

Abstract
BACKGROUND AND PURPOSE
Drugs used clinically usually have a primary mechanism of action, but additional effects on other biological targets can contribute to their effects. A potentially useful additional target is the endocannabinoid metabolizing enzyme monoacylglycerol lipase (MGL). We have screened a range of drugs for inhibition of MGL and compared the observed potencies using different MGL enzyme assays.
EXPERIMENTAL APPROACH
MGL activity was screened using recombinant human MGL (cell lysates and purified enzyme) with 4-nitrophenyl acetate (NPA) as substrate. 2-Oleolyglycerol metabolism by rat cerebellar cytosolic MGL and by recombinant MGL was also investigated.
KEY RESULTS
Among the 96 compounds screened in the NPA assay, troglitazone, CP55,940, N-arachidonoyl dopamine and AM404 inhibited NPA hydrolysis by the lysates with IC50 values of 1.1, 4.9, 0.78 and 3.1 µM, respectively. The potency for troglitazone is in the same range as its primary pharmacological activity, activation of peroxisome proliferator-activated receptor (PPAR) γ. Among PPARγ ligands, the potency order towards human MGL was troglitazone > ciglitazone > rosiglitazone > 15-deoxy-Δ12,14-prostaglandin J2≈ CAY 10415 > CAY 10514. In contrast to the time-dependent inhibitor JZL184, the potency of troglitazone was dependent upon the enzyme assay system used. Thus, troglitazone inhibited rat cytosolic 2-oleoylglycerol hydrolysis less potently (IC50 41 µM) than hydrolysis of NPA by the human MGL lysates.
CONCLUSIONS AND IMPLICATIONS
‘Hits’ in screening programmes for MGL inhibitors should be assessed in different MGL assays. Troglitazone may be a useful lead for the design of novel, dual action MGL inhibitors/PPARγ activators.
Keywords: monoacylglycerol lipase, cannabinoid, troglitazone, rosiglitazone, peroxisome proliferator-activated receptor γ
Introduction
The endocannabinoid system, comprising the endogenous cannabinoids (CB) anandamide (AEA, arachidonoyl ethanolamide) and 2-arachidonoylglycerol (2-AG), their target CB receptors, and their synthetic and degradative enzymes, are involved in a wide number of physiological processes in the body, ranging from cognition to bone metabolism (review, see Pacher et al., 2006). AEA is primarily metabolized by the enzyme fatty acid amide hydrolase (FAAH, also known as FAAH1, Alexander et al., 2009), and selective inhibitors of this enzyme such as URB597 (3′-(aminocarbonyl)[1,1′-biphenyl]-3-3-yl)-cyclohexylcarbamate, Kathuria et al. 2003) have shown potentially useful activity in models of pain, inflammation, anxiety and depression (see, e.g. Kathuria et al., 2003; Gobbi et al., 2005; Holt et al., 2005; Jayamanne et al., 2006). 2-AG is also a substrate for FAAH, cyclooxygenase-2, and the hydrolytic enzymes α/β-hydrolase domain (ABHD)12 and ABHD6, but in the mouse brain, the most important hydrolytic enzyme is monoacylglycerol lipase (MGL), which accounts for ∼85% of the hydrolysis of this substrate (Goparaju et al., 1998; Dinh et al., 2002; Blankman et al., 2007). JZL184 (4-nitrophenyl-4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate, Long et al., 2009a), a selective irreversible inhibitor of MGL, has been shown to produce anti-nociceptive effects in the chronic constriction injury model of neuropathic pain (Kinsey et al., 2009).
The wide range of effects produced by the endocannabinoid system in the body raises the possibility that many drugs that are, or have been, used clinically could interact with this system and that this interaction may contribute to their biological effects in humans over and above their primary pharmacological effect. Compounds directly activating CB1 receptors would presumably have been discovered by recreational drug abusers, but compounds with inhibitory effects upon FAAH and MGL are not likely to be of interest in this respect since they do not produce cannabis-like behaviours, unless both enzymes are inhibited by the compound in question (Kathuria et al., 2003; Long et al., 2009a,b;). There is evidence that non-steroidal anti-inflammatory drugs inhibit FAAH in vitro and that these drugs can influence endocannabinoid-mediated responses in vivo (Fowler et al., 1997; Gühring et al., 2002; Guindon et al., 2006; Naidu et al., 2009; Bishay et al., 2010). The intravenous anaesthetic propofol is also a FAAH inhibitor, a property that may contribute to its pharmacological effects in vivo (Patel et al., 2003).
