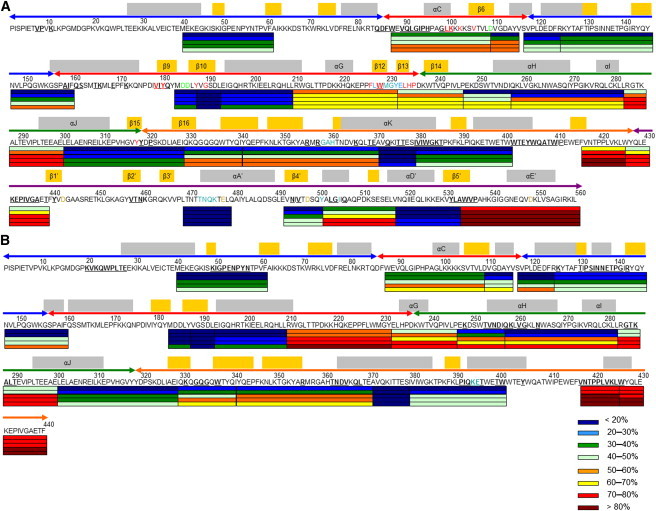
Percent exchange of peptides in RT-EFV complex. (A) p66 and (B) p51 subunits. Gray and orange bars above sequence represent α-helices and β-strands, with noted secondary structure labeled. Secondary structure represents the consensus of 1FK9 and 1IKW supplemented by the program STRIDE. Colored arrows above sequence represent polymerase subdomains: (blue) fingers, (red) palm, (green), thumb, (orange) connection; and (magenta) RNase H domain. Colored bars below sequence from top to bottom give exchange at 5, 60, 900, 3000, and 7200 s. Contact residues: (bold and underlined) dimer interface, (blue) polymerase and RNase H primer grips, (green) polymerase active site, (tan) RNase H active site, and (red) efavirenz.
Half of the exchangeable amide hydrogens in this peptide are located in an α-helix in p51, and yet the peptide shows no protection at even the shortest labeling times. Exchange that occurs at early times (5 s) after dilution into D2O primarily reflects solvent accessibility of the amide backbone; regions that are intrinsically unstructured and solvent-exposed or that are in a rapid equilibrium that favors an unfolded solvent-exposed form will undergo extensive exchange within a few seconds. Differences in exchange at 5 s in the presence and absence of ligand can therefore reveal folding/unfolding that occurs in response to ligand binding (31). Exchange at longer times (minutes to hours) reflects the flexibility of secondary and tertiary structure.
Efavirenz binding region
The consensus NNRTI binding pocket is formed by two β-sheets, β6-β10-β9 and β12-β13-β14, in the palm and Y318 at the thumb/connection junction of p66 as well as residue E138 of p51 (32). Efavirenz makes contact with 14 residues in p66 and none in p51 (29,30,33). Peptides 88–109, 187–192, 210–231, 232–246, and 301–328 contain 12 of the 14 residues in the binding site; V179 and Y181 in β-strand 9 are missing coverage. All but one of these peptides show significant reductions in H/D exchange in RT-EFV complex. Peptide 187–192 is so rigid that it exchanges only one amide hydrogen at long times in unliganded RT (24) and less than one amide hydrogen in the complex.
Three of the other peptides in p66 show modest reductions in exchange at long times (Fig. 3 A); 88–109, 210–231, and 232–246 exchange 2–4 fewer amide hydrogens in RT-EFV complex compared to unliganded RT. As in unliganded RT (24), peptide 232–246 undergoes slow cooperative unfolding/refolding leading to EX1 exchange. However, the rate of unfolding is dramatically slowed in the presence of efavirenz. This peptide includes β-strands 13 and 14 of β12-β13-β14 that forms the lid of the binding pocket. The EX1 exchange causes a double isotopic envelope due to two slowly interconverting conformations in solution, as discussed in the Introduction (34). Once the unfolded state is sampled in solution, it becomes irreversibly labeled with deuterium.
Figure 3.
