

Abstract
Initial identification of populations at high risk of gastric cancer (GC) is important for endoscopic screening of GC. As serum pepsinogen (PG) test-positive subjects with progression of chronic atrophic gastritis (CAG) show a high likelihood of future cancer development, this population warrants careful follow-up observation as a high-risk GC group. By combining the PG test with Helicobacter pylori (HP) antibody titers, the HP-related chronic gastritis stage can be classified, thus identifying not only a GC high-risk group but also a low-risk group. Among PG test-negative patients without CAG, those with high serum PG II levels and HP antibody titers are thought to have severe gastric mucosal inflammation and the risk of diffuse-type GC is also high. Meanwhile, in gastric mucosae obtained by endoscopic biopsy, HP infection induces aberrant DNA methylation in CpG islands in multiple gene regions and the extent of methylation clearly correlates with GC risk. By quantifying aberrant DNA methylation in suitable gene markers, we can determine the extent of the epigenetic field for cancerization. These novel concepts and risk markers will have many clinical applications in gastrointestinal endoscopy, including more efficient endoscopic GC screening and a strategic approach to metachronous multiple GCs after endoscopic treatment.
Keywords: Gastric cancer, Screening, Risk, Pepsinogen, Helicobacter pylori, DNA methylation
INTRODUCTION
Owing to the recent advances in minimally invasive and radical endoscopic treatments including endoscopic mucosal resection (EMR) and endoscopic submucosal dissection (ESD), early gastric cancers (GCs) have been endoscopically resected, especially in Japan[1-3]. Following advances in new endoscopic treatment, early detection and accurate diagnosis of GC has been increasing in importance. In particular, advances in endoscopic equipment and developments in endoscopic image enhancement technology have greatly contributed to improved diagnosis for early GC[4-6]. Furthermore, identifying which populations are at high risk for GC plays a key role in endoscopic GC diagnosis. This not only assists in endoscopic diagnosis but can also contribute greatly to other aspects of endoscopic management of GC, including the current problem of identifying populations who should be targeted for GC screening[7] and strategic approaches to metachronous multiple GC after EMR or ESD[8].
Helicobacter pylori (HP) infection is a major risk factor in GC development[9]. However, in countries like Japan with high HP infection rates, the existence of HP infection alone offers inadequate specificity for the assessment of GC risk. Novel risk markers to identify GC high-risk groups based on a detailed natural history of GC have thus long been awaited. In this paper, we discuss the emerging significance of serum pepsinogen (PG) as a GC risk marker for more precise identification of GC high-risk groups. We also discuss our research on DNA methylation in gastric mucosae obtained at endoscopic biopsy as a molecular biological marker to evaluate GC risk.
SERUM PG TEST FOR IDENTIFICATION OF GC HIGH-RISK GROUPS
Theoretical considerations of the serum PG test
PG is the inactive precursor of pepsin, a gastrointestinal enzyme specifically produced in the gastric mucosae[10]. PG is mainly excreted into the stomach lumen but about 1% of the total enters into the blood stream and is measurable as serum PG. Changes in serum PG levels reflects gastric mucosal morphology and exocrine function[11,12]. In an endoscopic study with Congo red staining, an increase in glandular boundary, associated with diagnosed progression of gastric mucosal atrophy, correlated strongly with stepwise reductions in serum PG I levels and the PG I/II ratio[13]. In other words, measuring serum PG I and the PG I/II ratio offers the opportunity to evaluate the progression of chronic atrophic gastritis (CAG), a precursor of GC[14].
As criteria for the serum PG test used for GC screening, the combination of PG I ≤ 70 ng/mL and PG I/II ≤ 3.0 is widely accepted as a reference value (PG index 1+)[14,15]. Low values based on this reference are considered PG test-positive. In addition, to identify more severe CAG progression, criteria of PG I ≤ 50 ng/mL and PG I/II ≤ 3.0 (PG index 2+), and PG I ≤ 30 ng/mL and PG I/II ≤ 2.0 (PG index 3+) are also used. Since 1992, when PG assay kits became commercially available, a number of screening services provided by work place or community health services have adopted this serum test as a filter test[16-22].
Accuracy of GC detection using the serum PG test
We conducted a 10 year follow-up observation study of GC occurrence in a cohort of middle-aged healthy men[23-25]. Based on the results, we evaluated the accuracy of each serum PG test index for detecting GC during the observation period[25]. Table 1 summarizes the accuracy for each PG test index. For the most lenient criteria (PG index 1+), sensitivity was 58.7%, specificity was 73.4% and positive predictive value was 2.6%. Overall, the results showed obviously low sensitivity. Compared to a recently reported meta-analysis of PG test sensitivity[26], these results were clearly poor, particularly in terms of low sensitivity.
Table 1.