It is not known, however, whether MGL is also inhibited by drugs that are either currently or previously in our therapeutic arsenal or used experimentally, for pharmacological properties other than MGL inhibition. One way of identifying such compounds is the use of an assay permitting the characterization of a large number of compounds. One such assay, whereby the hydrolysis of 4-nitrophenyl acetate (NPA) by recombinant MGL can be followed spectrophotometrically, has recently been reported (Muccioli et al., 2008). In the present study, we investigated a series of drugs with primary effects in therapeutic areas known to be sensitive to CB actions, such as non-steroidal anti-inflammatory drugs, antidepressants and drugs for the treatment of type 2 diabetes. Active compounds have been characterized further both in the NPA assay and in a standard radiochemical assay using 2-oleoylglycerol (2-OG) as substrate. We report that the peroxisome proliferator-activated receptor (PPAR) γ ligand troglitazone and the endogenous vanilloid receptor agonist, N-arachidonoyl dopamine, are relatively potent MGL inhibitors using the NPA assay, but that the potency of troglitazone is dependent upon the MGL assay used.
Methods
Spectrophotometric assay for MGL activity using recombinant human MGL
Assays were carried out in a 96-well microtiter plate (100 µL total volume) using a method based on the assay of Muccioli et al. (2008). Human recombinant MGL (either clear lysates or purified enzyme, as indicated) in 10 mM Tris–HCl, 1 mM EDTA, pH 7.4 (added volume 70 µL) was added to each well which also contained test compounds [3 µL in vehicle, DMSO except for N-arachidonoyl dopamine (ethanol), 15-deoxy-Δ12,14-prostaglandin J2 and CAY 10541 (methyl acetate)] or vehicle alone. Buffer (7 µL) was added to each well. To start the hydrolysis, NPA (20 µL, final concentration of 0.25 mM, unless otherwise shown) was added rapidly. Blanks contained buffer alone. The concentration of MGL was chosen to ensure that initial velocities were measured. The samples were incubated at room temperature. The absorbance was measured at 405 nm after 0 min (to rule out effects of the compounds per se on the absorbance) and at least two 20 min intervals, thereafter using a Thermomax Microplate Reader (ThermoMax Kinetic Microplate Reader, Molecular Devices, Sunnyvale, CA, USA). For the experiments reported here, the 20 min time point was used to ensure that initial activities were measured but at the same time allow for sufficient product formation. In a typical experiment, the OD values for the blanks were ∼0.045 (with very little variation between assays), while controls were two- to threefold higher than blanks (with a small intra-assay variation). Measurements at 40 min were also made to confirm the inhibition at a higher control : blank ratio.
Radiochemical assay for 2-OG hydrolysis by rat brain extracts
All animal care and experimental procedures complied with national guidelines and laws and were approved by the local animal ethical committee. Assays were carried out essentially as described in Ghafouri et al. (2004), but with a charcoal separation (Boldrup et al., 2004) in place of a chloroform : methanol extraction. Briefly, cerebella from adult Wistar or Sprague-Dawley rats that had been obtained previously and stored frozen at −80°C were thawed and homogenized in 0.32 M sucrose containing 50 mM sodium phosphate, pH 8. Homogenates were centrifuged at 100 000 g for 60 min at 4°C and the supernatants (‘cytosolic fractions’) were collected and stored at −80°C in aliquots until used for assay. Protein concentrations for the assays were 3 µg/assay, the fractions being diluted with 10 mM Tris–HCl, 1 mM EDTA, pH 7.4.
The radiochemical assays contained enzyme source (cytosol or recombinant MGL), 10 mM Tris–HCl, 1 mM EDTA and test compound (10 µL, in vehicle [DMSO for troglitazone and JZL184; ethanol for N-arachidonoyl dopamine) in a volume of 175 µL. After pre-incubation, when appropriate, 25 µL of a solution of [3H]2-OG (4 µM, to give an assay concentration of 0.5 µM) in 10 mM Tris buffer, pH 7.4 containing 1% (w v−1) fatty acid-free bovine serum albumin (BSA, assay concentration 0.125% w v−1) was added and the samples were incubated for 10 min at 37°C. Thereafter, reactions were stopped by the addition of 400 µL of active charcoal mixture (80 µL charcoal + 320 µL 0.5 M HCl). After vortex mixing and phase separation, aliquots of the aqueous phase were counted for tritium content by liquid scintillation spectroscopy with quench correction. Blanks were prepared in the same manner but without enzyme source. FAAH assays with JZL184 were carried out using homogenates of whole brain (minus cerebella, 0.6 µg protein per assay) from adult Wistar or Sprague-Dawley rats using the method of Boldrup et al. (2004) and [3H]AEA (labelled in the ethanolamine part of the molecule, final concentration 0.5 µM) as substrate.