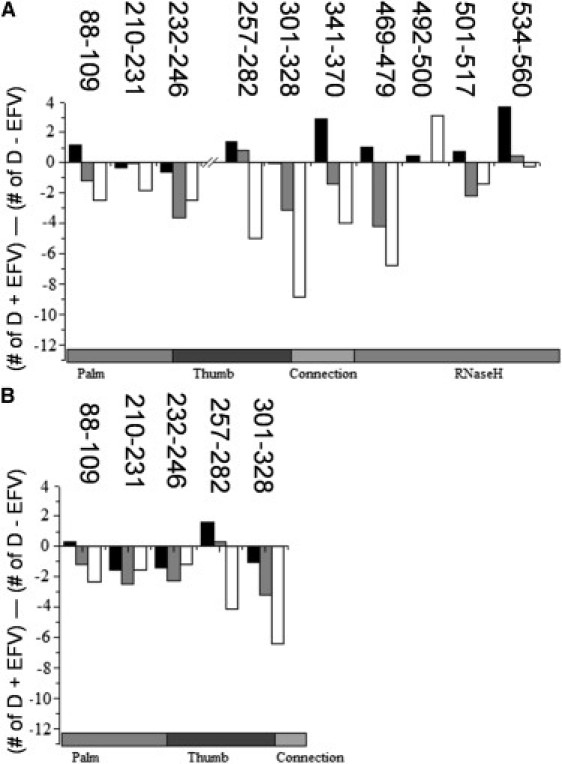
Difference in number of deuteria exchanged in RT-EFV complex and unliganded RT. Differences calculated by subtracting the exchange of unliganded RT (24) from the exchange of RT-EFV complex. Differences are shown for (A) p66 and (B) p51 subunits for exchange at (black) 5 s, (gray) 60 s, and (white) 3000 s. Only peptides with a difference >1.2 deuteria are shown.
Fig. 4 A shows the mass spectra of the doubly charged ion of peptide 232–246 at various times after dilution of RT-EFV complex into D2O. The 5 s time point shows that the folded population of β12-β13-β14 is the dominant form in solution with ∼80% of the molecules in the folded conformation. By 8 h, 60% of the folded population still remains, indicating that efavirenz locks the β-sheet in the folded conformation in the majority of the complexes. At the concentrations of RT and efavirenz used in the H/D exchange experiments, the heterodimer is saturated with efavirenz before and after dilution into D2O. Thus, RT-EFV complex populates at least two conformations with either a folded or unfolded β-sheet. The behavior of β12-β13-β14 in RT-EFV complex is in stark contrast to the rapid unfolding in unliganded RT, where the folded population has entirely disappeared after 30 s (24). Ligand binding has been shown before to slow the rate of unfolding observed by HXMS (23). The dramatic effect on the unfolding of β-strands 13 and 14, together with the decreased exchange at long times in the four peptides containing 10 of 14 drug contact residues, indicate that efavirenz binding stabilizes the entire efavirenz-binding region.
Figure 4.
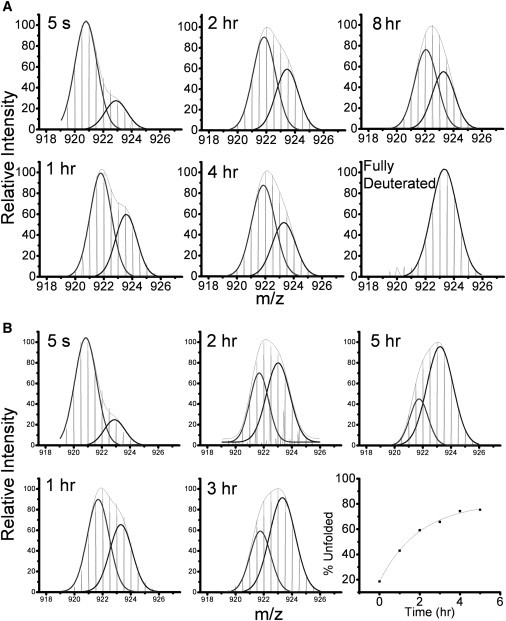
Fourier transform ion cyclotron resonance (FT-ICR) mass spectra of peptide 232–246 in RT-EFV complex. (Upper) p66 and (lower) p51 subunits after various incubation times in D2O (vertical lines). The low and high m/z peaks for both subunits are fit to (black) Gaussian distributions. The (gray) envelope is the sum of these two distributions. The percent unfolded for p51 subunit was calculated from the ratio of the areas of the high m/z black envelope to the gray envelope (bottom right panel).
Allosteric effects in p66
Fig. 3 shows differences in deuteria exchanged in RT-EFV complex relative to unliganded RT (24) for the 10 of 26 peptides in p66 and the five of 21 peptides in p51 that show significant changes in deuterium uptake at early, intermediate, or long times. Efavirenz contacts account for the three peptides in the p66 palm discussed above: 88–109, 210–231, and 232–246. The remaining seven peptides in the p66 thumb and connection and the RNase H domain show altered deuterium uptake due to allosteric effects of efavirenz binding (Fig. 3 A). The allosteric changes in p66 can be divided into three regions based on the effects of efavirenz on H/D exchange (Fig. 3 A; Table 1). The peptides in these three allosteric regions are contiguous in the three-dimensional structure of RT (Fig. 1 B).
Table 1.
