

Abstract
Intrinsic antiviral resistance mediated by constitutively expressed cellular proteins is one arm of defence against virus infection. Promyelocytic leukaemia nuclear bodies (PML-NBs, also known as ND10) contribute to host restriction of herpes simplex virus type 1 (HSV-1) replication via mechanisms that are counteracted by viral regulatory protein ICP0. ND10 assembly is dependent on PML, which comprises several different isoforms, and depletion of all PML isoforms decreases cellular resistance to ICP0-null mutant HSV-1. We report that individual expression of PML isoforms I and II partially reverses the increase in ICP0-null mutant HSV-1 plaque formation that occurs in PML-depleted cells. This activity of PML isoform I is dependent on SUMO modification, its SUMO interaction motif (SIM), and each element of its TRIM domain. Detailed analysis revealed that the punctate foci formed by individual PML isoforms differ subtly from normal ND10 in terms of composition and/or Sp100 modification. Surprisingly, deletion of the SIM motif from PML isoform I resulted in increased colocalisation with other major ND10 components in cells lacking endogenous PML. Our observations suggest that complete functionality of PML is dependent on isoform-specific C-terminal sequences acting in concert.
Keywords: HSV-1, ICP0, ND10, PML
Introduction
Promyelocytic leukaemia protein (PML) nuclear bodies (PML-NBs, referred to herein as ND10) are discrete, dynamic punctate structures within the nucleus of mammalian cells. ND10 components undergo continuous and in some cases rapid exchange with the surrounding nucleoplasm (Boisvert et al., 2001; Everett and Murray, 2005; Weidtkamp-Peters et al., 2008; Wiesmeijer et al., 2002), and their number, composition and morphology alter in response to stress and virus infection (reviewed in Dellaire and Bazett-Jones, 2004; Eskiw et al., 2003; Everett, 2001; Everett and Chelbi-Alix, 2007; Maul, 1998). They undergo significant changes during the cell cycle, particularly during mitosis (Dellaire et al., 2006; Everett et al., 1999; Sternsdorf et al., 1997), and they are modified in cells in a variety of disorders, including cancers such as promyelocytic leukaemia. The complexity of ND10 is exemplified by the reported presence of 70 or more ND10- or PML-associated proteins (Negorev and Maul, 2001) (see also the nuclear protein database, http://npd.hgu.mrc.ac.uk/user/).
PML is the prototype member of a family of proteins containing the so-called tripartite motif (TRIM), which comprises a RING finger, two additional zinc-stabilised domains known as B-Boxes, and a coiled-coil motif (Jensen et al., 2001). The primary PML gene transcript undergoes extensive alternative splicing, resulting in the expression of six major nuclear isoforms, as well as a cytoplasmic variant lacking the nuclear localisation signal. The nuclear isoforms have the first six exons in common, and these include all the TRIM elements. The different isoforms include combinations of the last three exons, which themselves can be spliced in alternative ways (Jensen et al., 2001). PML isoforms are modified by phosphorylation (Chang et al., 1995; Everett et al., 1999; Hayakawa and Privalsky, 2004; Scaglioni et al., 2006) and conjugation to members of the SUMO family of ubiquitin-like proteins (Kamitani et al., 1998b; Sternsdorf et al., 1997), with the extent of these modifications varying during the cell cycle (Everett et al., 1999). PML and the other ND10 component proteins have connections with several important biological processes, including regulation of transcription, chromatin assembly and modification, DNA repair, apoptosis, stress and the response to virus infection, and a number of cancers (Bernardi and Pandolfi, 2003; Bernardi and Pandolfi, 2007; Borden, 2002; Dellaire and Bazett-Jones, 2004; Eskiw and Bazett-Jones, 2002; Everett and Chelbi-Alix, 2007; Maul, 1998; Reineke and Kao, 2009; Zhong et al., 2000b). Given this plethora of functional involvements, it is not surprising that work on PML and ND10 has generated a wealth of published literature.
Our interests lie in the role of PML in intrinsic resistance to DNA virus infection. It has been established that the genomes of herpes simplex virus type 1 (HSV-1) and human cytomegalovirus (HCMV) are subject to transcriptional repression during the initial stages of infection via mechanisms involving PML and other ND10 components (Cantrell and Bresnahan, 2006; Everett et al., 2008a; Everett et al., 2006; Lukashchuk et al., 2008; Lukashchuk et al., 2010; Negorev et al., 2006; Preston and Nicholl, 2006; Saffert and Kalejta, 2006; Tavalai et al., 2006; Tavalai et al., 2008). This intrinsic cellular antiviral defence is counteracted by viral regulatory proteins, namely pp71 and IE1 of HCMV, and ICP0 of HSV-1. These proteins affect ND10 in a variety of ways; for example, ICP0-induced degradation of PML (Everett et al., 1998), pp71-mediated displacement of ND10 components hDaxx and ATRX (Cantrell and Bresnahan, 2006; Lukashchuk et al., 2008; Preston and Nicholl, 2006; Saffert and Kalejta, 2006; Woodhall et al., 2006), and induction of the loss of SUMO-modified forms of PML by IE1 (Lee et al., 2004). Consistent with the hypothesis that these activities counteract aspects of cellular antiviral defence, the works cited above have demonstrated that depletion of these major ND10 proteins using RNA interference allows improved replication of HSV-1 and HCMV mutants that lack the above-named viral regulatory proteins.
Given that depletion of all PML isoforms increases the replication of ICP0-null mutant HSV-1 (Everett et al., 2006), we set out to investigate whether the apparent repressive effect of PML could be attributed to a specific PML isoform. We developed methods to express individual PML isoforms at close to endogenous levels in human cell lines that either were or were not highly depleted of endogenous PML. Our core finding is that the major PML isoforms (PML.I and PML.II) partially reverse the improved replication of ICP0-null mutant HSV-1 that is observed in PML-depleted cells, but no single PML isoform could fully reverse the effect of endogenous PML depletion. The inhibitory effect of PML.I was dependent on SUMO modification, the presence of the SUMO interaction motif (SIM), all the elements of its TRIM motif, and proper SUMO modification. In parallel, we found that no single major nuclear PML isoform can re-establish completely authentic ND10 in terms of composition and/or modification of Sp100, despite nucleating punctuate PML foci. These observations suggest that the isoform-specific C-terminal sequences of the various PML isoforms are required to act in collaboration to establish fully functional ND10 structures.
Results
A system for expression of individual PML isoforms at close to endogenous levels in human cells depleted of endogenous PML
This study arose from the observation that depletion of PML from human fibroblasts results in increased infection efficiency by ICP0-null mutant HSV-1 (Everett et al., 2006), and also by other herpesviruses (Kyratsous and Silverstein, 2009; Tavalai et al., 2006). Because of the complex nature of the expression pattern of PML isoforms in normal cells (Condemine et al., 2006; Jensen et al., 2001), we set out to determine whether any of the individual PML isoforms could reverse the effect of depletion of all PML isoforms and thereby re-establish full intrinsic resistance to ICP0-null mutant HSV-1.
Preliminary experiments were impeded by difficulties in achieving expression of individual PML isoforms at the necessary high frequency in an appropriate cell type, and by the problem common to many vector systems of expression at levels far exceeding that of the endogenous protein. Therefore, we developed a system with the following characteristics: (1) efficient depletion of all isoforms of endogenous PML from human cells suitable for studies on ICP0-null mutant HSV-1, using lentiviral-mediated expression of an anti-PML short hairpin RNA (shRNA); (2) lentiviral-mediated reconstitution of individual PML isoform expression using modified PML sequences resistant to the shRNA; (3) use of a weak promoter for PML isoform expression to avoid excessive expression levels; and (4) use of EYFP–PML fusion proteins to allow enrichment by fluorescence-activated cell sorting (FACS) of cells positive for PML isoform expression. A map of the EYFP–PML expression lentivirus vector and the exon structures of the PML isoforms studied is presented in Fig. 1. This system was adopted for expression of the major nuclear PML isoforms I to VI in both normal and PML-depleted cells.
Fig. 1.