Analysis of data
Kmapp, Vmaxapp, pI50 and IC50, values, linear regressions and confidence limits were determined using the GraphPad Prism computer programme (GraphPad Software Inc., San Diego, CA, USA). The pI50, and thereby IC50 values, were calculated using the built-in programme ‘sigmoidal dose–response (variable slope)’ from the data expressed as % of vehicle controls using top (i.e. uninhibited) values of 100% and bottom (residual activity) values that were either set to zero or allowed to float. The two curves were compared using Akaike's informative criteria and the preferred model thereafter used.
Compounds
2-Oleoylglyceerol [glycerol – 1,2,3-3H] (1.48 TBq.mmol−1) and AEA [ethanolamine-1-3H] (specific activity 2.22 TBq.mmol−1) were purchased from American Radiolabeled Chemicals, Inc (St. Louis, MO, USA). Human recombinant His-tagged MGL expressed in Escherichia coli (hMGL), either as clear lysates, catalogue nos. #10008354 and #705194; (according to the manufacturers, the two lysate preparations are the same enzyme preparation, but are used as part of different enzyme assay kits. The #10008354 is now discontinued. We have indicated the catalogue number of the lysate used in the Figure legends) or as the enzyme that had been further purified using a nickel column, catalogue no. #10007812; non-radiolabelled AEA, troglitazone, rosiglitazone, ciglitazone, 15-deoxy-Δ12,14-prostaglandin J2, CAY10415 (5-[[4-[2-(5-ethyl-2-pyridinyl)-2-oxoethoxy]phenyl]methyl]-2,4-thiazolidinedione, compound 10 of Tanis et al. 1996), CAY10514 (8-hydroxy-8-[2-(pentyloxy)phenyl]-5-octynoic acid, methyl ester, Compound 14a of Caijo et al. 2005), JZL184 (4-nitrophenyl-4-(dibenzo[d][1,3]dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate) and N-arachidonoyl dopamine were obtained from the (Cayman Chemical Co., Ann Arbor, MI, USA). AM404 (N-(4-hydroxyphenyl)-5Z,8Z,11Z,14Z-eicosatetraenamide), CP 55 940 ((-)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol), JWH015 ((2-methyl-1-propyl-1H-indol-3-yl)-1-naphthalenylmethanone) and WIN55,212-2 ((R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate) were obtained from Tocris Bioscience (Ellisville, MO, USA). NPA, fatty acid-free BSA, 4-nitrophenol and non-radioactive 2-OG were obtained from the Sigma Chemical Co., St. Louis, MO, USA.
Results
Preliminary screen of selected compounds for inhibition of MGL using the NPA assay
The NPA assay was used with the clear lysates of recombinant human His-tagged MGL expressed in E. coli (‘hMGL lysate’) as enzyme source. Initial experiments indicated that the OD405 for the NPA hydrolysis product, 4-nitrophenol, was linear over the range 0.01–0.15 mM (r2 = 0.998, seven concentrations tested) and that the OD405 measured when the hMGL lysates were incubated with NPA showed the appropriate dependency upon the incubation time and enzyme concentration (data not shown). Over a range of NPA concentrations (0.08–0.64 mM), the hydrolysis produced by the hMGL lysates was saturable, with a Km value of 0.23 mM (data not shown), consistent with the study of Muccioli et al. (2008) who used a different hMGL preparation. Finally, the rate of NPA hydrolysis showed the expected pH optimum (∼7), consistent with the literature (Di Marzo et al., 1999; Goparaju et al., 1999), and was reduced by the alternative substrates, 2-AG and 2-OG, but not by AEA, which is not a substrate for this enzyme (Dinh et al., 2002) (data not shown).