Allosteric peptides and functional residues
| Function | Peptides/residues | Subunit |
|---|---|---|
| Efavirenz binding region | 88–109, 210–231, 232–246 | p66 |
| Allosteric region 1 | 257–282, 301–328 | p66 |
| Allosteric region 2 | 341–370, 469–479, 501–517 | p66 |
| Allosteric region 3 | 492–500, 534–560 | p66 |
| Allosteric region 4 | 88–109, 210–231, 232–246 | p51 |
| Allosteric region 5 | 257–282, 301–328 | p51 |
| Polymerase active site | D110, D185, D186 | p66 |
| Polymerase primer grip | F227, L228, W229, M230, G231, Y232, E233, L234 | p66 |
| RNase H active site | D443, E478, D498, D549 | p66 |
| RNase H primer grip | G359, A360, H361, T473, N474, Q475, K476, Y501, I505 | p66 |
| K395, E396 | p51 |
Region-1 peptides exchange fewer deuteria at long times in the presence of efavirenz: 257–282 in the thumb and 301–328 in the thumb/connection junction. Peptide 257–282 shows minor increases in exchange at early times, but large decreases at long times. These decreases indicate that the existing helices in the thumb, αH and αI, become more rigid as a result of efavirenz binding (Fig. 2 A). Peptide 301–328 contains a single drug contact at Y318. As with peptide 257–282, this peptide shows a large decrease in exchange at long times. Additionally, there are modest decreases at intermediate times. Although peptide 301–328 contains a drug contact residue, the fact that nine amide hydrogens show reduced exchange indicates allosteric stabilization of β-strands 15 and 16 and helix αJ (Fig. 2 A).
Region-2 peptides exchange fewer deuteria at intermediate and long times: 341–370 in the connection and 469–479 and 501–517 in the RNase H domain, indicating stabilization of three α-helices, αK, αA′, and αB′, and two β-strands, β17 and β18 (Fig. 2 A).
Region-3 peptides become slightly more flexible in RT-EFV complex: 492–500 and 534–560 also in the RNase H domain. This is the only region that is primarily destabilized by efavirenz binding. Peptide 492–500 contains the seven-residue β-strand 4′ (Fig. 2 A). All seven amide hydrogens are protected at all times in unliganded RT, and at early and intermediate times in RT-EFV complex. However, the increased exchange of three amide hydrogens at long times indicates that efavirenz binding makes the five-strand β-sheet β3′-β2′-β1′-β4′-β5′ in the RNase H domain slightly more flexible. Peptide 534–560 shows exchange of four more amide hydrogens at 5 s, which indicates unfolding. One crystal structure of RT-EFV complex contains the entire C-terminal helix αE′ (Fig. 2 A), which spans 11 amino acids (29). At early times in unliganded RT, peptide 534–560 has 11 protected amide hydrogens, dropping to four amide hydrogens at intermediate and long times (24). The RT-EFV complex has only seven amide hydrogens protected at early times, likewise dropping to four amide hydrogens at intermediate and long times. The early time data for this peptide indicate that helix αE′ is shortened by four amino acids in solution. Two of the four catalytic residues in the RNase H active site are contained in the region-3 peptides, and another single residue lacking coverage is adjacent to and possibly part of the putatively flexible β-strand 1′. Destabilization of secondary structure around the active site residues may account for the constellation of effects of efavirenz on RNase H activity (35).
Allosteric effects in p51
Five peptides in the palm and thumb subdomains of p51 show significant stabilization in RT-EFV complex (Fig. 3 B; Table 1).
Region-4 peptides exchange fewer deuteria at intermediate and long times in the presence of efavirenz: 88–109 and 210–231 in the palm, and 232–246 in the palm/thumb junction. Peptide 88–109 exchanges two fewer deuteria at long times, implying modest stabilization of β-strand 6 and helix αC due to efavirenz binding (Fig. 2 B). Peptide 210–231 exchanges approximately two fewer deuteria at all times, indicating the formation of two stable hydrogen bonds in RT-EFV complex. The crystal structures are missing electron density in the latter half of this peptide, and no secondary structure is reported for the first half in either the absence or presence of efavirenz. Nevertheless, about seven and five amide hydrogens are protected in RT-EFV complex and about five and three are protected in unliganded RT at intermediate and long times.
Surprisingly, peptide 232–246 in region 4 of p51 shows EX1 exchange in RT-EFV complex (Fig. 4 B). In p51 of unliganded RT, this peptide is fully exchanged by 5 s. In p66, this same peptide, which contains β-strands 13 and 14, shows EX1 exchange in both the presence and absence of efavirenz, but the rate of local cooperative unfolding is drastically slowed in the presence of efavirenz, as described above. Also in p66, this peptide is part of the NNRTI binding pocket. Equilibrium dialysis studies of p66/p66 and p51/p51 homodimers showed that one efavirenz binds per dimer in solution (16), consistent with the binding stoichiometry in all the crystal structures of RT-NNRTI complexes (11). Therefore, the EX1 exchange in Fig. 4 B is not due to efavirenz binding to the p51 subunit in solution.