Comparison of accuracy of gastric cancer detection by each serum pepsinogen test index
| Serum PG test |
Our results[25] |
Meta-analysis of reported cases[26] |
||
| Sensitivity (95% CI) | Specificity (95% CI) | Pooled sensitivity (95% CI) | Pooled specificity (95% CI) | |
| PG I ≤ 70 and PG I/II ≤ 3 (PG index 1+) | 58.70% (45.6-70.8) | 73.40% (72.1-74.6) | 77.30% (69.8-83.8) | 73.20% (72.8-73.6) |
| PG I ≤ 50 and PG I/II ≤ 3 (PG index 2+) | 49.20% (36.5-62.0) | 80.50% (79.4-81.6) | 68.40% (59.1-76.8) | 69.30% (66.6-70.0) |
| PG I ≤ 30 and PG I/II ≤ 2 (PG index 3+) | 27.00% (16.9-39.9) | 92.00% (91.3-92.8) | 51.90% (40.3-63.5) | 84.40% (83.7-85.0) |
PG: pepsinogen.
One interpretation of these results is that some GCs are easier to detect by barium X-ray and some GCs are easier to detect by the serum PG test[22]. In the above-mentioned meta-analysis, many of the reviewed reports were studies of populations in whom GC was diagnosed over a long period by barium X-rays. Targeting a population with a concentration of GC cases difficult to detect by barium X-ray, or in other words, GC easy to detect by the serum PG test, these studies analyzed results of GC detection just after introduction of the serum PG test and over a short period. On the other hand, in our study, GC cases just after introduction of the serum PG test were excluded and follow-up was continued over a period of 10 years. The results of detecting GC occurring during the observation period were thus examined more rigorously, better depicting the accuracy of GC detection using the serum PG test. Based on these results, the serum PG test has limitations when used alone for GC screening. This shows the need for more in-depth systematic screening, including in PG test-negative GC.
GC risk diagnosis using the serum PG test
Previous studies have examined the accuracy of serum PG as a filter test for endoscopy. Recently, as part of an investigation into the natural history of GC occurrence, we examined GC risk in each population identified using each serum PG test index[25]. The annual incidence of GC was 0.07% in the atrophy-negative group, compared to 0.28% in the atrophy-positive (PG index 1+) group, 0.32% in the PG index 2+ group and 0.42% in the PG index 3+ group. The incidence of GC thus increased in a stepwise and significant manner with CAG progression (Figure 1). These results clearly indicate that PG test-positive subjects are a high-risk GC group, have a higher future likelihood of developing GC and represent a population requiring careful follow-up observation.
Figure 1.
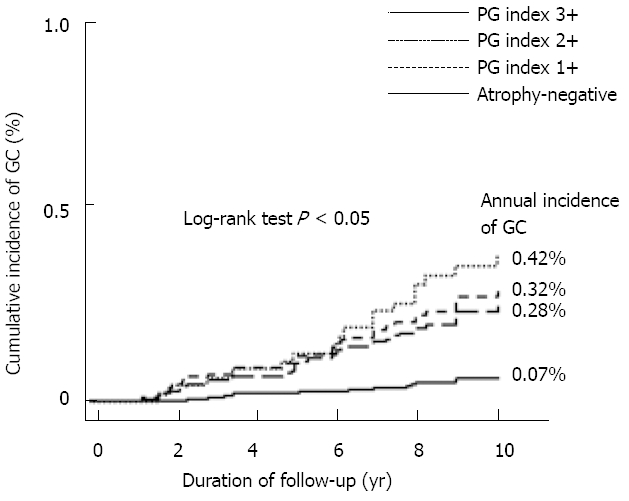
Kaplan-Meier analysis of gastric cancer development in subjects classified using the index of the pepsinogen test (modified from Yanaoka et al[25]). This shows the annual incidence of gastric cancer (GC) in each population identified based on serum pepsinogen test index criteria in middle-aged healthy men. With chronic atrophic gastritis progression, incidence of GC increased in a stepwise and significant manner. PG: pepsinogen.
Identification of GC high-risk groups using a combination of the serum PG test and HP infection diagnosis
Next, in the same populations, the relationship between HP infection, a major cause of chronic gastritis, and GC risk was also examined[23,24]. To diagnose HP infection, we used anti-HP antibody titers which, like serum PG, are easily measured using blood samples. The stage of HP-related chronic gastritis was classified into 4 stages based on the combination of both test results: Group A [HP(-), PG(-)]; Group B [HP(+), PG(-)]; Group C [HP(+), PG(+)]; and Group D [HP(-), PG(+)] (Figure 2). Group A comprised of HP non-infected healthy men. Group B showed established HP infection but without CAG. Group C had CAG. Group D had severe intestinal metaplasia due to progression of CAG but HP had been spontaneously eliminated, representing so-called metaplastic gastritis. Annual incidences of GC were: Group A, 0%; Group B, 0.11%; Group C, 0.24%; and Group D, 1.31%. Thus, with HP infection and CAG progression, the rate increased in a stepwise and significant manner. Moreover, in the non-infected healthy Group A, GC did not occur in a single case during 10 years of follow-up observation. Based on the above results, using a combination of the serum PG test and HP infection diagnosis, not only high-risk groups, but also a low-risk group, can theoretically be identified.