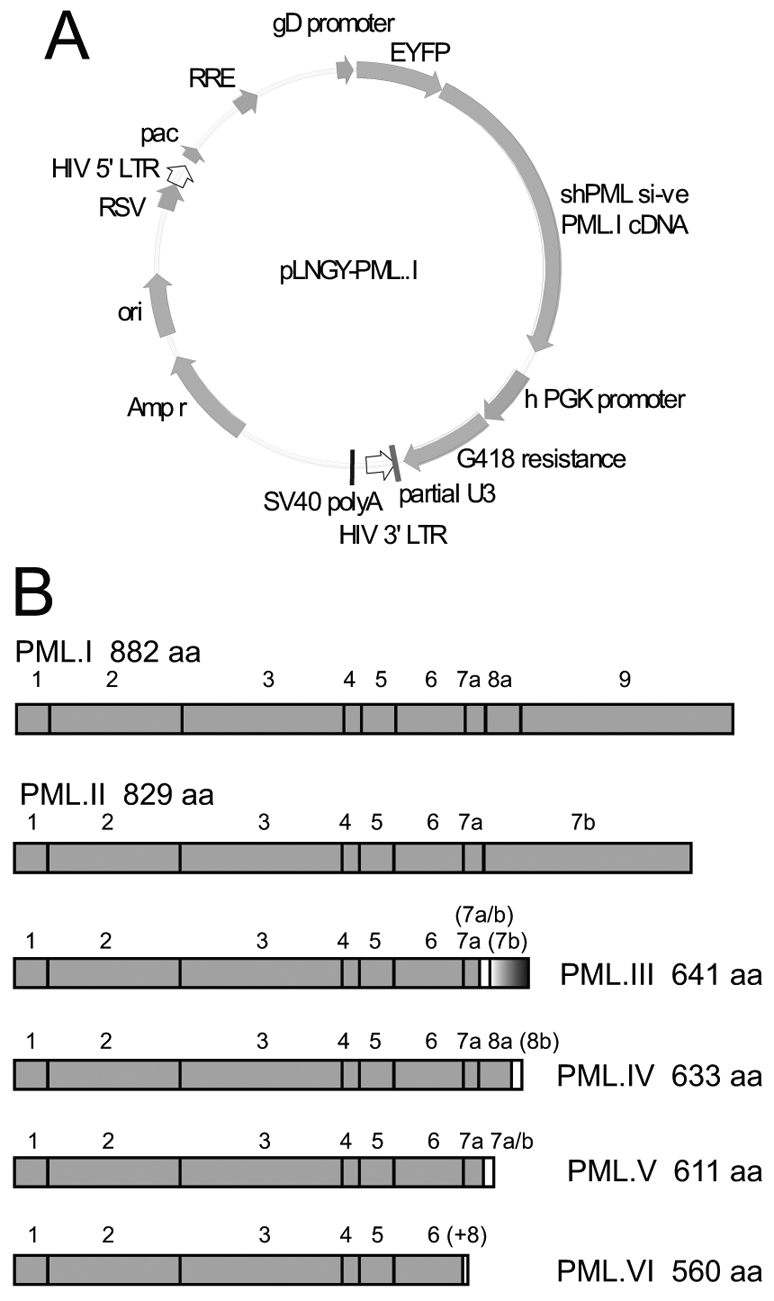
Map of the lentiviral vector used for PML expression and a summary of the PML isoforms studied. (A) Lentivirus plasmid vector pLNGY-PML.I. The key features of the lentivirus vector are noted: pac, HIV packaging sequence; RRE, REV response element; RSV, RSV promoter; ori, bacterial origin of replication; Amp r, ampicillin resistance; shPML1 si-ve, position of target sequence of shPML1 and relevant non-coding mutations (other labels are self-explanatory). (B) PML isoforms I to VI, noting the exons included in each isoform cDNA and the size of the translated product. The bracketed exon labels indicate the use of alternative reading frames compared to unbracketed exons of the same name. The (+8) label of PML.VI indicates that an additional eight residues follow exon 6 as a result of an alternative splicing event that deletes exon 7a.
Analysis of control and PML-depleted cells expressing EYFP–PML isoform fusion proteins
The cells used predominantly in these studies were HepaRG hepatocytes (Gripon et al., 2002) and derivatives expressing control and anti-PML shRNAs (Everett et al., 2008a), named HALL and HALP cells, respectively. Endogenous PML and Sp100 colocalised strongly in HALL cells (supplementary material Fig. S1). ATRX and hDaxx exhibited considerable diffuse nuclear fluorescence, but both also colocalised with PML, albeit less distinctly and to a variable extent between cells. On average, around 50–70% of PML foci contained detectable accumulations of hDaxx or ATRX (supplementary material Fig. S1). As observed previously, depletion of endogenous PML caused the dispersal of other ND10 components, including Sp100, hDaxx and ATRX, into mainly nuclear diffuse staining patterns (Everett et al., 2007; Everett et al., 2008a; Everett et al., 2006; Ishov et al., 1999; Shen et al., 2006; Zhong et al., 2000a). Some HALP cells contained foci of these proteins, but these were less distinct than in the control cells and they did not colocalise with each other (supplementary material Fig. S1) (Everett et al., 2007; Everett et al., 2008a).
HALL and HALP cells transduced with lentiviruses expressing the EYFP–PML isoform fusion proteins were enriched by FACS and all expressed the relevant PML isoforms in greater than 95% of the cells. Analysis of these sets of cells indicated that all EYFP–PML isoforms colocalised with Sp100 in the great majority of the foci in both cell type backgrounds (Fig. 2). However, some cell-to-cell variability was evident, and although Sp100 was present in most PML.I foci in HALP.EYFP–PML.I cells (Fig. 2), this was not true in all cells, resulting in an overall reduced proportion of colocalising foci (see later for quantification of the data). These data were consistent with many previous studies demonstrating colocalisation of exogenously expressed PML isoforms with endogenous ND10, and with Sp100 in the absence of endogenous PML (e.g. Ishov et al., 1999; Shen et al., 2006; Zhong et al., 2000a).
Fig. 2.

Colocalisation of Sp100 and EYPF-tagged PML isoforms in HALL and HALP cells. Immunofluorescence staining of endogenous Sp100 in HALL (left-hand set of images) and HALP cells (right-hand set of images) expressing PML isoforms I to VI as EYFP fusion proteins (detected by EYFP autofluorescence). Images are representative of the majority phenotype in each cell line. The images are single plane projections of short z-stacks. Scale bars: 5 μm.
Western blot analysis of extracts of the series of HALL.EYFP–PML.I to HALL.EYFP–PML.VI cells using an antibody that recognises all endogenous PML isoforms indicated that each EYFP fusion protein isoform gives a family of multiple bands that are likely to represent modification by SUMO family proteins, as occurs with endogenous PML (Fig. 3A, upper panel). Steady state expression levels varied between the isoforms, with isoforms I and VI being routinely more abundant than the others, and isoform V being apparently the least well expressed. Because the promoter driving expression was the same in all cases, these differences were probably a consequence of post-transcriptional regulation. The most abundant unmodified form of endogenous PML corresponds to a combination of isoforms I and II, with the PML.I-specific mRNA being the most highly expressed under natural circumstances, comprising 40–80% of total PML-encoding mRNA (Condemine et al., 2006). Despite these variations, all EYFP-linked PML isoforms were expressed at levels of a similar order to that of endogenous PML, in that both endogenous and exogenous protein could be visualised on a single western blot exposure, resulting in complicated band patterns consequent on the overlay of the endogenous and exogenous signals (Fig. 3A, upper panel). Analysis of the same samples using an anti-EGFP antibody revealed the bands that were specific to the introduced EYFP–PML isoform fusion proteins (Fig. 3A, lower panel).
Fig. 3.
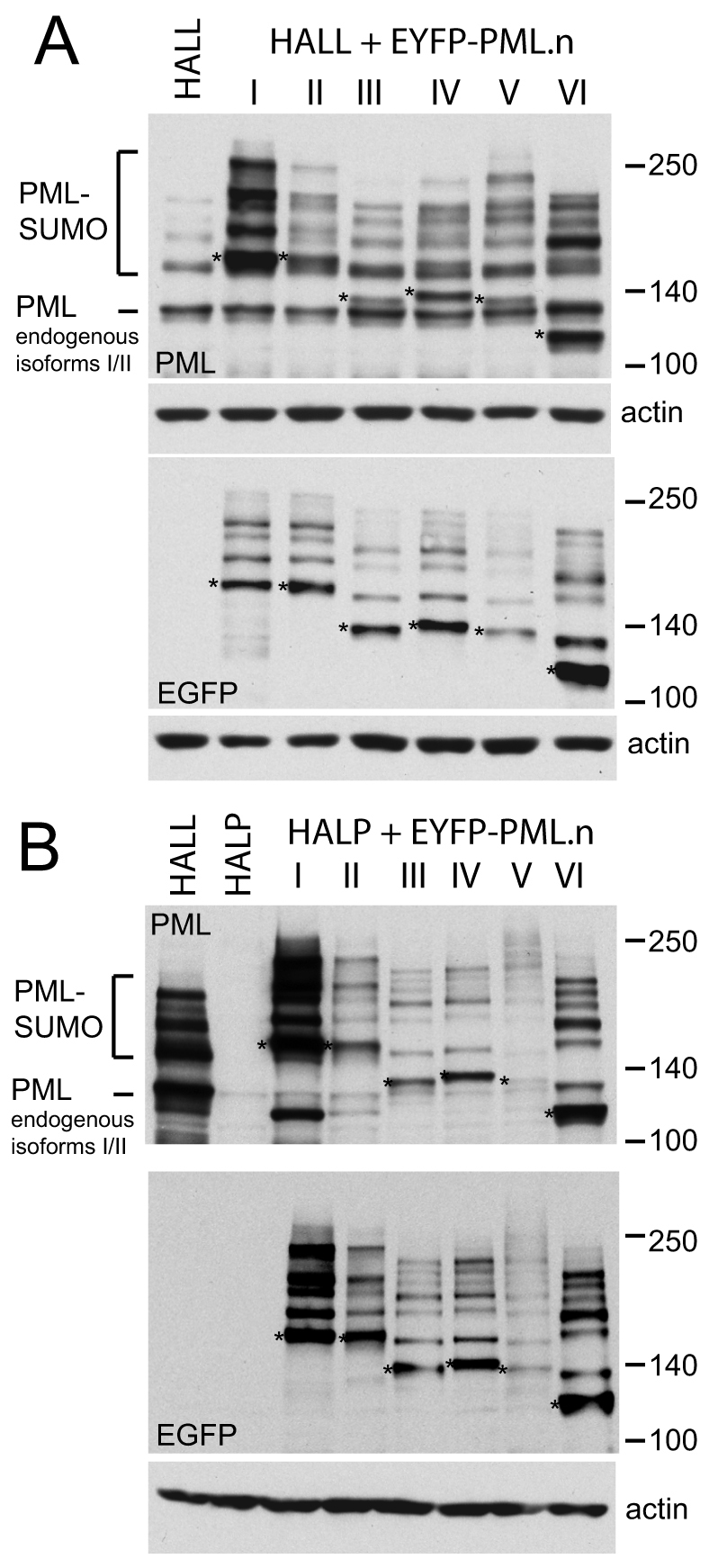
Expression of EYFP-linked PML isoforms as shown by western blotting. (A) Western blot analysis of HALL and HALL.EYFP–PML.I to PML.VI cells. The upper panel probed with anti-PML mAb 5E10 detects all endogenous PML isoforms and the introduced EYFP–PML fusion proteins. The most prominent band in HALL cells corresponds to the endogenous PML isoforms I and II, and their SUMO modified forms are identified above. The positions of the major unmodified bands of the EYFP–PML isoforms I to VI are indicated with asterisks, with their SUMO modified forms of lower mobility overlaying the bands derived from endogenous PML. The lower panel shows the same samples analyzed using an anti-EGFP antibody. The major unmodified forms of the EYFP–PML fusion proteins are marked by asterisks. (B) Western blot analysis of HALL, HALP and HALP.EYFP–PML.I to PML.VI cells. The upper and lower panels show bands detected by anti-PML mAb 5E10 and an anti-EGFP antibody, respectively. The major unmodified forms of the EYFP–PML fusion proteins are marked by asterisks.