The double isotopic envelope in p51 was fitted to two Gaussian distributions and the mass difference between the centroids corresponds to a difference of four amide hydrogens between the folded and unfolded peaks (Fig. 4 B). In the crystal structures of RT-EFV complex (29,30), this peptide contains helix αG with five exchangeable amide hydrogens (Fig. 2 B); helix αG is absent in unliganded RT. The EX1 behavior suggests helix αG undergoes slow cooperative unfolding with a t1/2 of 2 h in the presence of efavirenz. Formation of the small helix αG in the presence of efavirenz shortens the linker connecting the p51 thumb to the palm and could constrain the thumb motion, which would change the interaction between the p51 thumb and the RNase H domain. The tighter subunit interactions would stabilize helices αH, αI, and αJ in region 5 (Fig. 2 B). This scenario is consistent with the large enhancement of RT dimerization by efavirenz (25,36).
Region-5 peptides exchange fewer amide hydrogens at long times in RT-EFV complex: 257–282 in the thumb and 301–328 in the thumb/connection junction. At early times, peptide 257–282 exchanges two more deuteria in RT-EFV complex, which suggests slight unfolding upon efavirenz binding. At long times, this peptide exchanges four fewer amide hydrogens, consistent with stabilization of helix αH or αI (Fig. 2 B). Peptide 301–328 shows large decreases in exchange at intermediate and long times, indicating substantial stabilization of helix αJ. Region 5 is spatially adjacent to region 3 in the RNase H domain, and intersubunit allosteric effects may propagate through contacts between the p51 thumb and the RNase H domain.
Discussion
Efavirenz binding rigidifies the structure of RT. In the absence of inhibitor, the structure in the palm, thumb, and connection subdomains of both subunits as well as the RNase H domain is significantly more flexible than expected from crystal structures (24). The allosterically coupled regions identified here, which extend throughout the heterodimer, represent a subset of these more flexible peptides (Fig. 1 B). The large-scale rigidification in the presence of efavirenz is circumstantial evidence that the flexibility observed in the absence of inhibitor is critical for normal function of the enzyme. The multiple conformational states observed in crystallographic studies also provide circumstantial evidence for the importance of structural flexibility in the activity of RT and other polymerases (2,37,38). Although the p51 subunit is essential for RT function, it contains only one residue in the consensus NNRTI binding pocket (0 in the efavirenz binding site) and two residues in the RNase H primer grip (3,32). This, combined with the fact that p51 undergoes miniscule structural changes upon ligand binding, has led to the conjecture that p51 plays only a structural role. The HXMS results indicate otherwise. Figs. 1 B and 3 B show that two regions of p51 are part of the allosteric network.
How do the observed changes in flexibility correlate with the proposed models for NNRTI inhibition? The arthritic thumb model states that inhibitor binding restricts the motions of the thumb subdomain. HXMS results show that efavirenz binding rigidifies secondary structure in region 1 of the allosteric network, indicating that helices αH, αI, and αJ, which form the core of the p66 thumb, can no longer flex.
In the primer grip model, NNRTI binding distorts the residues which make up the polymerase primer grip, preventing proper positioning of the primer 3′-end in the polymerase active site. Peptides covering the efavirenz binding region include not only the drug contact residues, but also the polymerase primer grip (residues 227–235). The polymerase primer grip spans two peptides 210–231 and 232–246. The latter peptide, containing β-strands 13 and 14, undergoes slow cooperative unfolding in the absence of inhibitor, but efavirenz binding dramatically stabilizes β12-β13-β14. Peptide 210–231 exchanges two fewer deuteria at long times, implying that β-strand 12, which forms the other half of the polymerase primer grip, is also stabilized. Together these results clearly indicate that the flexibility of the polymerase primer grip is perturbed by efavirenz binding.
The active site distortion model asserts that NNRTI binding restricts conformational changes in the YMDD loop necessary for DNA polymerization. Peptide 183–187 covers the YMDD loop, which contains two of the three aspartate residues in the catalytic triad; the third aspartate of the triad is in peptide 110–115. H/D exchange rates of both peptides are similar in the presence and absence of efavirenz. The peptide containing the YMDD loop, like the adjacent peptide 187–192, is rigid, exchanging less than one amide hydrogen at long times in unliganded RT and at most one amide hydrogen in RT-EFV complex. The central β-strand 10 of β6-β10-β9 spans the rigid peptides 183–187 and 187–192, suggesting that this β-sheet is stable in both free and bound RT. The biochemical data and proposed inhibition mechanisms focus on the p66 polymerase domain, whereas the HXMS results point to a view of NNRTI action in which inhibition involves global suppression of protein dynamics in multiple domains and both subunits.