Figure 2.

Gastric cancer incidence and Helicobacter pylori-related chronic gastritis stage classification based on a combination of the serum pepsinogen test and helicobacter pylori-infection diagnosis (modified from Ohata et al[23]). This shows percentages in each group, among middle-aged healthy men, based on the serum pepsinogen test and Helicobacter pylori (HP) antibody titers. As HP-related chronic gastritis stage progressed from Group A to Group D, annual incidence of gastric cancer increased in a stepwise and significant manner. PG: pepsinogen.
Points to consider in the serum PG test-negative GC
The serum PG test is highly useful as a GC risk marker but the occurrence of GC (particularly diffuse-type GC) in the PG test-negative group (Group B in HP-related chronic gastritis stage) cannot be ignored. In our study, even using the most balanced PG test criteria in terms of test accuracy (PG index 1+), about 40% of GCs that occurred represented PG test-negative GC. This point must be clearly kept in mind when assessing GC risk using the serum PG test.
We therefore evaluated the occurrence of GC in the PG test-negative group in further detail. Specifically, we examined the incidence of GC in 3 PG test-negative subgroups: α group (PG I ≤ 70 ng/mL and PG I/II > 3); β group (PG I > 70 ng/mL and PG I/II > 3); and γ group (PG I > 70 ng/mL and PG I/II ≤ 3). In the γ group, with a higher serum PG II and presumably severe gastric mucosal inflammation, the incidence of GC was 0.2%, thus identifying a new GC high-risk group mainly at risk of developing diffuse-type GC[25]. The rate in the γ group, although not high among the serum PG test-negative group, does indicate a subgroup to which careful attention should be paid. In addition, the group with high HP antibody titers (a marker which, like serum PG II levels, reflects the degree of gastric mucosal inflammation) had a higher incidence of GC compared to a group with lower titers[24]. Furthermore, in this group, HP eradication therapy can be highly effective in preventing GC[27].
ABERRANT DNA METHYLATION AND GC RISK
Aberrant DNA methylation in cancers
Epigenetic abnormalities are also important as cancer gene abnormalities in addition to gene structural abnormalities such as mutations and chromosomal deletions. DNA methylation represents one type of epigenetic information. DNA methylation occurs physiologically and is observed at CpG sites where cytosine (C) is located adjacent to guanine (G) in gene sequences. CpG sites occur with low frequency in the genome but areas with a high density of CpG sites are occasionally encountered as so-called CpG islands (CGIs). When a CGI is in a gene promotor region and is entirely methylated, transcription of downstream genes to mRNA is potently inhibited (silencing). DNA methylation together with mutations and chromosomal deletions is a major factor in gene inactivation in many cancers[28-31].
In cancer cells, compared to normal cells, genome-overall hypomethylation and regional hypermethylation are observed. Genome-overall hypomethylation is involved in carcinogenesis by causing chromosomal instability[32]. Regional hypermethylation refers to aberrant methylation of a specific CGI that is normally unmethylated. If hypermethylation is induced in a promotor region CGI of a tumor suppressor gene, gene inactivation occurs. This causes cell cycle abnormalities, growth signaling abnormalities and mutation accumulation, thus playing a role in cancer onset and progression.
In gastrointestinal cancers, including GC, silencing of several important tumor suppressor genes has been reported. In particular, in GC, inactivation of CDKN2A, MLH1 and CDH1 due to methylation is more frequent than inactivation due mutations or chromosomal deletions[33].
Induction of aberrant DNA methylation in non-cancerous gastric mucosae by HP infection
Aberrant DNA methylation is important in GC but the mechanisms of induction have remained unknown. Using gastric mucosae obtained by endoscopic biopsy from both HP-positive healthy volunteers (individuals without GC) and HP-negative healthy volunteers, we used quantitative methylation-specific PCR (qMSP) to measure the percentage of DNA molecules with aberrant methylation (methylation level, reflecting the percentage of cells with aberrant methylation)[34]. As genes for analysis, we selected CGIs from 8 regions of 7 genes found to be methylated at high frequency in GC[35]. All of the eight regions showed a similar tendency in terms of methylation levels. Among healthy volunteers, methylation levels were 5.4- to 303-fold higher in HP-positive individuals than HP-negative individuals. This suggests that HP infection can potently induce aberrant DNA methylation.
Accumulation of aberrant DNA methylation in gastric mucosa and GC risk
In addition, to correlate the extent of aberrant DNA methylation in the gastric mucosae with GC risk, we analyzed gastric mucosae in healthy volunteers and non-cancerous gastric mucosae in patients with well-differentiated GC. In a comparison among HP-negative cases, methylation levels were 2- to 32-fold higher in non-cancerous gastric mucosae of GC patients than in gastric mucosae of healthy volunteers. We also newly collected non-cancerous gastric mucosae of patients with a single GC and those with multiple GCs and compared methylation levels in the gastric mucosae of patients with multiple GC (very high risk of GC) and patients with single GC. In HP-negative cases, specific gene methylation levels were increased in the order of healthy individual gastric mucosae → single GC patient non-cancerous gastric mucosae → multiple GC patient non-cancerous gastric mucosae[36]. These findings suggested a correlation between gastric mucosae methylation levels and GC risk in HP-negative cases. However, in HP-positive cases, both GC patients and healthy individuals showed potent induction of aberrant DNA methylation with almost no difference in methylation levels.