Similar analysis of HALL, HALP and HALP.EYFP–PML.I to HALP.EYFP–PML.VI cells demonstrated the loss of endogenous PML from HALP cells and the expression of the re-introduced EYFP–PML isoforms at close to endogenous levels in each HALP-derived cell line (Fig. 3B). As in the HALL series of cells, unmodified PML.V appeared to be expressed less efficiently than the other isoforms, but note that the most of the protein was in the form of modified species, which might give a misleading impression of the level of total PML.V expression. Interestingly, the pattern of modified PML species was more complex than could be explained by conjugation of a single SUMO moiety at only the three mapped SUMO conjugation sites, especially with isoforms V and VI. Although it is possible that some of these modified bands could be due to SUMO modification of lysine residues in the EYFP moiety of the fusion protein, a large number of modified PML isoform bands were also observed in human fibroblasts expressing FLAG-tagged versions of these proteins (Sykes, 2007). One possible explanation of these bands is the presence of oligomeric SUMO-2/3 chains.
PML isoforms I and II participate in PML-dependent restriction of HSV-1 replication
The motivation behind this study was to determine whether any particular individual PML isoform could reverse the increased replication of ICP0-null mutant HSV-1 in PML-depleted cells. Therefore, plaque formation by wild type and ICP0-null mutant HSV-1 was assessed in control and PML-depleted HepaRG cells expressing individual PML isoforms. As expected from previous work (Everett et al., 2008a; Everett et al., 2006), the plaque-forming efficiency of wild-type HSV-1 was similar in all cell lines, whether or not they expressed endogenous PML (Fig. 4A,B, upper panels). These data are in accord with previous studies that found that increased expression of selected PML isoforms does not impede wild-type HSV-1 infection (Chelbi-Alix and de The, 1999; Everett and Zafiropoulos, 2004; Lopez et al., 2002).
Fig. 4.
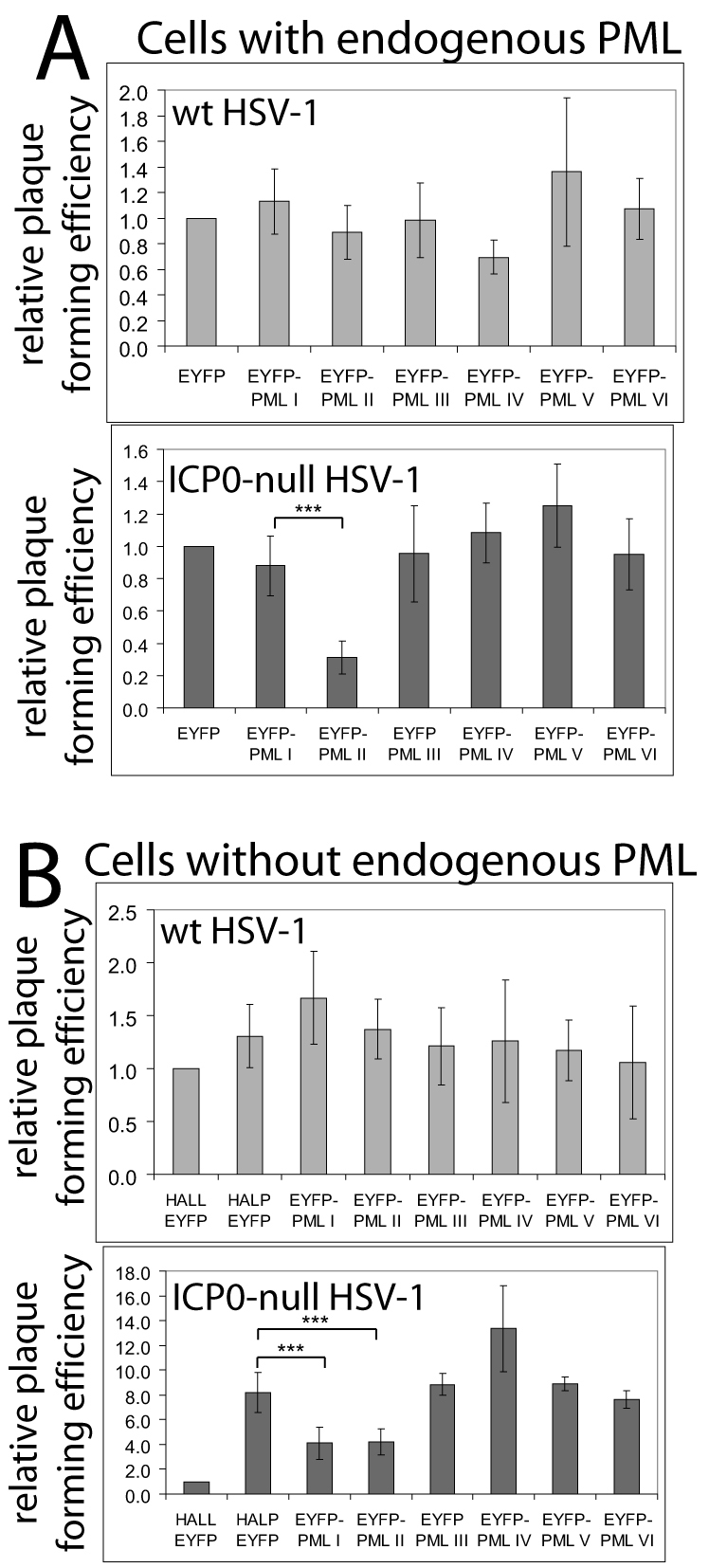
Effects of EYFP-PML isoform expression on wild-type and ICP0-null mutant HSV-1 plaque formation. (A) The relative plaque formation of wild-type and ICP0-null mutant HSV-1 in HALL cells expressing endogenous PML and EYFP or individual EYFP-PML isoforms, as indicated. (B) As A, except using HALP cells depleted of endogenous PML. The effects of the EYFP–PML isoforms were compared with control (HALL–EYFP) and PML-depleted (HALP–EYFP) cells expressing EYFP. The average increase in ICP0-null mutant HSV-1 plaque formation in HALP–EYFP over HALL–EYFP cells was 8.2±1.63-fold. Data from several independent experiments were averaged then plotted as mean ± s.d.. Calculations were as described in Materials and Methods. The asterisks indicate statistical significance of the difference between the indicated data sets (***P=0.001 or less, Student's paired two-tailed t-test).
Plaque formation efficiency of ICP0-null mutant HSV-1 was also unaffected in most HALL-derived cells expressing individual EYFP–PML isoform fusions, with the exception of cells expressing PML.II (Fig. 4A, lower panel). As expected, significantly increased plaque formation of ICP0-null mutant HSV-1 was observed in PML-depleted cells (Fig. 4B, lower panel). Expression of individual PML isoforms in HALP-derived cells was unable to reverse completely the effect of PML depletion, but mutant virus plaque formation was about half as efficient as that in the HALP parents in cells expressing PML.I or PML.II (Fig. 4B, lower panel). These data indicate that the naturally most abundantly expressed PML isoforms play a role in the mechanism by which PML contributes to repression of ICP0-null mutant HSV-1 infection.
To test whether increased expression of PML isoforms I or II might further inhibit ICP0-null mutant HSV-1 plaque formation, we developed cell lines in which these EYFP-linked proteins were expressed at very high levels from a truncated HCMV promoter. The increase in plaque-forming efficiency of ICP0-null mutant virus in PML-depleted cells was also partially reversed in the PML overexpressing cells, but had no effect on plaque formation by wild-type virus (supplementary material Fig. S2). These data independently confirm the results of Fig. 4B and demonstrate that increased expression of the isoforms above endogenous levels does not have a further repressive effect.