In contrast to the effects of NNRTIs on polymerase activity, the effects of inhibitors on RNase H activity have been less extensively studied. Efavirenz enhances the polymerase-dependent RNase H activity and inhibits the polymerase-independent activity (39,40). The RNase H inhibitor dihydroxy benzoyl napthyl hydrazone (DHBNH) provides another example of allosteric communication between the polymerase and RNase H domains (41). DHBNH binds between the NNRTI binding pocket and the polymerase active site, >50 Å from the RNase H active site. However, this inhibitor has little effect on polymerase activity. HXMS indicates that four of the five peptides in the RNase H domain undergo changes in flexibility upon EFV binding (Fig. 3 A). These peptides include nine of 11 residues in the RNase H primer grip, and two of four residues in the RNase H active site (1,3). Efavirenz binding stabilizes the RNase H primer grip and loosens the RNase H active site slightly. Although dramatic, the functional significance of these allosteric changes is not obvious. One possibility is that stabilization of the RNase H primer grip residues has differential effects on polymerase-dependent and -independent binding modes. Interestingly, in p66 monomer complexed with efavirenz, the RNase H domain does not undergo allosteric changes in regions 2 and 3 (42), indicating an active role for the p51 subunit in communication between the polymerase and RNase H domains of the p66 subunit.
The long-range effects of efavirenz binding identified by H/D exchange have implications for NNRTI resistance in RT. Most of the common clinical NNRTI resistance mutations, such as K103N and Y181C, are located near or in the NNRTI binding pocket. More recently, drug resistance mutations located in the connection and RNase H domain have been identified in patient populations (43). In particular, mutations N348I and Q509L confer resistance to some NNRTIs both alone and in combination with NRTI resistance mutations. It is puzzling how these mutations confer NNRTI resistance, given their locations remote from the binding pocket. Both mutations are located in region 2 of the allosteric network, whose conformational dynamics are altered by efavirenz binding. If the effects of NNRTI binding can propagate to these remote regions, as the HXMS data clearly demonstrate, then it is possible that mutational perturbations in these regions can propagate to the NNRTI binding site (as well as elsewhere in the allosteric network). Such bidirectional allosteric coupling has been observed in other proteins such as integrins and α2A/D adrenoceptors (44,45).
Ligands that preferentially bind to the folded state will stabilize a protein against unfolding. If a protein folds/unfolds as a single cooperative unit, then it is clear that ligand binding will stabilize the entire structure including regions distant from the binding site. Although this mechanism may account for the reduced exchange in the p66 polymerase domain, it cannot explain all of the long-range effects of efavirenz binding on RT. RT contains multiple cooperative folding units. For example, the isolated RNase H domain can be expressed as a folded and functional protein (46,47). Additionally, monomeric forms of the p66 and p51 subunits are folded in solution and bind efavirenz (16). Therefore some of the long-range alterations in H/D exchange induced by efavirenz must involve allosteric coupling between cooperative domains (48).
Classical studies of allostery emphasized discrete conformational changes, but it is now appreciated that allosteric communication can also occur through changes in structural dynamics (19). One view states that allostery originates in the ligand-induced stabilization of the residues around the binding site (20). This stabilization propagates by reducing the mobility of amino-acid side chains and amide backbone, forming numerous pathways through the protein. It eventually reaches other sites, either activating or inhibiting them. According to this model, upon efavirenz binding in the pocket, each region of the allosteric network would stabilize or destabilize contiguous regions, mechanically propagating the effects of binding throughout the heterodimer.
Alternately, an ensemble-based approach has been used to demonstrate energetic coupling of remote regions of a protein (21,49). In this model, the energy propagates through changes in the distribution of intra- and intermolecular interactions, including solvent, rather than by discrete pathways. In the case of RT, the subensemble with stabilized efavirenz binding and allosteric regions (Fig. 1 B) represents the protein population that binds efavirenz. This subensemble is sparsely populated in the absence of efavirenz, but shifts to the dominant population in RT-EFV complex. This picture is consistent with recent studies suggesting that RT binds NNRTIs by a conformational selection mechanism (16,17).
For efficient propagation of binding effects to distant regions, the ensemble-based approach predicts that the binding site must possess a dual character, including both stable and unstable regions (50). The NNRTI binding pocket is not present in crystal structures of RT in the absence of NNRTIs (2). Of the two β-sheets that form the efavirenz binding site, HXMS data show that β6-β10-β9 is almost completely stable, whereas β12-β13-β14 is quite unstable and rapidly samples an unfolded conformation in the absence of NNRTIs. Binding of efavirenz shifts the conformational ensemble of RT to a population in which β12-β13-β14 is greatly stabilized. This redistribution of the ensemble affects the efavirenz binding region as well as regions 1–5 of the allosteric network. Efavirenz binding to RT may therefore represent a particularly dramatic example of binding site stabilization coupled to allostery.