When evaluated by each gene, mean methylation levels for the tumor suppressor genes CDKN2A and MLH1 were very low, so evaluating the correlation with GC risk was difficult (Figure 3)[34,37]. However, LOX, a tumor suppressor gene, showed relatively high methylation levels. Similarly, the microRNA gene, with tumor suppressor activity, also showed high methylation levels[38]. Methylation of non-tumor suppressor genes like THBD was observed in a relatively large number of cells. These levels correlated with GC risk (Figure 3). Genes methylated by HP infection show specificity. With HP infection, resistant genes show no methylation at all while susceptible genes display a high frequency of methylation[39]. Important in this mechanism is a lower expression of methylation-susceptible genes in the gastric mucosae of healthy individuals[39,40]. Thus, with HP infection, gene-specific regional hypermethylation occurs in non-cancerous gastric mucosa. Furthermore, recent study showed that regional (Alu and Sat) hypomethylation is induced in gastric mucosae by HP infection during gastric carcinogenesis[41].
Figure 3.

Relationship between gastric mucosae methylation levels and Helicobacter pylori infection/gastric cancer (modified from Maekita et al[34]). This shows mean methylation levels for the THBD and CDKN2A genes as measured by quantitative methylation-specific PCR in endoscopically biopsied gastric mucosae. Among Helicobacter pylori (HP)-negative cases, non-cancerous gastric mucosae in gastric cancer (GC) patients showed higher methylation levels than gastric mucosa in healthy volunteers. Among HP-positive cases, methylation levels were high regardless of the presence or absence of GC. Methylation susceptibility differed among genes. Compared to THBD, CDKN2A showed very little induction of methylation. The error bar denotes standard error. HV: healthy volunteers; Pt: patients.
DNA methylation levels after spontaneous elimination and eradication of HP infection
As most patients with intestinal-type GC have a past history of HP infection[42], the following changes in methylation levels are postulated to occur in the natural history of GC development. Firstly, methylation levels in the gastric mucosae are low in HP-non-infected individuals (near 0%). Secondly, with HP infection, DNA methylation of the gastric mucosae is potently induced. Thirdly, with progression of atrophic gastritis, spontaneous elimination of HP infection decreases methylation levels (Figure 4).
Figure 4.
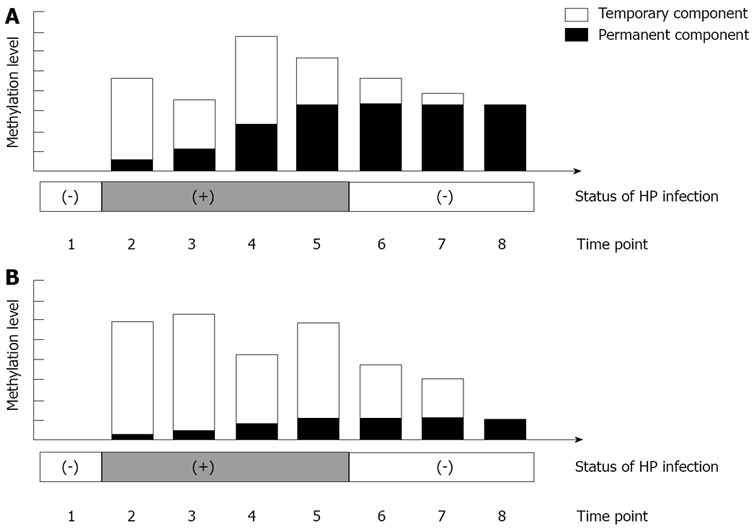
Temporary DNA methylation and permanent DNA methylation of gastric mucosae. DNA methylation includes temporary methylation, which is induced only during Helicobacter pylori (HP) infection, and permanent methylation, which persists even after elimination of HP infection. From time points 2 to 5, when HP infection was positive, overall methylation levels changed, with increases in permanent methylation, and increases and decreases in temporary methylation. At this stage, temporary methylation showed large fluctuations, so distinguishing differences in gastric cancer (GC) risk between cases was difficult. However, after spontaneous elimination of HP or HP eradication, at time point 8, at which time only permanent DNA methylation was present, GC risk was clearly higher in Figure 4A.
In addition, decreased methylation levels after HP eradication have been confirmed in specific genes and different kinetics for each gene have been shown[43,44]. Once methylation has occurred in a cell, it is difficult to conceive that demethylation would again occur in the same region. The decrease in methylation levels observed after HP eradication is thus probably due to cell turnover (temporary methylation). Residual aberrant methylation even after eradication is thought to reflect methylation in gastric gland stem cells (permanent methylation).