PML.I-mediated restriction requires SUMO modification and all elements of the TRIM
We analysed the characteristics of PML.I that are required for its restrictive effect on ICP0-null mutant HSV-1 plaque formation by constructing cells expressing proteins with mutations in conserved cysteine residues in the RING finger, B-Box 1 and B-Box 2 (PML.I.ΔRING, PML.I.ΔBB1 and PML.I.ΔBB2, respectively), and a deletion of the coiled-coil element of the TRIM (PML.I.ΔCC), and with lysine to arginine substitutions at the two major SUMO modification sites, lysine residues 160 and 490 (PML.I.KK) (Fig. 5A). We also included a mutant of PML.I lacking the short exon 7a (PML.I.Δ7a), which includes the SUMO interaction motif (SIM) (see Fig. 8). In the HALL cell background, wild-type EYFP–PML.I foci mostly colocalised with Sp100 and, surprisingly, so did the RING finger, SUMO modification site and SIM deletion mutants (supplementary material Fig. S3, left; data not shown for PML.I.Δ7a). The B-Box 1 mutant also colocalised with Sp100, but the mutant protein clearly affected the appearance of endogenous ND10. The B-Box 2 mutant formed many punctate foci, some colocalising with endogenous ND10 but most not (supplementary material Fig. S3, left). The coiled-coil deletion mutant was entirely nuclear diffuse, but again extensive evidence of disturbance of ND10 integrity was present (supplementary material Fig. S3, left). These results imply that PML–PML interactions through the coiled-coil are dominant in determining the localisation of mutant proteins when expressed at close to endogenous levels. However, it is clear that the presence of a mutant PML protein can affect the structure of endogenous ND10, even when not highly expressed.
Fig. 5.

All elements of the PML.I TRIM motif, the SIM and the major SUMO modification sites are required for PML.I-dependent reduction in ICP0-null mutant HSV-1 plaque formation. (A) The conserved exons 1-6 of PML and the mutations that were analysed. (B) Wild-type and mutant EYFP–PML.I expression detected using an anti-EGFP antibody. The upper set of images shows expression in HALP PML-depleted cells and the lower set in HALL cells expressing endogenous PML. (C) Relative plaque-forming efficiency of wild-type HSV-1 (upper panel) and ICP0-null mutant HSV-1 (lower panel) in the various cell lines, analysed as described in Fig. 4. In these experiments, the average increase in ICP0-null mutant plaque formation in HALP–EYFP over HALL–EYFP cells was 11.55±2.43-fold. The asterisks indicate statistical significance of the difference between the indicated data sets (***P<0.001, Student's paired two-tailed t-test).
Fig. 8.

Analysis of PML.I lacking the SIM. (A) The structure of PML.I, highlighting exon 7a and noting the extent of the deletion in PML.I.Δ7a. (B) Western blot analysis of wild-type and Δ7a mutant PML.I in the indicated cell types, probing for EYFP (left) and Sp100 (right). (C) Immunofluorescence analysis of HALP cells expressing PML.I.Δ7a. Colocalisation of Sp100, hDaxx and ATRX with EYFP-PML.IΔ7a is shown. Scale bars: 5 μm. (D) Immunofluorescence data were quantified, presented and analysed statistically as described for Fig. 6.
The results were substantially different in the PML-depleted background. As noted in Fig. 2, Sp100 was more diffuse even in cells reconstituted with wild-type EYFP–PML.I, and this was true of all cell lines expressing mutant forms of PML.I (supplementary material Fig. S3, right), with the exception of PML.I.Δ7a (see later). Colocalisation with Sp100 occurred, albeit to a reduced extent, with wild-type EYFP–PML.I, but not with the RING finger, B-Box 1, B-Box-2 and coiled coil mutants, even when these formed distinct foci (supplementary material Fig. S3, right). The SUMO modification mutant formed intense nuclear aggregates, some of which colocalised with Sp100 (supplementary material Fig. S3, right). By contrast, the SIM deletion mutant formed foci that colocalised extremely well with Sp100 (see Fig. 8).
The mutant PML proteins were expressed at broadly similar levels in both control and PML-depleted backgrounds, but the modification of all except PML.I.Δ7a was compromised in the PML-depleted background, indicating that all the elements of the TRIM are required for proper PML.I modification (Fig. 5B, upper pair of images). In cells expressing endogenous PML, however, all the mutant proteins except the coiled-coil deletion expressed one additional modified band (Fig. 5B, lower pair of images), indicating that the endogenous protein can influence not only the localisation of mutant forms of PML (supplementary material Fig. S3), but also their modification. The modified form of the PML.I.KK mutant might be due to SUMO modification (possibly with a dimeric SUMO chain, given the difference in size shift compared to the others) at either the previously characterised lysine 65 (Kamitani et al., 1998a) or at a different lysine residue.
None of the mutant proteins had any marked effect on the plaque-forming efficiency of wild-type HSV-1 (Fig. 5C, upper panel). Although wild-type EYFP–PML.I reproduced the twofold reduction in ICP0-null mutant HSV-1 plaque formation seen in earlier experiments, none of the mutant proteins had a significant inhibitory effect (Fig. 5C, lower panel). These data confirm the restrictive effect of PML.I and demonstrate that the integrity of the TRIM, the major SUMO modification sites and the SIM are required for PML.I activity.
All EYFP–PML isoforms nucleate foci that are distinguishable from normal ND10
One possible explanation of the observation that no single PML isoform can re-establish the level of repression of ICP0-null mutant cells that occurs in the presence of endogenous PML is that the PML foci formed are in some way different from normal ND10. This hypothesis was supported by detailed examination of other ND10 components. For example, although all re-introduced EYFP–PML isoforms colocalised with Sp100 in the HALP-PML series of cells (Fig. 2), recruitment of hDaxx (supplementary material Fig. S4) and ATRX (supplementary material Fig. S5) was isoform-dependent. These two latter proteins were mainly diffusely distributed in the nucleus of EYFP–PML.I and EYFP–PML.II reconstituted cells, and there was a lower proportion of PML.III and PML.IV foci that contained ATRX than in the corresponding endogenous PML-positive cells. On the other hand, both hDaxx and ATRX were efficiently recruited into PML.V and PML.VI foci to an extent that, in the case of PML.VI, was even greater than that seen with endogenous PML in HALL cells. The data were quantified and presented as box plots in Fig. 6. For ease of visualisation, the means of these data are shown in supplementary material Fig. S6. Statistical analysis was performed to compare the colocalisation of Sp100, hDaxx and ATRX with endogenous PML in HALL cells (supplementary material Fig. S1).
Fig. 6.

Quantification of PML isoform colocalisation with Sp100, hDaxx and ATRX. Box plots of percentages of colocalising and separate foci of the indicated pairs of proteins in individual cells containing endogenous PML (HALL background, upper sets of data) and PML-depleted HALP cells (lower sets of data). The EYFP–PML foci in six to ten cells (totalling over 100 foci) were scored for the presence or absence of Sp100, hDaxx or ATRX. Separate foci of the other ND10 protein were also recorded. The presence of colocalising Sp100 was always distinct, whereas it was more variable with hDaxx and ATRX. Colocalisation was scored if there was an increase above background, even if faint, of signal colocalising with PML. Cell-to-cell variability occurred in terms of both proportion of colocalising foci and the intensity of colocalisation. The fields of view in Fig. 2 and in supplementary material Figs S4 and S5 were chosen to reflect these variations. The shaded boxes represent the range of 50% of the values, with the line extensions indicating the range of all values. The thin line within the box is the median value, the thicker line is the mean. The data of the colocalising foci were compared with those of colocalisation with endogenous PML in HALL cells (supplementary material Fig. S1) using the Mann–Whitney U-test. Statistically different distributions are indicated by asterisks (*P<0.05, **P<0.02). Statistical analysis was performed only on the data sets of colocalising foci, because the data on non-colocalising foci were related to the degree of colocalisation.
Analysis of these observations in the light of the exon structure of the various PML isoforms (Fig. 1) did not reveal any obvious isoform-specific sequences that are specifically required for the recruitment of hDaxx and ATRX into PML foci. It is likely that other factors, such as interactions with other proteins not analysed here, are also involved. It is surprising that PML.VI recruited all the tested ND10 components so efficiently, even in the PML-depleted background, because this isoform lacks the SUMO interaction motif that has been implicated in the mechanism of ND10 assembly (Shen et al., 2006). However, our data are consistent with a previous finding of increased recruitment of hDaxx into foci containing overexpressed PML.VI (Block et al., 2006). The efficient recruitment of Sp100, hDaxx and ATRX into PML.V foci is consistent with the suggestion that this isoform plays a structural role in nucleating ND10 (Brand et al., 2010; Weidtkamp-Peters et al., 2008). Another surprising feature of these results was that expression of PML.I in HepaRG cells appeared to disturb normal ND10 integrity, because colocalisation with hDaxx and ATRX was poor even in the PML-positive background (Fig. 6; supplementary material Figs S4, S5). This might be a cell-type-dependent effect because this result was not observed in analogous human fibroblast-derived cells (data not shown). These data suggest that the simple presence of PML foci after exogenous expression cannot be taken as evidence of formation of structures identical to ND10 in normal cells, and more detailed analysis of other ND10 components is necessary to characterise such foci completely.