A series of 96 compounds available at the department were tested at two concentrations, 3 and 10 µM. The compounds tested included a number of agents known to interact with the CB system, non-steroidal anti-inflammatory agents, compounds with different actions upon targets in the brain (such as, for example, antipsychotic and antidepressant drugs) and some naturally occurring compounds. The initial screen was carried out with single assays, albeit with separate controls for the 3 µM and 10 µM concentrations, to allow the identification of ‘hits’, and the values should be considered in this light. Four compounds were found to produce more than 60% inhibition at concentrations of both 3 and 10 µM: the CB receptor agonist CP55,940, the endogenous transient receptor potential vanilloid 1 (TRPV1) agonist N-arachidonoyl dopamine, the FAAH inhibitor and TRPV1 antagonist, N-arachidonoyl serotonin, and the PPARγ ligand, troglitazone (see Table 1 for a selection of the compounds). Three compounds produced >60% inhibition at 10 but not 3 µM concentrations: the AEA uptake inhibitor AM404, the lipase (including diacylglycerol lipase) inhibitor tetrahydrolipstatin, and the CB2 receptor-selective agonist JWH015. Among other compounds tested, it was noted that in some cases, the compounds increased the observed rate of NPA hydrolysis, a case in point being JWH133 (see Table 1). The high value for JWH133 was not due to assay interference, as this compound, at the concentrations tested, did not affect the OD at the 0 min time point. In a repeat experiment, activation was also seen when the compound was pre-incubated with the enzyme for 30 or 60 min prior to addition of substrate, but the degree of activation was very variable, and it was not seen in the absence of a pre-incubation phase (see legend to Table 1). This property of the compound was not studied further, given its limited robustness and given that the concentrations used are much higher than needed to interact with CB2 receptors (Huffman et al., 1999), but it may reflect some form of allosteric action on the enzyme. The raw data for the compounds corresponding to the major groups described above are given for reference purposes in Table 1.
Table 1.
Effects of 76 compounds with different biological actions upon the activity of human recombinant MGL (lysate assay, 0.25 mM NPA as substrate)
| MGL activity (% of control) | |||
|---|---|---|---|
| Compound | Primary pharmacological action | 3 µM | 10 µM |
| CB/TRPV1 receptor ligands and related compounds | |||
| ACEA | CB1 receptor agonist | 126 | 81 |
| AM251 | CB1 receptor inverse agonist | 69 | 62 |
| AM404** | Blocks endocannabinoid reuptake | 63 | 18 |
| AM630 | CB2 receptor inverse agonist | 90 | 55 |
| N-arachidonoyl serotonin | FAAH and TRPV1 inhibitor | 37 | 32 |
| N-arachidonoyl dopamine** | Endogenous TRPV1 ligand | 15 | −6 |
| Capsazepine | TRPV1 antagonist | 90 | 63 |
| CP 55,940** | Non-selective CB receptor agonist | 28 | 26 |
| JWH015** | CB2 receptor agonist | 53 | 37 |
| JWH133a | CB2 receptor agonist | 172 | 170 |
| (±)-2-Methylarachidonyl-2′-fluoroethylamide | CB1 receptor agonist | 89 | 99 |
| Resveratrol | Antioxidant, CB1 receptor ligand | 101 | 86 |
| RHC-80267 | DAG lipase inhibitor | 102 | 71 |
| (±)SLV 319 | CB1 receptor inverse agonist | 99 | 79 |
| Tetrahydrolipstatin | Lipase inhibitor (incl. DAG lipase) | 95 | 37 |
| Win 55,212-2 mesylate** | Non-selective CB receptor agonist | 67 | 25 |
| Non-steroidal anti-inflammatory drugs | |||
| Acetylsalicylic acid*, diclofenac, dipyrone, fenbufen*, fenoprofen, flufenamic acid, ketoprofen*, meloxicam*, nabumetone, niflumic acid, nimesulide, sulindac, suprofen | Inhibition of cyclooxygenase | range 76–147 | range 95–161 |