Advantages of DNA methylation as a marker of a field for cancerization
Individuals with low residual methylation levels (permanent methylation levels) after HP elimination or eradication have a low risk of GC. Conversely, those with high levels have a higher risk of GC (Figure 4). Using methylation-susceptible genes like THBD that are easily methylated at high frequency by HP infection, the GC risk in patients with high methylation levels is 2- to 3-fold higher than that in patients with low methylation levels, if appropriate cut-off values are established. Moreover, in the case of recently discovered genes such as miR124a-1, -2 and -3, the GC risk is 5- to 20-fold higher[38].
Aberrant DNA methylation of the gastric mucosae has been strongly suggested to play an important role in the formation of an epigenetic field for cancerization, as the so-called epigenetic field defect[38,45,46]. These have similarly been found for esophageal cancer[47], colon cancer[48], hepatocellular carcinoma[49] and renal cancer[50]. Specific clinical applications of an epigenetic field for cancerization include measurement of methylation levels after HP eradication in healthy individuals to predict the risk of GC and measurement of methylation levels in patients who have undergone endoscopic treatment such as ESD to predict the risk of metachronous multiple GC. Large-scale prospective clinical trials are currently underway to confirm these concepts.
CONCLUSION
In conclusion, we have discussed identifying groups at high risk of developing GC using the serum PG test and predicting GC risk based on the accumulation of aberrant DNA methylation in the gastric mucosae from endoscopically biopsied tissue (Figure 5). Gastrointestinal endoscopists are aiming to improve diagnostic and treatment technology in GC but at the same time, as discussed in this paper, a thorough awareness of new concepts and risk markers of GC is also important. This is anticipated to have clinical applications such as in more effective endoscopic GC screening, and in establishing appropriate follow-up intervals for endoscopy based on individual GC risk.
Figure 5.
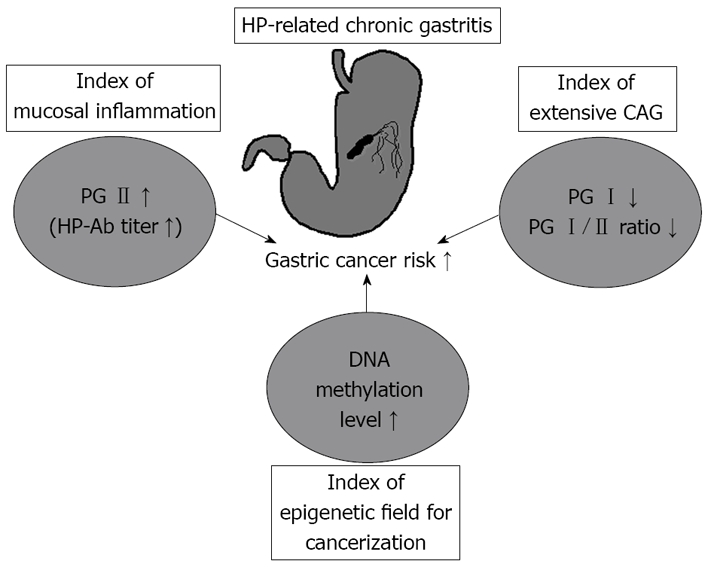
Schematic presentation of novel risk markers for gastric cancer screening. HP: Helicobacter pylori; CAG: chronic atrophic gastritis; PG: pepsinogen.