Modification of Sp100 in cells with and without endogenous PML
Immunofluorescence analysis suggested that the foci formed by different PML isoforms in cells depleted of endogenous PML might differ in detail from normal ND10. This hypothesis was supported by analysis of the modification profile of Sp100. In normal cells, Sp100 exhibited a number of bands (Fig. 7A), the most prominent of which represented unmodified Sp100 isoform A (Sp100A) and a derivative of this conjugated to a single SUMO-1 moiety (Sternsdorf et al., 1997). The identity of fainter, slower mobility bands was less certain, because they could represent less abundantly expressed Sp100 isoforms such as Sp100B, Sp100C and Sp100HMG, or further modified forms of Sp100A, or a combination of all isoforms with or without SUMO modifications. Whatever the identity of these slower mobility bands, depletion of PML resulted in their loss and a concomitant increase in unmodified Sp100A (Fig. 7A) (Everett et al., 2008a; Everett et al., 2006). In contrast to its effect on SUMO modification of Sp100, PML depletion did not result in major global differences in SUMO-modified proteins, as detected by the high molecular weight smear of proteins using anti-SUMO-1 or SUMO-2/3 antibodies (Fig. 7C).
Fig. 7.

Analysis of SUMO modification of Sp100 in HALL, HALP and HALP.EYFP–PML.I to PML.VI cells. (A) Western blot showing endogenous Sp100A and its major SUMO-modified isoform in the indicated cell lines. (B) The relative intensities of the SUMO-modified and unmodified Sp100A were determined by densitometry. The data show mean ± s.d. from scans of three independent gels. The asterisks indicate those values that were statistically significantly different from those of HALP cells, Student's two-tailed t-test, *P<0.05. (C) Depletion of PML does not have any substantial effect on the pattern of endogenous SUMO-conjugated proteins. Whole-cell extracts of HALL and HALP cells were analyzed by western blotting for SUMO-1, SUMO-2/3 and actin, as indicated. Endogenous SUMO conjugates were detected as a high molecular weight smear. Parallel samples on the same gel were analysed for PML and Sp100.
All PML isoforms enabled an increase in SUMO modification of Sp100A and an increase in the lower mobility bands compared to that in PML-depleted cells, but none reproduced completely the Sp100 profile in HALL cells (Fig. 7A,B). We also noted a slight shift in mobility of unmodified Sp100A in HALL compared to HALP cells (Fig. 7C), which was not reversed in any of the PML reconstituted cells (Fig. 7A). PML isoforms V and VI produced the most marked increase in SUMO modification of Sp100A, a result that might be related to their efficient recruitment of other ND10 proteins (Fig. 6; supplementary material Figs S4, S5). The other PML isoforms were all substantially defective in reproducing normal levels of SUMO modification of Sp100A, despite their colocalisation with Sp100 (Fig. 2).
The data demonstrated that SUMO modification of Sp100 is PML-dependent, and that the increased levels of SUMO-modified Sp100A in PML.V and PML.VI reconstituted cells raises the possibility that these isoforms act as SUMO E3 ligases for Sp100. The situation is likely to be more complex, however, because the PML.VI sequence is almost entirely represented in all other isoforms. Furthermore, consistent with a previous report utilising PML.III (Sternsdorf et al., 1995), we could not detect any interaction between these proteins in immunoprecipitation studies (data not shown). Although RanBP2 has been found to stimulate SUMO modification of Sp100 (Pichler et al., 2002; Tatham et al., 2005), the proposal that Sp100 is SUMO-modified during nuclear entry (Pichler et al., 2002) is unlikely to be correct because our data clearly demonstrate that Sp100 within the nucleus is not SUMO-modified unless PML is present.
A surprising feature of our analysis concerns the SIM deletion mutant, PML.I.Δ7a. We noted that it forms foci that colocalised with Sp100 to a greater degree than the wild-type protein, and this also extended to colocalisation with hDaxx and ATRX (Fig. 8C). In fact, PML.I.Δ7a behaved in these respects very much like PML.VI, which also lacked the SIM (compare Fig. 8C,D with Figs 2, 6 and supplementary material Figs S4, S5). Furthermore, the ratio of SUMO-modified to unmodified Sp100A in HALP cells expressing PML.I.Δ7a was much greater than that in cells expressing wild-type PML.I (Fig. 8B). These results are consistent with a role for the SIM in the regulation of ND10 composition, but not with a requirement for the SIM for ND10 assembly. It is possible that the SIM is involved in transitory interactions and component exchange within ND10, such that in its absence certain proteins are recruited more stably, allowing greater levels of SUMO modification of Sp100. However, it is also formally possible that the presence of PML.I.Δ7a alters the relative stabilities of SUMO-modified and unmodified Sp100.
Discussion
There is increasing evidence that several ND10 components contribute to intrinsic cellular resistance to a variety of herpesvirus infections (Cantrell and Bresnahan, 2006; Everett et al., 2008a; Kyratsous and Silverstein, 2009; Ling et al., 2008; Lukashchuk et al., 2008; Lukashchuk et al., 2010; Negorev et al., 2006; Preston and Nicholl, 2006; Saffert and Kalejta, 2006; Tavalai et al., 2006; Tavalai et al., 2008; Woodhall et al., 2006). We have shown that depletion of PML increases the replication efficiency of ICP0-null mutant HSV-1 (Everett et al., 2008a; Everett et al., 2006; Everett et al., 2008b), a finding that implicates PML in a repression mechanism that targets HSV-1 genomes and which is inactivated by ICP0 during the course of a wild-type virus infection. Consistent with these results, decreased expression of PML in a variety of tumour cells correlates with increased permissiveness for ICP0-null mutant HSV-1 replication, and disruption of ND10 by expression of the PML–RARα fusion protein also increases replication of the mutant virus (Sobol et al., 2009). Given that PML is expressed as a complex family of proteins as a result of both alternative splicing and post-translational modification (Jensen et al., 2001), we set out to determine which PML isoforms are most significant in terms of restricting ICP0-null mutant HSV-1 infection. We developed an experimental approach that provides a robust methodology for the investigation of the functions of PML, and which in principle is applicable to many other systems.
As pointed out previously (Bernardi and Pandolfi, 2007), many studies involving exogenous PML expression have used overexpressed proteins in a background containing a full complement of endogenous proteins. In preliminary experiments using vector systems with a stronger promoter than utilised herein, we found that highly expressed PML forms abnormally large nuclear aggregates in many cells, sometimes with additional cytoplasmic foci, perhaps because the normal trafficking system and interaction partners of the protein had been saturated. When exogenous PML was expressed at close to endogenous levels, all EYFP-linked PML isoform proteins colocalised with Sp100, and all except PML.I colocalised frequently with both hDaxx and ATRX in normal cells. In PML-depleted cells, however, significant differences were observed in the distributions of some PML isoforms and their recruitment of other ND10 proteins. For example, PML.I and PML.II recruit Sp100 but neither hDaxx nor ATRX into colocalising foci (Figs 2, 6; supplementary material Figs S4, S5). These observations indicate that differences in the properties of the various PML isoforms become evident more readily in the absence of endogenous PML, whereas in its presence the ability of PML isoforms to interact through the coiled-coil domain might enable a dominant influence of the endogenous protein.
Our results are consistent with previous studies of single human PML isoforms in transfected PML−/− mouse fibroblasts. For example, PML.VI recruited endogenous Sp100, Daxx and SUMO-1 into colocalising foci (Ishov et al., 1999), and isoforms III and IV recruited Daxx and SUMO-1 (Shen et al., 2006; Zhong et al., 2000a). Our study expanded these observations to a systematic analysis of all major nuclear PML isoforms, and the comparative analysis of the efficiencies with which a range of ND10 proteins are recruited into the PML isoform foci. The results reveal subtleties that might have been missed in previous analyses, particularly those utilising transfection-mediated high-level expression of PML.
We realise that the differences observed between the various re-introduced PML isoforms in PML-depleted HepaRG cells (Figs 2, 6; supplementary material Figs S4, S5) might vary according to cell type and expression level. During preliminary work, we investigated different cell types and vector systems that gave much higher levels of PML expression. In HEp-2 cells depleted of endogenous PML, we found that highly expressed PML isoforms IV and VI strongly recruited Sp100 into colocalising foci, whereas with isoforms II and V it was variable, and recruitment of Sp100 was not observed in cells expressing isoforms I and III (Sykes, 2007). On the other hand, recruitment of both Sp100 and hDaxx into ND10-like foci occurred in PML-depleted human fibroblasts expressing each individual PML isoform, although with the high-level expression system employed the foci were larger and more diffuse than normal ND10, particularly with cells expressing PML.VI (Sykes, 2007). Such cell type and expression level variations could explain differences in PML isoform localisation reported in previous papers (e.g. Beech et al., 2005; Condemine et al., 2007; Condemine et al., 2006). The specifics of these cell type differences are not as important as the general principles that PML isoform behaviour is complicated by many factors, and that individual PML isoforms cannot be relied upon to re-form truly authentic ND10 in a PML-depleted background. This latter conclusion is supported by a previous study that found that PML.IV could induce premature senescence in PML-positive but not PML-negative mouse fibroblasts, implying that PML.IV cannot substitute for all other isoforms (Bischof et al., 2002).
The failure of any single individual major PML isoform to re-establish ND10 that are indistinguishable from the authentic structures on the basis of fluorescence analysis and/or Sp100 modification might be an important factor in their inability to reverse completely the effect of PML depletion on ICP0-null mutant HSV-1 plaque formation. Interestingly, PML.I is able partially to reverse the effect of PML depletion on ICP0-null mutant plaque formation and is naturally the most highly expressed in human cells (Condemine et al., 2006). Recently, PML.I expression in PML-negative mouse fibroblasts was found to decrease the robust replication of murine γ-herpesvirus 68 normally observed in these cells in comparison to control PML-positive fibroblasts (Ling et al., 2008). Given that PML.II also had a repressive effect in our assays, it is of interest that this isoform interacts specifically with adenovirus E4orf3 protein, thereby enabling the mechanism by which E4orf3 disrupts ND10 during adenovirus infection (Hoppe et al., 2006; Leppard et al., 2009).