Conclusions
The HXMS studies of RT-EFV complex reveal an extended network of allosterically coupled sites that includes the NNRTI binding pocket, the p66 thumb and connection, and the RNase H domain, as well as the p51 palm and thumb. This network suggests a possible mechanism by which efavirenz binding in the NNRTI binding pocket might alter the function of both the polymerase active site 10 Å away and the RNase H active site 50 Å away. Moreover, it suggests an expanded view of inhibition, in which inhibitor binding rigidifies multiple regions of RT, encompassing regions beyond the p66 polymerase domain. Our results support both the arthritic thumb and primer grip models of inhibition, but incorporate an allosteric population shift from the flexible unliganded ensemble to a more rigid efavirenz-bound ensemble. The HXMS results reveal five regions in p66 and p51 which together form an extended allosterically coupled network. These allosteric regions are potential new drug targets (51).
Acknowledgments
We are grateful to Kathryn J. Howard for preparation of protein samples.
This work was supported by National Institutes of Health grant No. GM071267.
Contributor Information
Mary D. Barkley, Email: mdb4@case.edu.
Patrick L. Wintrode, Email: patrick.wintrode@case.edu.
References
- 1.Jacobo-Molina A., Ding J., Arnold E. Crystal structure of human immunodeficiency virus type 1 reverse transcriptase complexed with double-stranded DNA at 3.0 Å resolution shows bent DNA. Proc. Natl. Acad. Sci. USA. 1993;90:6320–6324. doi: 10.1073/pnas.90.13.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sarafianos S.G., Marchand B., Arnold E. Structure and function of HIV-1 reverse transcriptase: molecular mechanisms of polymerization and inhibition. J. Mol. Biol. 2009;385:693–713. doi: 10.1016/j.jmb.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sarafianos S.G., Das K., Arnold E. Crystal structure of HIV-1 reverse transcriptase in complex with a polypurine tract RNA:DNA. EMBO J. 2001;20:1449–1461. doi: 10.1093/emboj/20.6.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shen L., Peterson S., Siliciano R.F. Dose-response curve slope sets class-specific limits on inhibitory potential of anti-HIV drugs. Nat. Med. 2008;14:762–766. doi: 10.1038/nm1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abbondanzieri E.A., Bokinsky G., Zhuang X. Dynamic binding orientations direct activity of HIV reverse transcriptase. Nature. 2008;453:184–189. doi: 10.1038/nature06941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu S., Abbondanzieri E.A., Zhuang X. Slide into action: dynamic shuttling of HIV reverse transcriptase on nucleic acid substrates. Science. 2008;322:1092–1097. doi: 10.1126/science.1163108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Figueiredo A., Moore K.L., Tachedjian G. Potent nonnucleoside reverse transcriptase inhibitors target HIV-1 Gag-Pol. PLoS Pathog. 2006;2:e119. doi: 10.1371/journal.ppat.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kohlstaedt L.A., Wang J., Steitz T.A. Crystal structure at 3.5 Å resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- 9.Hsiou Y., Ding J., Arnold E. Structure of unliganded HIV-1 reverse transcriptase at 2.7 Å resolution: implications of conformational changes for polymerization and inhibition mechanisms. Structure. 1996;4:853–860. doi: 10.1016/s0969-2126(96)00091-3. [DOI] [PubMed] [Google Scholar]
- 10.Rodgers D.W., Gamblin S.J., Harrison S.C. The structure of unliganded reverse transcriptase from the human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. USA. 1995;92:1222–1226. doi: 10.1073/pnas.92.4.1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ren J., Stammers D.K. Structural basis for drug resistance mechanisms for non-nucleoside inhibitors of HIV reverse transcriptase. Virus Res. 2008;134:157–170. doi: 10.1016/j.virusres.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 12.Ivetac A., McCammon J.A. Elucidating the inhibition mechanism of HIV-1 non-nucleoside reverse transcriptase inhibitors through multicopy molecular dynamics simulations. J. Mol. Biol. 2009;388:644–658. doi: 10.1016/j.jmb.2009.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen L., Shen J., Jiang H. Steered molecular dynamics simulation on the binding of NNRTI to HIV-1 RT. Biophys. J. 2003;84:3547–3563. doi: 10.1016/S0006-3495(03)75088-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Temiz N.A., Bahar I. Inhibitor binding alters the directions of domain motions in HIV-1 reverse transcriptase. Proteins Struct. Funct. Bioinf. 2002;49:61–70. doi: 10.1002/prot.10183. [DOI] [PubMed] [Google Scholar]