Footnotes
Peer reviewers: Perminder Phull, MD, FRCP, FRCPE, Gastrointestinal and Liver Service, Room 2.58, Ashgrove House, Aberdeen Royal Infirmary, Foresterhill, Aberdeen AB25 2ZN, United Kingdom; Jiang-Fan Zhu, MD, Professor of Surgery, Department of General Surgery, East Hospital of Tongji University, Pudong 200120, Shanghai, China
S- Editor Zhang HN L- Editor Roemmele A E- Editor Liu N
References
- 1.Kakushima N, Fujishiro M. Endoscopic submucosal dissection for gastrointestinal neoplasms. World J Gastroenterol. 2008;14:2962–2967. doi: 10.3748/wjg.14.2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yamamoto H. Technology insight: endoscopic submucosal dissection of gastrointestinal neoplasms. Nat Clin Pract Gastroenterol Hepatol. 2007;4:511–520. doi: 10.1038/ncpgasthep0906. [DOI] [PubMed] [Google Scholar]
- 3.Gotoda T. Endoscopic resection of early gastric cancer. Gastric Cancer. 2007;10:1–11. doi: 10.1007/s10120-006-0408-1. [DOI] [PubMed] [Google Scholar]
- 4.Kodashima S, Fujishiro M. Novel image-enhanced endoscopy with i-scan technology. World J Gastroenterol. 2010;16:1043–1049. doi: 10.3748/wjg.v16.i9.1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Curvers WL, van den Broek FJ, Reitsma JB, Dekker E, Bergman JJ. Systematic review of narrow-band imaging for the detection and differentiation of abnormalities in the esophagus and stomach (with video) Gastrointest Endosc. 2009;69:307–317. doi: 10.1016/j.gie.2008.09.048. [DOI] [PubMed] [Google Scholar]
- 6.Polkowski M. Endoscopic diagnosis and treatment of upper gastrointestinal tumors. Endoscopy. 2008;40:862–867. doi: 10.1055/s-2008-1077588. [DOI] [PubMed] [Google Scholar]
- 7.Mukoubayashi C, Yanaoka K, Ohata H, Arii K, Tamai H, Oka M, Ichinose M. Serum pepsinogen and gastric cancer screening. Intern Med. 2007;46:261–266. doi: 10.2169/internalmedicine.46.6181. [DOI] [PubMed] [Google Scholar]
- 8.Nakajima T, Oda I, Gotoda T, Hamanaka H, Eguchi T, Yokoi C, Saito D. Metachronous gastric cancers after endoscopic resection: how effective is annual endoscopic surveillance? Gastric Cancer. 2006;9:93–98. doi: 10.1007/s10120-006-0372-9. [DOI] [PubMed] [Google Scholar]
- 9.Organization IafrocWH. Schistosomes, liver flukes and Helicobacter pylori. Lyon: France; 1994. pp. 177–241. [Google Scholar]
- 10.Kageyama T, Ichinose M. Diversity of structure and function of pepsinogens and pepsins. Rescent Research Developments and Biophysics and Biochemistry. 2003;3:159–178. [Google Scholar]
- 11.Hirschowitz BI. Pepsinogen: its origins, secretion and excretion. Physiol Rev. 1957;37:475–511. doi: 10.1152/physrev.1957.37.4.475. [DOI] [PubMed] [Google Scholar]
- 12.Samloff IM, Varis K, Ihamaki T, Siurala M, Rotter JI. Relationships among serum pepsinogen I, serum pepsinogen II, and gastric mucosal histology. A study in relatives of patients with pernicious anemia. Gastroenterology. 1982;83:204–209. [PubMed] [Google Scholar]
- 13.Miki K, Ichinose M, Shimizu A, Huang SC, Oka H, Furihata C, Matsushima T, Takahashi K. Serum pepsinogens as a screening test of extensive chronic gastritis. Gastroenterol Jpn. 1987;22:133–141. doi: 10.1007/BF02774209. [DOI] [PubMed] [Google Scholar]
- 14.Ichinose M, Yahagi N, Oka M, Ikeda H, Miki K, Omata M. Screening for gastric cancer in Japan. In: Wu GY, Aziz K, editors. Cancer screening for common malignancies. Totowa, New Jersey: Humana Press; 2001. pp. 87–102. [Google Scholar]
- 15.Watanabe Y, Kurata JH, Mizuno S, Mukai M, Inokuchi H, Miki K, Ozasa K, Kawai K. Helicobacter pylori infection and gastric cancer. A nested case-control study in a rural area of Japan. Dig Dis Sci. 1997;42:1383–1387. doi: 10.1023/a:1018833819860. [DOI] [PubMed] [Google Scholar]
- 16.Miki K, Ichinose M, Ishikawa KB, Yahagi N, Matsushima M, Kakei N, Tsukada S, Kido M, Ishihama S, Shimizu Y. Clinical application of serum pepsinogen I and II levels for mass screening to detect gastric cancer. Jpn J Cancer Res. 1993;84:1086–1090. doi: 10.1111/j.1349-7006.1993.tb02805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kodoi A, Yoshihara M, Sumii K, Haruma K, Kajiyama G. Serum pepsinogen in screening for gastric cancer. J Gastroenterol. 1995;30:452–460. doi: 10.1007/BF02347560. [DOI] [PubMed] [Google Scholar]