The mechanism by which PML exerts its negative effect on ICP0-null mutant HSV-1 plaque-forming efficiency is yet to be determined, but our mutational analysis demonstrates that the integrity of the entire TRIM structure is required. Note that an intact TRIM is also required for normal SUMO modification of PML.I. Although we have not analysed any mutants of PML.II, it would be expected that its TRIM is also required for the inhibitory effects documented in Fig. 4. Why PML.II alone amongst the isoforms increases the repression of ICP0-null mutant plaque formation in PML-positive cells is not clear. It is possible that the PML.II specific exon 7b interacts with repressive factors that further reduce the probability that cells infected with the mutant virus will commit to lytic infection.
The analysis indicates that each element of the TRIM is involved in PML.I modification and localisation, but that endogenous PML can influence both these factors providing the coiled-coil motif is present. This is graphically illustrated by the difference in localisation of the SUMO modification mutant, EYFP–PML.I.KK, whose distribution is entirely different in the presence and absence of endogenous PML. Mutations in the individual TRIM elements affected PML.I localisation in distinguishable ways, whereas both wild-type PML.I (Fig. 2; supplementary material Figs S4, S5) and some of the mutant forms (supplementary material Fig. S3) affected even endogenous ND10 composition or appearance. These observations further underline the complexity of the inter-relationships between endogenous and introduced PML proteins.
Because up to 11 PML isoforms have been described, we cannot exclude that some of them, even minor splice and/or cytoplasmic variants, could contribute to intrinsic cellular defence against HSV-1. Generally, their endogenous expression levels are low in comparison to the major PML isoforms. It has been observed that the cytoplasmic PML isoform lacking exons 5 and 6 (called PML.Ib) is enriched in HSV-1-infected human fibroblasts, and it was proposed that PML.Ib mediates intrinsic cellular defence against wild-type HSV-1 via cytoplasmic sequestration of ICP0 (McNally et al., 2008). Although PML.Ib might have an inhibitory effect on wild-type virus when very highly expressed, it could not influence ICP0-null mutant infection by this mechanism.
The failure of individual PML isoforms to restore full ND10 function is consistent with an emerging view that the isoform-specific C-terminal regions of PML contribute distinct functions that combine for normal ND10 functionality (Weidtkamp-Peters et al., 2008). It has been proposed that PML.V plays a structural role in nucleating ND10 (Brand et al., 2010; Weidtkamp-Peters et al., 2008), but it is also important to consider how the isoform-specific sequences might affect partner recruitment and exchange. Thus, the strong colocalisation of PML.V, PML.VI and PML.I.Δ7a with Sp100, hDaxx and ATRX could be influenced not only by the efficiency of recruitment of these other ND10 proteins, but also by the dynamic stability of that recruitment. It is likely that ND10 function is a complex synthesis of several component proteins and their various isoforms, and in retrospect it is perhaps unsurprising that no single major PML isoform can fully substitute for the combination of them all. A consequence of this conclusion is that deciphering the functions of PML, both during virus infection and in its various functions in uninfected cells, is an extremely complex issue. Nonetheless, the methodology of depletion and re-introduction of a given protein at close to endogenous levels should enable detailed analysis of the functions of wild-type and mutant cellular proteins in a more robust manner than achieved by methods based on transfection and/or overexpression in the presence of the endogenous protein.
Materials and Methods
Cells
U2OS, HEK-293T and human diploid fibroblast cells were grown in Dulbecco's modified Eagles' medium supplemented with 10% foetal calf serum. BHK cells were grown in Glasgow modified Eagles' medium supplemented with 10% new-born calf serum and 10% tryptose phosphate broth. HepaRG hepatocyte cells (Gripon et al., 2002) were grown in William's medium E supplemented with 10% foetal bovine serum Gold (PAA Laboratories), 2 mM glutamine, 5 μg/ml insulin and 0.5 μM hydrocortisone. All cell growth media contained 100 units/ml penicillin and 100 μg/ml streptomycin. Lentivirus transduced cells were maintained with continuous antibiotic selection, as appropriate.
Plasmids
Lentivirus vector plasmids expressing anti-PML and control anti-luciferase shRNAs (pLKO-shPML1 and pLKO-shLuc) were as described (Everett et al., 2008a). The anti-PML shRNA coding strand sequence was 5′-AGATGCAGCTGTATCCAAG-3′, which lies in conserved exon 4. The BamHI-KpnI fragment encoding puromycin resistance was removed from pLKO-shPML1 and replaced with a PCR fragment encoding G418 resistance flanked by the same restriction sites, to create pLKOneo-shPML1. Lentivirus vector plasmids expressing proteins from the HSV-1 gD (US6) gene promoter were constructed by removing the RNA polymerase III and shRNA sequences from pLKOneo.shPML1 and replacing them with a fragment from −122 to +11 of the HSV-1 gD promoter (Everett, 1983) linked to the EYFP coding region. Plasmids with this backbone were designated with the prefix pLNGY. The open reading frames of PML isoforms I to VI, as defined (Jensen et al., 2001), were inserted downstream of the EYFP sequences to give plasmids pLNGY-PML.I to pLNGY-PML.VI. All cDNAs encoding the PML isoforms were made resistant to the anti-PML shRNA by the introduction of five silent point mutations in the relevant sequence. The altered sequence was 5′-AGATGCTGCAGTTAGCAAG-3′. High-level expression of PML isoforms I and II was accomplished by use of lentivirus vectors pLNDY-PML.I and pLNDY-PML.II, in which the gD promoter of the pLNGY series plasmids was replaced with the 372 bp NdeI-AgeI fragment of pEGFP-C1 (Clontech) containing the proximal part of the HCMV IE promoter/enhancer.
PML mutants
Derivatives of pLNGY-PML.I with point mutations in the RING finger (ΔRING; C57A, C60A), the first B-Box (ΔBB1; C139A, C142A), and the second B-Box (ΔBB2; C211A, C214A) were constructed by site-directed mutagenesis or a PCR splicing approach using mutagenic oligonucleotides. Deletion of the coiled-coil domain (ΔCC, deletion of E239 to L322) and exon 7a (Δ7a, deletion of E553 to S570) were accomplished by PCR splicing. Mutant PML.I.KK with lysine to arginine mutations in the major SUMO modification sites (K160R, K490R) was constructed by fragment transfer from a previously characterised plasmid (Boutell et al., 2003).
Lentivirus transduction
Lentivirus supernatants were prepared after co-transfection into HEK-293T cells of a lentivirus vector plasmid with pVSV-G (expressing the VSV envelope protein) and pCMV.DR8.91 (expressing lentivirus helper functions), as described previously (Everett et al., 2008a). HepaRG cells were transduced with lentiviruses expressing shRNAs directed against either luciferase (as a control) or PML. Stable cell lines were then selected with puromycin (initially 1 μg/ml, then reduced to 0.5 μg/ml during subsequent passage). The cell populations so generated, named HALL (for hepatocyte lentivirus luciferase shRNA) and HALP (for hepatocyte lentivirus PML shRNA), respectively, were subsequently transduced with lentiviruses derived from the pLNGY-PML series to create HALL.EYFP–PML.I to HALL.EYFP–PML.VI and HALP.EYFP–PML.I to HALP.EYFP–PML.VI cells, which were selected with G418 (initially 1 mg/ml, then reduced to 0.5 mg/ml during subsequent passage). For further controls, HALL and HALP cells were also transduced with lentiviruses made from the pLNGY backbone vector that expresses EYFP alone.
Cells depleted of endogenous PML and expressing high levels of PML.I and PML.II were constructed by transduction with lentiviruses derived from pLNDY-PML.I and pLNDY-PML.II in an analogous manner.
Human fibroblast-derived cells transduced with the pLNGY-PML plasmids were also isolated, as were human fibroblast and HEp-2 derived cells transduced with lentiviruses made from vectors of the pLND.FLAG–PML.I to pLND.FLAG–PML.VI series. In all cases, these were made in both normal and endogenous PML-depleted cell lines. These additional cells are not described in detail but are referred to for control or confirmatory evidence when necessary.
Fluorescence-activated cell sorting
Enrichment of the HALL.EYFP–PML.I to HALL.EYFP–PML.VI and HALP.EYFP–PML.I to HALP.EYFP–PML.VI series of cells was achieved by FACs. Cells were trypsinised, washed twice and resuspended in medium containing 200 units/ml penicillin, 200 μg/ml streptomycin and 1% foetal bovine serum Gold. EYFP-positive cells were sorted using a Becton Dickinson SORP FACSaria (Beatson Institute for Cancer Research, Glasgow), then transferred into fresh complete medium and expanded into cell lines.