| Antidepressants | |||
| Amitriptyline, clomipramine, desipramine, imipramine, nortriptyline, maprotiline | Monoamine reuptake inhibitors | range 82–120 | range 78–122 |
| Phenelzine | Monoamine oxidase inhibitor | ||
| Other CNS-active drugs and related compounds | |||
| Haloperidol, cis-(Z)-flupenthixol | Dopamine receptor antagonists | range 86–116 | range 72–116 |
| Spiperone* | DA/5-HT1A receptor antagonists | ||
| Ketanserin, pirenperone* | 5-HT2 receptor antagonists | ||
| Promethazine | Histamine H1 receptor antagonist | ||
| Diazepam, flurazepam, temazepam | Benzodiazepines | ||
| Sodium barbital | Barbiturate | ||
| Nipecotic acid | GABA reuptake inhibitor | ||
| (+)MK801 | NMDA receptor antagonist | ||
| Naloxone | Opioid receptor antagonist | ||
| Physostigmine sulphate | Acetylcholinesterase inhibitor | ||
| Other drugs and related compounds | |||
| Lidocaine, tetracaine | Na+-channel blockers | range 87–170 | range 90–114 |
| Verapamil | L-type Ca2+ channel blocker | ||
| Lovastatin | HMG-CoA reductase inhibitor | ||
| S(−)Atenolol, R(+)atenolol | ß-Adrenoceptor antagonists (S>R) | ||
| Captopril | ACE inhibitor | ||
| Theophylline, 8-phenyltheophylline | Adenosine receptor antagonists | ||
| Cromolyn sodium | ‘Mast cell stabiliser’ | ||
| Progesterone | Sex steroid hormone | ||
| Troglitazone** | PPARγ ligand | 21 | 2 |
| Naturally occurring, biologically active compounds | |||
| Coumestrol | Non-steroidal phytoestrogen | range 74–99 | range 45–143 |
| Baicalein, fisetin, luteolin, morin, phloretin, quercetin | Flavonoids; antioxidant and other properties | ||
| Other compounds (a selection) | |||
| 3-Hydroxytyramine | Range 92–126 | range 76–103 | |
| Benzamide | |||
| Hydrocinnamic acid | |||
| N-Acetyl-D-sphingosine | |||
| 8-Methyl-N-vanillyl-6-noneamide | |||
| Thimerosal | 61 | 45 | |
Data are for single experiments, albeit run on different plates for the 3 µM and the 10 µM test concentrations, with ‘hits’ (>60% inhibition at the test concentration shown) given in bold text. Selected compounds (marked with asterisks) were retested at a later date at a concentration of either 10 µM (inactive compounds, *) or over a concentration range (**, compounds shown in Figure 1A) to confirm the reliability, but the values shown here are from the initial experiments.
JWH133 was also retested at a later stage, and the % of control activities seen after 0, 30 and 60 min pre-incubation, respectively, were: 3 µM, 100 ± 17, 129 ± 17 and 133 ± 25; 10 µM, 98 ± 20, 146 ± 8 and 159 ± 21 (means ± SEM, n = 3).
Inhibition of MGL by troglitazone and the other ‘hits’ in the primary screen
Six of the ‘hits’ described above were characterized further using hMGL lysates and NPA as substrate. Concentration–response curves for troglitazone, CP55,940, N-arachidonoyl dopamine and AM404, over the range 0.1–10 µM were undertaken and pI50 values (IC50 values in brackets) of 5.95 ± 0.04 (1.1 µM), 5.31 ± 0.07 (4.9 µM), 6.11 ± 0.08 (0.78 µM) and 5.51 ± 0.09 (3.1 µM), respectively, were found (Figure 1A). For WIN55,212-2 and JWH015, there was a large variability at the highest concentration tested, presumably related to the solubility of the compounds, precluding accurate determinations of their IC50 values. The inhibition of MGL by troglitazone was compared with that for other PPARγ ligands (Figure 1B; note that the datasets for troglitazone in the two panels of Figure 1 are different, but show very similar potencies), and the potency order was found to be troglitazone > ciglitazone > rosiglitazone > 15-deoxy-Δ12,14-prostaglandin J2≈ CAY 10415 > CAY 10514 (Table 2). This differs considerably from their potency order as inhibitors of FAAH or as activators of PPARγ (see Table 2).
Figure 1.