- 15.Zhou Z., Madrid M., Madura J.D. Effect of a bound non-nucleoside RT inhibitor on the dynamics of wild-type and mutant HIV-1 reverse transcriptase. J. Am. Chem. Soc. 2005;127:17253–17260. doi: 10.1021/ja053973d. [DOI] [PubMed] [Google Scholar]
- 16.Braz V.A., Holladay L.A., Barkley M.D. Efavirenz binding to HIV-1 reverse transcriptase monomers and dimers. Biochemistry. 2010;49:601–610. doi: 10.1021/bi901579y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geitmann M., Unge T., Danielson U.H. Biosensor-based kinetic characterization of the interaction between HIV-1 reverse transcriptase and non-nucleoside inhibitors. J. Med. Chem. 2006;49:2367–2374. doi: 10.1021/jm0504048. [DOI] [PubMed] [Google Scholar]
- 18.Weikl T.R., von Deuster C. Selected-fit versus induced-fit protein binding: kinetic differences and mutational analysis. Proteins Struct. Funct. Bioinf. 2009;75:104–110. doi: 10.1002/prot.22223. [DOI] [PubMed] [Google Scholar]
- 19.Tsai C.-J., del Sol A., Nussinov R. Allostery: absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol. 2008;378:1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.del Sol A., Tsai C.-J., Nussinov R. The origin of allosteric functional modulation: multiple pre-existing pathways. Structure. 2009;17:1042–1050. doi: 10.1016/j.str.2009.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu T., Whitten S.T., Hilser V.J. Ensemble-based signatures of energy propagation in proteins: a new view of an old phenomenon. Proteins Struct. Funct. Bioinf. 2006;62:728–738. doi: 10.1002/prot.20749. [DOI] [PubMed] [Google Scholar]
- 22.Brier S., Engen J.R. Hydrogen exchange mass spectrometry: principles and capabilities. In: Chance M., editor. Mass Spectrometry Analysis for Protein-Protein Interactions and Dynamics. Wiley; Hoboken, NJ: 2008. pp. 11–43. [Google Scholar]
- 23.Engen J.R., Smithgall T.E., Smith D.L. Identification and localization of slow, natural, cooperative unfolding in the hematopoietic cell kinase SH3 domain by amide hydrogen exchange and mass spectrometry. Biochemistry. 1997;36:14384–14391. doi: 10.1021/bi971635m. [DOI] [PubMed] [Google Scholar]
- 24.Seckler J.M., Howard K.J., Wintrode P.L. Solution structural dynamics of HIV-1 reverse transcriptase heterodimer. Biochemistry. 2009;48:7646–7655. doi: 10.1021/bi900790x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Venezia C.F., Howard K.J., Barkley M.D. Effects of efavirenz binding on the subunit equilibria of HIV-1 reverse transcriptase. Biochemistry. 2006;45:2779–2789. doi: 10.1021/bi051915z. [DOI] [PubMed] [Google Scholar]
- 26.Tsutsui Y., Liu L., Wintrode P.L. The conformational dynamics of a metastable serpin studied by hydrogen exchange and mass spectrometry. Biochemistry. 2006;45:6561–6569. doi: 10.1021/bi060431f. [DOI] [PubMed] [Google Scholar]
- 27.Zhang Z., Smith D.L. Determination of amide hydrogen exchange by mass spectrometry: a new tool for protein structure elucidation. Protein Sci. 1993;2:522–531. doi: 10.1002/pro.5560020404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weis D.D., Engen J.R., Kass I.J. Semi-automated data processing of hydrogen exchange mass spectra using HX-Express. J. Am. Soc. Mass Spectrom. 2006;17:1700–1703. doi: 10.1016/j.jasms.2006.07.025. [DOI] [PubMed] [Google Scholar]
- 29.Lindberg J., Sigurdsson S., Unge T. Structural basis for the inhibitory efficacy of efavirenz (DMP-266), MSC194 and PNU142721 towards the HIV-1 RT K103N mutant. Eur. J. Biochem. 2002;269:1670–1677. doi: 10.1046/j.1432-1327.2002.02811.x. [DOI] [PubMed] [Google Scholar]
- 30.Ren J., Milton J., Stammers D.K. Structural basis for the resilience of efavirenz (DMP-266) to drug resistance mutations in HIV-1 reverse transcriptase. Structure. 2000;8:1089–1094. doi: 10.1016/s0969-2126(00)00513-x. [DOI] [PubMed] [Google Scholar]
- 31.Tsutsui Y., Wintrode P.L. Hydrogen/deuterium exchange-mass spectrometry: a powerful tool for probing protein structure, dynamics and interactions. Curr. Med. Chem. 2007;14:2344–2358. doi: 10.2174/092986707781745596. [DOI] [PubMed] [Google Scholar]