- 18.Hattori Y, Tashiro H, Kawamoto T, Kodama Y. Sensitivity and specificity of mass screening for gastric cancer using the measurment of serum pepsinogens. Jpn J Cancer Res. 1995;86:1210–1215. doi: 10.1111/j.1349-7006.1995.tb03317.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoshihara M, Sumii K, Haruma K, Kiyohira K, Hattori N, Tanaka S, Kajiyama G, Shigenobu T. The usefulness of gastric mass screening using serum pepsinogen levels compared with photofluorography. Hiroshima J Med Sci. 1997;46:81–86. [PubMed] [Google Scholar]
- 20.Kitahara F, Kobayashi K, Sato T, Kojima Y, Araki T, Fujino MA. Accuracy of screening for gastric cancer using serum pepsinogen concentrations. Gut. 1999;44:693–697. doi: 10.1136/gut.44.5.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miki K, Morita M, Sasajima M, Hoshina R, Kanda E, Urita Y. Usefulness of gastric cancer screening using the serum pepsinogen test method. Am J Gastroenterol. 2003;98:735–739. doi: 10.1111/j.1572-0241.2003.07410.x. [DOI] [PubMed] [Google Scholar]
- 22.Ohata H, Oka M, Yanaoka K, Shimizu Y, Mukoubayashi C, Mugitani K, Iwane M, Nakamura H, Tamai H, Arii K, et al. Gastric cancer screening of a high-risk population in Japan using serum pepsinogen and barium digital radiography. Cancer Sci. 2005;96:713–720. doi: 10.1111/j.1349-7006.2005.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohata H, Kitauchi S, Yoshimura N, Mugitani K, Iwane M, Nakamura H, Yoshikawa A, Yanaoka K, Arii K, Tamai H, et al. Progression of chronic atrophic gastritis associated with Helicobacter pylori infection increases risk of gastric cancer. Int J Cancer. 2004;109:138–143. doi: 10.1002/ijc.11680. [DOI] [PubMed] [Google Scholar]
- 24.Yanaoka K, Oka M, Yoshimura N, Mukoubayashi C, Enomoto S, Iguchi M, Magari H, Utsunomiya H, Tamai H, Arii K, et al. Risk of gastric cancer in asymptomatic, middle-aged Japanese subjects based on serum pepsinogen and Helicobacter pylori antibody levels. Int J Cancer. 2008;123:917–926. doi: 10.1002/ijc.23571. [DOI] [PubMed] [Google Scholar]
- 25.Yanaoka K, Oka M, Mukoubayashi C, Yoshimura N, Enomoto S, Iguchi M, Magari H, Utsunomiya H, Tamai H, Arii K, et al. Cancer high-risk subjects identified by serum pepsinogen tests: outcomes after 10-year follow-up in asymptomatic middle-aged males. Cancer Epidemiol Biomarkers Prev. 2008;17:838–845. doi: 10.1158/1055-9965.EPI-07-2762. [DOI] [PubMed] [Google Scholar]
- 26.Dinis-Ribeiro M, Yamaki G, Miki K, Costa-Pereira A, Matsukawa M, Kurihara M. Meta-analysis on the validity of pepsinogen test for gastric carcinoma, dysplasia or chronic atrophic gastritis screening. J Med Screen. 2004;11:141–147. doi: 10.1258/0969141041732184. [DOI] [PubMed] [Google Scholar]
- 27.Yanaoka K, Oka M, Ohata H, Yoshimura N, Deguchi H, Mukoubayashi C, Enomoto S, Inoue I, Iguchi M, Maekita T, et al. Eradication of Helicobacter pylori prevents cancer development in subjects with mild gastric atrophy identified by serum pepsinogen levels. Int J Cancer. 2009;125:2697–2703. doi: 10.1002/ijc.24591. [DOI] [PubMed] [Google Scholar]
- 28.Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 29.Jones PA, Baylin SB. The epigenomics of cancer. Cell. 2007;128:683–692. doi: 10.1016/j.cell.2007.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ushijima T. Detection and interpretation of altered methylation patterns in cancer cells. Nat Rev Cancer. 2005;5:223–231. doi: 10.1038/nrc1571. [DOI] [PubMed] [Google Scholar]
- 31.Ushijima T, Asada K. Aberrant DNA methylation in contrast with mutations. Cancer Sci. 2010;101:300–305. doi: 10.1111/j.1349-7006.2009.01434.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaudet F, Hodgson JG, Eden A, Jackson-Grusby L, Dausman J, Gray JW, Leonhardt H, Jaenisch R. Induction of tumors in mice by genomic hypomethylation. Science. 2003;300:489–492. doi: 10.1126/science.1083558. [DOI] [PubMed] [Google Scholar]
- 33.Ushijima T, Sasako M. Focus on gastric cancer. Cancer Cell. 2004;5:121–125. doi: 10.1016/s1535-6108(04)00033-9. [DOI] [PubMed] [Google Scholar]
- 34.Maekita T, Nakazawa K, Mihara M, Nakajima T, Yanaoka K, Iguchi M, Arii K, Kaneda A, Tsukamoto T, Tatematsu M, et al. High levels of aberrant DNA methylation in Helicobacter pylori-infected gastric mucosae and its possible association with gastric cancer risk. Clin Cancer Res. 2006;12:989–995. doi: 10.1158/1078-0432.CCR-05-2096. [DOI] [PubMed] [Google Scholar]
- 35.Kaneda A, Kaminishi M, Yanagihara K, Sugimura T, Ushijima T. Identification of silencing of nine genes in human gastric cancers. Cancer Res. 2002;62:6645–6650. [PubMed] [Google Scholar]