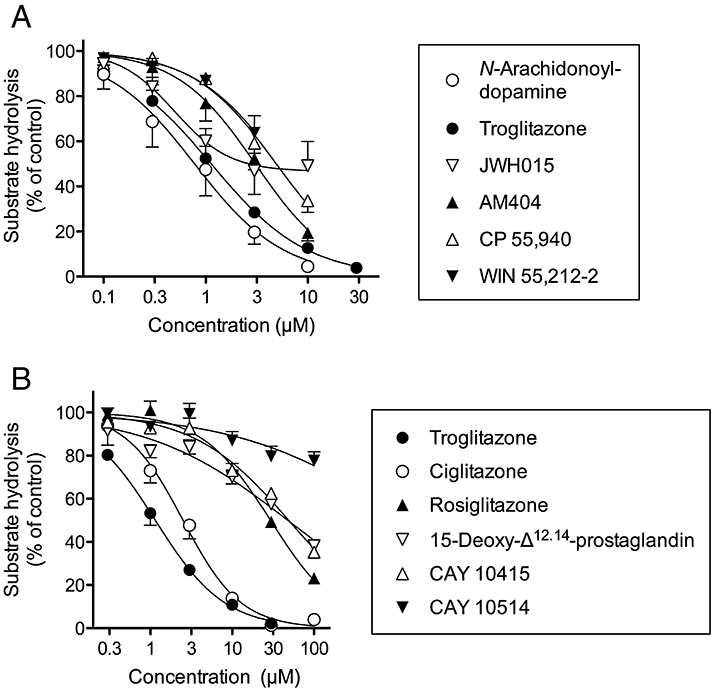
Inhibition of the hydrolysis of 4-nitrophenyl acetate by human recombinant monoacylglycerol lipase lysates by (A) ‘hits’ from the initial screen of a small compound library and (B) peroxisome proliferator-activated receptor (PPAR) γ agonists. Shown are means and SEM (when not enclosed by the symbols), n = 3–4. The catalogue numbers of the lysates were for Panel A, #705194 and for Panel B 10008354.
Table 2.
Inhibition of NPA hydrolysis in hMGL lysates by PPARγ ligands
| Compound | Structure | hMGL pI50[IC50 (µM)] | rat brain FAAH IC50 (µM) | PPARγ EC50 (µM) (h/m) |
|---|---|---|---|---|
| Troglitazone | 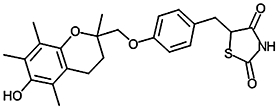 |
5.94 ± 0.03 (1.2) | ∼200 µMa | 0.55/0.78b |
| Ciglitazone | 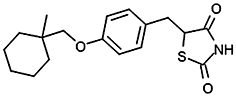 |
5.60 ± 0.05 (2.5) | 84 µMa | −/3c |
| Rosiglitazone | 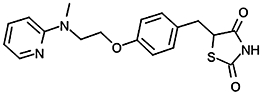 |
4.53 ± 0.04 (29) | ∼200 µMa | 0.043/0.076b |
| CAY 10415 | 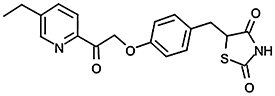 |
4.31 ± 0.04 (49) | ||
| 15-deoxy-Δ12,14-prostaglandin J2 | 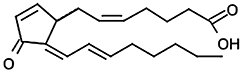 |
4.32 ± 0.06 (48) | 87 µMa | −/2d |
| CAY 10514 | 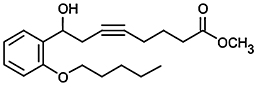 |
<4 (>100) | 0.64/−e |
The pI50 values and hence IC50 values were calculated from the raw data shown in Figure 1B, using hMGL lysates (catalogue no. #10008354) and 0.25 mM NPA.
The FAAH data are for rat brain homogenates, 0.5 µM [3H]AEA as substrate, and an assay pH of 7.4, and are taken from Lenman et al. (2007). The PPARγ values for troglitazone, ciglitazone, rosiglitazone, 5-deoxy-Δ12,14-prostaglandin J2 and CAY10514 are for transactivation assays using human (h) or mouse (m) chimeric receptors and are from
Note that CAY10514 showed a low efficacy compared with rosiglitazone, which in the same study reported to have an EC50 value of 0.004 µM. Comparable data are not (to our knowledge) reported for CAY10415. However, this compound increases insulin-stimulated inccorporation of [14C]acetate into lipid in differentiated mouse 3T3-L1 cells at 3–10 µM concentrations (Tanis et al., 1996).