Viruses and virus plaque assays
HSV-1 strain 17 was the wild-type virus used, from which the ICP0-null mutant dl1403 (Stow and Stow, 1986) was derived. HSV-1 strains in1863 and dl1403/CMVlacZ were derivatives of wild-type HSV-1 strain 17 and dl1403 that contain the lacZ gene under the control of the HCMV promoter/enhancer inserted into the tk gene. The viruses were grown in BHK cells and titrated in U2OS cells, in which ICP0 is not required for efficient replication of HSV-1. For plaque assays in HepaRG-derived lines, cells were seeded into 24-well dishes at 1×105 cells per well, then infected the following day with appropriate sequential threefold dilutions of in1863 or dl1403/CMVlacZ. After virus adsorption, the cells were incubated in medium containing 1% human serum for 24 hours before plaques were identified by β-galactosidase staining (Jamieson et al., 1995). Relative efficiencies of plaque formation at a given virus input were calculated as described (Everett et al., 2006). All experiments were performed at least three times. Where relevant, the paired two-tailed Student's t-test was used for statistical analysis.
Western blot analysis
Cells were seeded into 24-well dishes at 1×105 cells per well. The following day, the cells were washed twice with PBS before harvesting in SDS-PAGE loading buffer. Proteins were resolved on 7.5% SDS gels, then transferred to nitrocellulose membranes by western blotting. The following antibodies were used: anti-actin mAb AC-40 (Sigma-Aldrich), anti-PML mAb 5E10 (Stuurman et al., 1992), anti-Sp100 rabbit serum SpGH (Sternsdorf et al., 1997), and anti-EGFP rabbit serum ab290 (which also detects EYFP), anti-SUMO-1 mAb ab299 and SUMO-2/3 rabbit polyclonal ab3741-100 (all Abcam).
Immunofluorescence and confocal microscopy
Cells on 13-mm glass coverslips were fixed using 1.5 % (v/v) formaldehyde in PBS containing 2% sucrose, then treated with 0.5 % Nonidet P40 substitute (EuroClone) in PBS containing 10% sucrose. PML was detected with mAb 5E10, Sp100 with rabbit serum SpGH, ICP4 with mAb 58S, hDaxx with rabbit serum 07-471 (Upstate), and ATRX with rabbit serum H-300 (Santa Cruz Biotechnology). The secondary antibodies were FITC-conjugated goat anti-mouse IgG (Sigma) and Alexa-Fluor-633-conjugated goat anti-rabbit IgG (Molecular Probes). A glycerol-based mounting medium was used (Citifluor AF1). The samples were examined using a Zeiss LSM 510 confocal microscope, using the 488 nm and 633 nm laser lines and scanning each channel separately under image capture conditions that eliminated channel overlap. The lens was an oil immersion 63× Plan-Apochromat, NA 1.40. To ensure that all PML and ND10 foci were recorded, the images were acquired as short z-stacks (three or four slices covering 1.5–2 μm) then single plane projections were produced for export as tiff files using LSM 510 software. Exported images were processed using Adobe Photoshop with minimal adjustment, then assembled for presentation using Adobe Illustrator.
Supplementary Material
Acknowledgments
This work was funded by the Medical Research Council (R.D.E. group), and the DFG-financed Sonderforschungsbereich SFB841 (Hamburg groups). We thank Keith Leppard (University of Warwick, UK) for cDNAs encoding PML isoforms, Roel van Driel (University of Amsterdam, The Netherlands) for anti-PML antibody 5E10, PhilippeGripon (University of Rennes, France) for HepaRG cells, Hans Will (Heinrich-Pette Institute, Hamburg, Germany) for anti-Sp100 antibody SpGH, Didier Trono (University of Geneva, Switzerland) for pCMV.DR8.91, Chris Preston (MRC-University of Glasgow Centre for Vims Research, UK) for viruses in1863 and dl1403/CMVlacZ, and Tom Gilbey (Beatson Institute for Cancer Research, Glasgow, UK) for FACS enrichment of EYFP-positive cells. Other members of the R.D.E. laboratory, in particular Chris Boutell, were very helpful in discussions throughout the course of this work. Deposited in PMC for release after 6 months.
Footnotes
Supplementary material available online at http://jcs.biologists.org/cgi/content/full/124/2/280/DC1
References
- Beech S. J., Lethbridge K. J., Killick N., McGlincy N., Leppard K. N. (2005). Isoforms of the promyelocytic leukemia protein differ in their effects on ND10 organization. Exp. Cell Res. 307, 109-117 [DOI] [PubMed] [Google Scholar]
- Bernardi R., Pandolfi P. P. (2003). Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene 22, 9048-9057 [DOI] [PubMed] [Google Scholar]
- Bernardi R., Pandolfi P. P. (2007). Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8, 1006-1016 [DOI] [PubMed] [Google Scholar]
- Bischof O., Kirsh O., Pearson M., Itahana K., Pelicci P. G., Dejean A. (2002). Deconstructing PML-induced premature senescence. EMBO J. 21, 3358-3369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block G. J., Eskiw C. H., Dellaire G., Bazett-Jones D. P. (2006). Transcriptional regulation is affected by subnuclear targeting of reporter plasmids to PML nuclear bodies. Mol. Cell. Biol. 26, 8814-8825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisvert F. M., Kruhlak M. J., Box A. K., Hendzel M. J., Bazett-Jones D. P. (2001). The transcription coactivator CBP is a dynamic component of the promyelocytic leukemia nuclear body. J. Cell Biol. 152, 1099-1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borden K. L. (2002). Pondering the promyelocytic leukemia protein (PML) puzzle: possible functions for PML nuclear bodies. Mol. Cell. Biol. 22, 5259-5269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boutell C., Orr A., Everett R. D. (2003). PML residue lysine 160 is required for the degradation of PML induced by herpes simplex virus type 1 regulatory protein ICP0. J. Virol. 77, 8686-8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand P., Lenser T., Hemmerich P. (2010). Assembly dynamics of PML nuclear bodies in living cells. PMC Biophys. 3, 3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantrell S. R., Bresnahan W. A. (2006). Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 80, 6188-6191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang K. S., Fan Y. H., Andreeff M., Liu J., Mu Z. M. (1995). The PML gene encodes a phosphoprotein associated with the nuclear matrix. Blood 85, 3646-3653 [PubMed] [Google Scholar]
- Chelbi-Alix M. K., de The H. (1999). Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18, 935-941 [DOI] [PubMed] [Google Scholar]
- Condemine W., Takahashi Y., Zhu J., Puvion-Dutilleul F., Guegan S., Janin A., de The H. (2006). Characterization of endogenous human promyelocytic leukemia isoforms. Cancer Res. 66, 6192-6198 [DOI] [PubMed] [Google Scholar]
- Condemine W., Takahashi Y., Le Bras M., de The H. (2007). A nucleolar targeting signal in PML-I addresses PML to nucleolar caps in stressed or senescent cells. J. Cell Sci. 120, 3219-3227 [DOI] [PubMed] [Google Scholar]
- Dellaire G., Bazett-Jones D. P. (2004). PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. BioEssays 26, 963-977 [DOI] [PubMed] [Google Scholar]
- Dellaire G., Eskiw C. H., Dehghani H., Ching R. W., Bazett-Jones D. P. (2006). Mitotic accumulations of PML protein contribute to the re-establishment of PML nuclear bodies in G1. J. Cell Sci. 119, 1034-1042 [DOI] [PubMed] [Google Scholar]
- Eskiw C. H., Bazett-Jones D. P. (2002). The promyelocytic leukemia nuclear body: sites of activity? Biochem. Cell Biol. 80, 301-310 [DOI] [PubMed] [Google Scholar]
- Eskiw C. H., Dellaire G., Mymryk J. S., Bazett-Jones D. P. (2003). Size, position and dynamic behavior of PML nuclear bodies following cell stress as a paradigm for supramolecular trafficking and assembly. J. Cell Sci. 116, 4455-4466 [DOI] [PubMed] [Google Scholar]
- Everett R. D. (1983). DNA sequence elements required for regulated expression of the HSV-1 glycoprotein D gene lie within 83 bp of the RNA capsites. Nucleic Acids Res. 11, 6647-6666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R. D. (2001). DNA viruses and viral proteins that interact with PML nuclear bodies. Oncogene 20, 7266-7273 [DOI] [PubMed] [Google Scholar]
- Everett R. D., Zafiropoulos A. (2004). Visualization by live-cell microscopy of disruption of ND10 during herpes simplex virus type 1 infection. J. Virol. 78, 11411-11415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R. D., Chelbi-Alix M. K. (2007). PML and PML nuclear bodies: Implications in antiviral defence. Biochimie 89, 819-830 [DOI] [PubMed] [Google Scholar]
- Everett R. D., Freemont P., Saitoh H., Dasso M., Orr A., Kathoria M., Parkinson J. (1998). The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72, 6581-6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R. D., Murray J. (2005). ND10 components relocate to sites associated with herpes simplex virus type 1 nucleoprotein complexes during virus infection. J. Virol. 79, 5078-5089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R. D., Lomonte P., Sternsdorf T., van Driel R., Orr A. (1999). Cell cycle regulation of PML modification and ND10 composition. J. Cell Sci. 112, 4581-4588 [DOI] [PubMed] [Google Scholar]