- 32.Das K., Lewi P.J., Arnold E. Crystallography and the design of anti-AIDS drugs: conformational flexibility and positional adaptability are important in the design of non-nucleoside HIV-1 reverse transcriptase inhibitors. Prog. Biophys. Mol. Biol. 2005;88:209–231. doi: 10.1016/j.pbiomolbio.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 33.Sobolev V., Sorokine A., Edelman M. Automated analysis of interatomic contacts in proteins. Bioinformatics. 1999;15:327–332. doi: 10.1093/bioinformatics/15.4.327. [DOI] [PubMed] [Google Scholar]
- 34.Deng Y., Smith D.L. Identification of unfolding domains in large proteins by their unfolding rates. Biochemistry. 1998;37:6256–6262. doi: 10.1021/bi972711o. [DOI] [PubMed] [Google Scholar]
- 35.Sluis-Cremer N., Tachedjian G. Mechanisms of inhibition of HIV replication by non-nucleoside reverse transcriptase inhibitors. Virus Res. 2008;134:147–156. doi: 10.1016/j.virusres.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tachedjian G., Orlova M., Goff S.P. Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc. Natl. Acad. Sci. USA. 2001;98:7188–7193. doi: 10.1073/pnas.121055998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steitz T.A. Visualizing polynucleotide polymerase machines at work. EMBO J. 2006;25:3458–3468. doi: 10.1038/sj.emboj.7601211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Svetlov V., Nudler E. Macromolecular micromovements: how RNA polymerase translocates. Curr. Opin. Struct. Biol. 2009;19:701–707. doi: 10.1016/j.sbi.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hang J.Q., Li Y., Klumpp K. Substrate-dependent inhibition or stimulation of HIV RNase H activity by non-nucleoside reverse transcriptase inhibitors (NNRTIs) Biochem. Biophys. Res. Commun. 2007;352:341–350. doi: 10.1016/j.bbrc.2006.11.018. [DOI] [PubMed] [Google Scholar]
- 40.Radzio J., Sluis-Cremer N. Efavirenz accelerates HIV-1 reverse transcriptase ribonuclease H cleavage, leading to diminished zidovudine excision. Mol. Pharmacol. 2008;73:601–606. doi: 10.1124/mol.107.038596. [DOI] [PubMed] [Google Scholar]
- 41.Himmel D.M., Sarafianos S.G., Arnold E. HIV-1 reverse transcriptase structure with RNase H inhibitor dihydroxy benzoyl napthyl hydrazone bound at a novel site. ACS Chem. Biol. 2006;1:702–712. doi: 10.1021/cb600303y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Braz V.A., Barkley M.D., Wintrode P.L. Efavirenz binding site in HIV-1 reverse transcriptase monomers. Biochemistry. 2010;49:10565–10573. doi: 10.1021/bi101480z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ehteshami M., Götte M. Effects of mutations in the connection and RNase H domains of HIV-1 reverse transcriptase on drug susceptibility. AIDS Rev. 2008;10:224–235. [PubMed] [Google Scholar]
- 44.Hynes R.O. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 45.Tian W.-N., Miller D.D., Deth R.C. Bidirectional allosteric effects of agonists and GTP at α2A/D-adrenoceptors. J. Pharmacol. Exp. Ther. 2000;292:664–671. [PubMed] [Google Scholar]
- 46.Pari K., Mueller G.A., London R.E. Solution structure of the RNase H domain of the HIV-1 reverse transcriptase in the presence of magnesium. Biochemistry. 2003;42:639–650. doi: 10.1021/bi0204894. [DOI] [PubMed] [Google Scholar]
- 47.Smith J.S., Roth M.J. Purification and characterization of an active human immunodeficiency virus type 1 RNase H domain. J. Virol. 1993;67:4037–4049. doi: 10.1128/jvi.67.7.4037-4049.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Luque I., Leavitt S.A., Freire E. The linkage between protein folding and functional cooperativity: two sides of the same coin? Annu. Rev. Biophys. Biomol. Struct. 2002;31:235–256. doi: 10.1146/annurev.biophys.31.082901.134215. [DOI] [PubMed] [Google Scholar]
- 49.Pan H., Lee J.C., Hilser V.J. Binding sites in Escherichia coli dihydrofolate reductase communicate by modulating the conformational ensemble. Proc. Natl. Acad. Sci. USA. 2000;97:12020–12025. doi: 10.1073/pnas.220240297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Freire E. The propagation of binding interactions to remote sites in proteins: analysis of the binding of the monoclonal antibody D1.3 to lysozyme. Proc. Natl. Acad. Sci. USA. 1999;96:10118–10122. doi: 10.1073/pnas.96.18.10118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goodey N.M., Benkovic S.J. Allosteric regulation and catalysis emerge via a common route. Nat. Chem. Biol. 2008;4:474–482. doi: 10.1038/nchembio.98. [DOI] [PubMed] [Google Scholar]
- 52.DeLano, W. L. 2002. The PyMOL Molecular Graphic System. DeLano Scientific, San Carlos, CA. http://www.pymol.org.