The mode of inhibition by troglitazone of NPA hydrolysis by hMGL lysates was investigated. The inhibition showed no time dependency (Figure 2A), but dilution experiments suggested that it was not rapidly reversible. In the latter experiments, troglitazone was incubated with the human MGL lysates (catalogue no. #705194) for 60 min at room temperature prior to addition of substrate and either a fivefold dilution or no dilution prior assay for activity. For a fully reversible compound, the inhibitory potency at, say, 10 µM prior to dilution should be the same as that seen for an undiluted sample using 2 µM inhibitor, whereas for an irreversible or tight-binding compound, the inhibitory potency should resemble that seen for an undiluted sample using 10 µM inhibitor. For the irreversible compound JZL184, used as a positive control, the % of vehicle control activity (means ± SEM, n = 3) seen after pre-incubation with 50 nM compound followed by fivefold dilution was 7 ± 0.8, which was similar to that seen with the undiluted samples incubated with 50 nM compound (5 ± 1) and much lower than seen with the undiluted samples incubated with 10 nM JZL184 (43 ± 3). A similar pattern was seen with troglitazone: the % of control activity (means ± SEM, n = 3) seen after pre-incubation with 10 µM compound followed by fivefold dilution was 34 ± 3, which was similar to that seen with the undiluted samples incubated with 10 µM compound (33 ± 2) and much lower than seen with the undiluted samples incubated with 2 µM troglitazone (77 ± 4).
Figure 2.
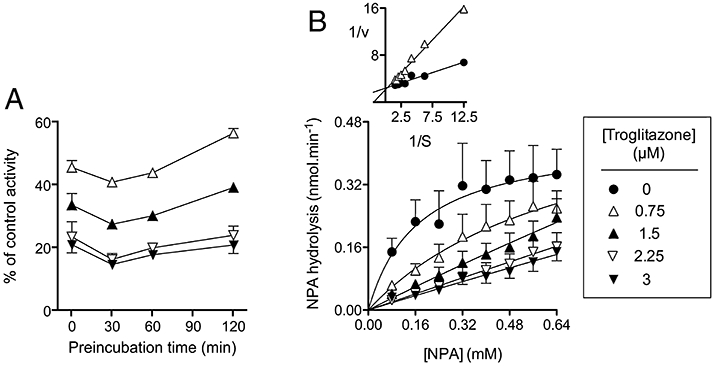
Mode of inhibition by troglitazone of 4-nitrophenyl acetate (NPA) hydrolysis by human monoacylglycerol lipase (MGL) lysates (catalogue number #10008354). In Panel A, the lysates were pre-incubated with troglitazone for the times shown at room temperature prior to addition of NPA (0.25 mM) and assay for activity. In Panel B, no pre-incubation phase was used. Shown are means and SEM (when not enclosed by the symbols), n = 3, except for the data in Panel B with 0.32 mM NPA, where n = 2. The inset in Panel B shows a double reciprocal plot of the mean data with 0 and 0.75 µM troglitazone, to illustrate the competitive nature of the inhibition.
The saturation curves for NPA incubated with troglitazone (no pre-incubation phase) are shown in Figure 2B. The Kmapp values calculated from the mean data with 0 and 0.75 µM troglitazone were 0.17 and 0.70 mM, respectively. The corresponding Vmaxapp values (calculated using a post hoc calibration curve for 4-nitrophenol) were 0.44 and 0.57 nmol substrate hydrolysed per minute and assay tube, respectively, each assay tube containing 0.01 µL of the lysate preparation. At higher troglitazone concentrations, the Kmapp values were greater than the highest substrate concentration tested, and so could not be determined with accuracy. A slope re-plot with 0 and 0.75 µM troglitazone gave an approximate Ki value of 0.34 µM. This is in good agreement with the value of Ki 0.45 calculated from the IC50 value of 1.1 µM (Table 2) assuming a competitive interaction and a Km value of 0.17 mM.
Assay dependency of the inhibition of MGL by troglitazone, N-arachidonoyl dopamine and JZL184
In order to investigate whether the inhibition of MGL by the compounds was assay, substrate and/or species dependent, two assay systems were compared: (i) NPA as substrate, hMGL lysates or purified hMGL as enzyme sources, assays run at room temperature; (ii) 2-OG as substrate, rat cytosol, hMGL lysates or purified hMGL as enzyme sources; 0.125% w·v−1 fatty acid-free BSA present, assays run at 37°C.