- Everett R. D., Rechter S., Papior P., Tavalai N., Stamminger T., Orr A. (2006). PML contributes to a cellular mechanism of repression of herpes simplex virus type 1 infection that is inactivated by ICP0. J. Virol. 80, 7995-8005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R. D., Murray J., Orr A., Preston C. M. (2007). Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 81, 10991-11004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R. D., Parada C., Gripon P., Sirma H., Orr A. (2008a). Replication of ICP0-null mutant herpes simplex virus type 1 is restricted by both PML and Sp100. J. Virol. 82, 2661-2672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett R. D., Young D. F., Randall R. E., Orr A. (2008b). STAT-1- and IRF-3-dependent pathways are not essential for repression of ICP0-null mutant herpes simplex virus type 1 in human fibroblasts. J. Virol. 82, 8871-8881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripon P., Rumin S., Urban S., Le Seyec J., Glaise D., Cannie I., Guyomard C., Lucas J., Trepo C., Guguen-Guillouzo C. (2002). Infection of a human hepatoma cell line by hepatitis B virus. Proc. Natl. Acad. Sci. USA 99, 15655-15660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa F., Privalsky M. L. (2004). Phosphorylation of PML by mitogen-activated protein kinases plays a key role in arsenic trioxide-mediated apoptosis. Cancer Cell 5, 389-401 [DOI] [PubMed] [Google Scholar]
- Hoppe A., Beech S. J., Dimmock J., Leppard K. N. (2006). Interaction of the adenovirus type 5 E4 Orf3 protein with promyelocytic leukemia protein isoform II is required for ND10 disruption. J. Virol. 80, 3042-3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishov A. M., Sotnikov A. G., Negorev D., Vladimirova O. V., Neff N., Kamitani T., Yeh E. T., Strauss J. F., Maul G. G. (1999). PML is critical for ND10 formation and recruits the PML-interacting protein daxx to this nuclear structure when modified by SUMO-1. J. Cell Biol. 147, 221-234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamieson D. R. S., Robinson L. H., Daksis J. I., Nicholl M. J., Preston C. M. (1995). Quiescent viral genomes in human fibroblasts after infection with herpes simplex virus Vmw65 mutants. J. Gen. Virol. 76, 1417-1431 [DOI] [PubMed] [Google Scholar]
- Jensen K., Shiels C., Freemont P. S. (2001). PML protein isoforms and the RBCC/TRIM motif. Oncogene 20, 7223-7233 [DOI] [PubMed] [Google Scholar]
- Kamitani T., Kito K., Nguyen H. P., Wada H., Fukuda-Kamitani T., Yeh E. T. (1998a). Identification of three major sentrinization sites in PML. J. Biol. Chem. 273, 26675-26682 [DOI] [PubMed] [Google Scholar]
- Kamitani T., Nguyen H. P., Kito K., Fukuda-Kamitani T., Yeh E. T. (1998b). Covalent modification of PML by the sentrin family of ubiquitin-like proteins. J. Biol. Chem. 273, 3117-3120 [DOI] [PubMed] [Google Scholar]
- Kyratsous C. A., Silverstein S. J. (2009). Components of nuclear domain 10 bodies regulate varicella-zoster virus replication. J. Virol. 83, 4262-4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. R., Kim D. J., Lee J. M., Choi C. Y., Ahn B. Y., Hayward G. S., Ahn J. H. (2004). Ability of the human cytomegalovirus IE1 protein to modulate sumoylation of PML correlates with its functional activities in transcriptional regulation and infectivity in cultured fibroblast cells. J. Virol. 78, 6527-6542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leppard K. N., Emmott E., Cortese M. S., Rich T. (2009). Adenovirus type 5 E4 Orf3 protein targets promyelocytic leukaemia (PML) protein nuclear domains for disruption via a sequence in PML isoform II that is predicted as a protein interaction site by bioinformatic analysis. J. Gen. Virol. 90, 95-104 [DOI] [PubMed] [Google Scholar]
- Ling P. D., Tan J., Sewatanon J., Peng R. (2008). Murine gammaherpesvirus 68 open reading frame 75c tegument protein induces the degradation of PML and is essential for production of infectious virus. J. Virol. 82, 8000-8012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez P., Jacob R. J., Roizman B. (2002). Overexpression of promyelocytic leukemia protein precludes the dispersal of ND10 structures and has no effect on accumulation of infectious herpes simplex virus 1 or its proteins. J. Virol. 76, 9355-9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashchuk V., McFarlane S., Everett R. D., Preston C. M. (2008). Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 82, 12543-12554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashchuk V., Orr A., Everett R. D. (2010). Regulation of ICP0 null mutant HSV-1 infection by ND10 components ATRX and hDaxx. J. Virol. 84, 4026-4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maul G. G. (1998). Nuclear domain 10, the site of DNA virus transcription and replication. BioEssays 20, 660-667 [DOI] [PubMed] [Google Scholar]
- McNally B. A., Trgovcich J., Maul G. G., Liu Y., Zheng P. (2008). A role for cytoplasmic PML in cellular resistance to viral infection. PLoS ONE 3, e2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negorev D., Maul G. G. (2001). Cellular proteins localized at and interacting within ND10/PML nuclear bodies/PODs suggest functions of a nuclear depot. Oncogene 20, 7234-7242 [DOI] [PubMed] [Google Scholar]
- Negorev D. G., Vladimirova O. V., Ivanov A., Rauscher F., 3rd, Maul G. G. (2006). Differential role of Sp100 isoforms in interferon-mediated repression of herpes simplex virus type 1 immediate-early protein expression. J. Virol. 80, 8019-8029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichler A., Gast A., Seeler J. S., Dejean A., Melchior F. (2002). The nucleoporin RanBP2 has SUMO1 E3 ligase activity. Cell 108, 109-120 [DOI] [PubMed] [Google Scholar]
- Preston C. M., Nicholl M. J. (2006). Role of the cellular protein hDaxx in human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 87, 1113-1121 [DOI] [PubMed] [Google Scholar]
- Reineke E. L., Kao H. Y. (2009). Targeting promyelocytic leukemia protein: a means to regulating PML nuclear bodies. Int. J. Biol. Sci. 5, 366-376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saffert R. T., Kalejta R. F. (2006). Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80, 3863-3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaglioni P. P., Yung T. M., Cai L. F., Erdjument-Bromage H., Kaufman A. J., Singh B., Teruya-Feldstein J., Tempst P., Pandolfi P. P. (2006). A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell 126, 269-283 [DOI] [PubMed] [Google Scholar]
- Shen T. H., Lin H. K., Scaglioni P. P., Yung T. M., Pandolfi P. P. (2006). The mechanisms of PML-nuclear body formation. Mol. Cell 24, 331-339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobol P. T., Hummel J. L., Rodrigues R. M., Mossman K. L. (2009). PML has a predictive role in tumor cell permissiveness to interferon-sensitive oncolytic viruses. Gene Ther. 16, 1077-1087 [DOI] [PubMed] [Google Scholar]
- Sternsdorf T., Guldner H. H., Szostecki C., Grotzinger T., Will H. (1995). Two nuclear dot-associated proteins, PML and Sp100, are often co-autoimmunogenic in patients with primary biliary cirrhosis. Scand. J. Immunol. 42, 257-268 [DOI] [PubMed] [Google Scholar]
- Sternsdorf T., Jensen K., Will H. (1997). Evidence for covalent modification of the nuclear dot-associated proteins PML and Sp100 by PIC1/SUMO-1. J. Cell Biol. 139, 1621-1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stow N. D., Stow E. C. (1986). Isolation and characterization of a herpes simplex virus type 1 mutant containing a deletion within the gene encoding the immediate early polypeptide Vmw110. J. Gen. Virol. 67, 2571-2585 [DOI] [PubMed] [Google Scholar]
- Stuurman N., de Graaf A., Floore A., Josso A., Humbel B., de Jong L., van Driel R. (1992). A monoclonal antibody recognizing nuclear matrix-associated nuclear bodies. J. Cell Sci. 101, 773-784 [DOI] [PubMed] [Google Scholar]
- Sykes A. K. (2007). An investigation into the roles of the major PML isoforms in HSV-1 infection. PhD thesis, University of Glasgow; [Google Scholar]
- Tatham M. H., Kim S., Jaffray E., Song J., Chen Y., Hay R. T. (2005). Unique binding interactions among Ubc9, SUMO and RanBP2 reveal a mechanism for SUMO paralog selection. Nat. Struct. Mol. Biol. 12, 67-74 [DOI] [PubMed] [Google Scholar]
- Tavalai N., Papior P., Rechter S., Leis M., Stamminger T. (2006). Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 80, 8006-8018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavalai N., Papior P., Rechter S., Stamminger T. (2008). Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J. Virol. 82, 126-137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidtkamp-Peters S., Lenser T., Negorev D., Gerstner N., Hofmann T. G., Schwanitz G., Hoischen C., Maul G., Dittrich P., Hemmerich P. (2008). Dynamics of component exchange at PML nuclear bodies. J. Cell Sci. 121, 2731-2743 [DOI] [PubMed] [Google Scholar]
- Wiesmeijer K., Molenaar C., Bekeer I. M., Tanke H. J., Dirks R. W. (2002). Mobile foci of Sp100 do not contain PML: PML bodies are immobile but PML and Sp100 proteins are not. J. Struct. Biol. 140, 180-188 [DOI] [PubMed] [Google Scholar]
- Woodhall D. L., Groves I. J., Reeves M. B., Wilkinson G., Sinclair J. H. (2006). Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 281, 37652-37660 [DOI] [PubMed] [Google Scholar]
- Zhong S., Muller S., Ronchetti S., Freemont P. S., Dejean A., Pandolfi P. P. (2000a). Role of SUMO-1-modified PML in nuclear body formation. Blood 95, 2748-2752 [PubMed] [Google Scholar]
- Zhong S., Salomoni P., Pandolfi P. P. (2000b). The transcriptional role of PML and the nuclear body. Nat. Cell Biol. 2, E85-E90 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.