- 36.Nakajima T, Maekita T, Oda I, Gotoda T, Yamamoto S, Umemura S, Ichinose M, Sugimura T, Ushijima T, Saito D. Higher methylation levels in gastric mucosae significantly correlate with higher risk of gastric cancers. Cancer Epidemiol Biomarkers Prev. 2006;15:2317–2321. doi: 10.1158/1055-9965.EPI-06-0436. [DOI] [PubMed] [Google Scholar]
- 37.Enomoto S, Maekita T, Tsukamoto T, Nakajima T, Nakazawa K, Tatematsu M, Ichinose M, Ushijima T. Lack of association between CpG island methylator phenotype in human gastric cancers and methylation in their background non-cancerous gastric mucosae. Cancer Sci. 2007;98:1853–1861. doi: 10.1111/j.1349-7006.2007.00625.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ando T, Yoshida T, Enomoto S, Asada K, Tatematsu M, Ichinose M, Sugiyama T, Ushijima T. DNA methylation of microRNA genes in gastric mucosae of gastric cancer patients: its possible involvement in the formation of epigenetic field defect. Int J Cancer. 2009;124:2367–2374. doi: 10.1002/ijc.24219. [DOI] [PubMed] [Google Scholar]
- 39.Nakajima T, Yamashita S, Maekita T, Niwa T, Nakazawa K, Ushijima T. The presence of a methylation fingerprint of Helicobacter pylori infection in human gastric mucosae. Int J Cancer. 2009;124:905–910. doi: 10.1002/ijc.24018. [DOI] [PubMed] [Google Scholar]
- 40.Takeshima H, Ushijima T. Methylation destiny: Moira takes account of histones and RNA polymerase II. Epigenetics. 2010;5:89–95. doi: 10.4161/epi.5.2.10774. [DOI] [PubMed] [Google Scholar]
- 41.Yoshida T, Yamashita S, Takamura-Enya T, Niwa T, Ando T, Enomoto S, Maekita T, Nakazawa K, Tatematsu M, Ichinose M, et al. Alu and Sata Hypomethylation in Helicobacter pylori-Infected Gastric Mucosae. Int J Cancer. 2010;45:37–44. doi: 10.1002/ijc.25534. [DOI] [PubMed] [Google Scholar]
- 42.Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, Taniyama K, Sasaki N, Schlemper RJ. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med. 2001;345:784–789. doi: 10.1056/NEJMoa001999. [DOI] [PubMed] [Google Scholar]
- 43.Niwa T, Tsukamoto T, Toyoda T, Mori A, Tanaka H, Maekita T, Ichinose M, Tatematsu M, Ushijima T. Inflammatory processes triggered by Helicobacter pylori infection cause aberrant DNA methylation in gastric epithelial cells. Cancer Res. 2010;70:1430–1440. doi: 10.1158/0008-5472.CAN-09-2755. [DOI] [PubMed] [Google Scholar]
- 44.Nakajima T, Enomoto S, Yamashita S, Ando T, Nakanishi Y, Nakazawa K, Oda I, Gotoda T, Ushijima T. Persistence of a component of DNA methylation in gastric mucosae after Helicobacter pylori eradication. J Gastroenterol. 2010;45:37–44. doi: 10.1007/s00535-009-0142-7. [DOI] [PubMed] [Google Scholar]
- 45.Ushijima T. Epigenetic field for cancerization. J Biochem Mol Biol. 2007;40:142–150. doi: 10.5483/bmbrep.2007.40.2.142. [DOI] [PubMed] [Google Scholar]
- 46.Nakajima T, Enomoto S, Ushijima T. DNA methylation: a marker for carcinogen exposure and cancer risk. Environ Health Prev Med. 2008;13:8–15. doi: 10.1007/s12199-007-0005-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ishii T, Murakami J, Notohara K, Cullings HM, Sasamoto H, Kambara T, Shirakawa Y, Naomoto Y, Ouchida M, Shimizu K, et al. Oesophageal squamous cell carcinoma may develop within a background of accumulating DNA methylation in normal and dysplastic mucosa. Gut. 2007;56:13–19. doi: 10.1136/gut.2005.089813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shen L, Kondo Y, Rosner GL, Xiao L, Hernandez NS, Vilaythong J, Houlihan PS, Krouse RS, Prasad AR, Einspahr JG, et al. MGMT promoter methylation and field defect in sporadic colorectal cancer. J Natl Cancer Inst. 2005;97:1330–1338. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 49.Kondo Y, Kanai Y, Sakamoto M, Mizokami M, Ueda R, Hirohashi S. Genetic instability and aberrant DNA methylation in chronic hepatitis and cirrhosis--A comprehensive study of loss of heterozygosity and microsatellite instability at 39 loci and DNA hypermethylation on 8 CpG islands in microdissected specimens from patients with hepatocellular carcinoma. Hepatology. 2000;32:970–979. doi: 10.1053/jhep.2000.19797. [DOI] [PubMed] [Google Scholar]
- 50.Arai E, Kanai Y, Ushijima S, Fujimoto H, Mukai K, Hirohashi S. Regional DNA hypermethylation and DNA methyltransferase (DNMT) 1 protein overexpression in both renal tumors and corresponding nontumorous renal tissues. Int J Cancer. 2006;119:288–296. doi: 10.1002/ijc.21807. [DOI] [PubMed] [Google Scholar]