In order to establish the degree of assay variation for a well-characterized compound, we used the selective irreversible MGL inhibitor JZL184 (Figure 3). In all assays, the compound inhibited the substrate hydrolysis in a time-dependent manner, consistent with its irreversible mode of action (Long et al., 2009a). JZL184 inhibited NPA hydrolysis by the hMGL lysates with pI50 values of 7.17 ± 0.04 (IC50 value 68 nM) and 8.33 ± 0.04 (IC50 value 4.7 nM) following 0 and 60 min of pre-incubation, respectively. When 2-OG was used as substrate, the potency was about fourfold lower (Figure 3A). In their original study, Long et al. (2009a) used recombinant mouse MGL with 100 µM 2-AG as substrate and reported IC50 values of 6 nM, following a 30 min pre-incubation period. For the 2-OG assays, the control activity was affected by the pre-incubation phase; thus, the rate of hydrolysis of 2-OG by the pre-incubated lysates was 36 ± 0.8% of the corresponding activity seen for the samples that were not pre-incubated. This was even more apparent when the purified hMGL was used: the rate of hydrolysis of the pre-incubated control lysates was only 14 ± 3% of that seen in the absence of pre-incubation. A reduction in activity was also seen with pre-incubation in the NPA assays, and the reduction was more variable. However, the remaining activity after the pre-incubation phase was sensitive to inhibition by JZL184, with pI50 values of 7.78 ± 0.12 (IC50 value 17 nM) and 8.02 ± 0.08 (IC50 value 9.6 nM) being found for NPA and 2-OG as substrates, respectively (Figure 3B). Finally, JZL184 inhibited rat cytosol 2-OG hydrolysis with pI50 values of 6.46 ± 0.04 (IC50 value 350 nM), 7.91 ± 0.10 (IC50 value 12 nM, residual activity 16 ± 6%) and 8.24 ± 0.02 (IC50 value 5.8 nM, residual activity 9 ± 1%) following 0, 30 and 60 min pre-incubation times, respectively (Figure 3C). In a single experiment, the effect of JZL184 upon 2-OG hydrolysis by the hMGL lysates was investigated at room temperature, rather than at 37°C. Approximate IC50 values of 850 and 14 nM were found following pre-incubation times of 0 and 60 min, respectively, in line with the data at 37°C (data not shown). Taken together, the results show there is a time dependency in the sensitivity of MGL to JZL184, but the potencies for a given pre-incubation time are reasonably consistent in the different assays.
Figure 3.
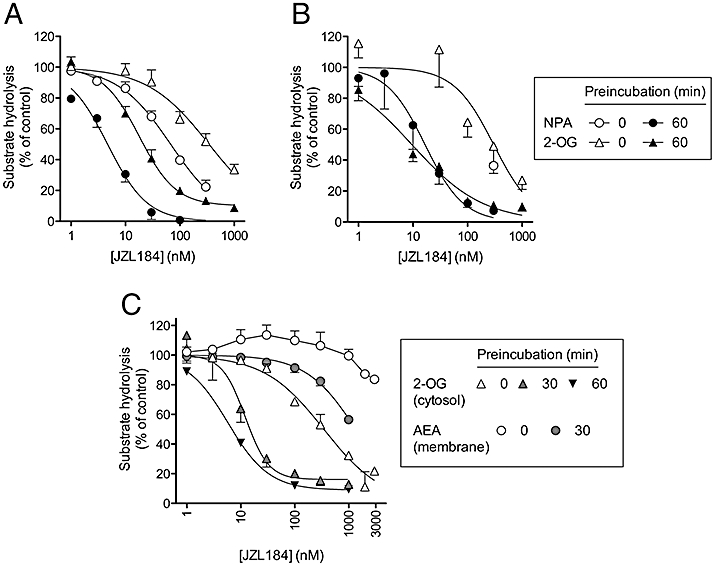
Inhibition by JZL184 of the hydrolysis of 4-nitrophenyl acetate (NPA) or 2-oleoylglycerol (2-OG) by A, human monoacylglycerol lipase (MGL) lysates (catalogue no. #705194); B, purified human MGL; C, rat cerebellar cytosol fractions. Shown are means and SEM (when not enclosed by the symbols), n = 3–9. For comparative purposes, the effect of the compound upon the hydrolysis of anandamide (AEA) (0.5 µM) by rat brain membrane preparations is shown to illustrate the MGL-selectivity of the compound.
In the absence of a pre-incubation phase, N-arachidonoyl dopamine inhibited the hydrolysis by the hMGL lysates of NPA and 2-OG with pI50 values of 6.11 ± 0.08 (IC50 value 0.78) and 5.66 ± 0.03 (IC50 value 2.2 µM), respectively), while the compound was less potent as an inhibitor of the hydrolysis of 2-OG by rat cytosol (pI50 value 4.70 ± 0.04, IC50 value 20 µM) (Figure 4A).
Figure 4.
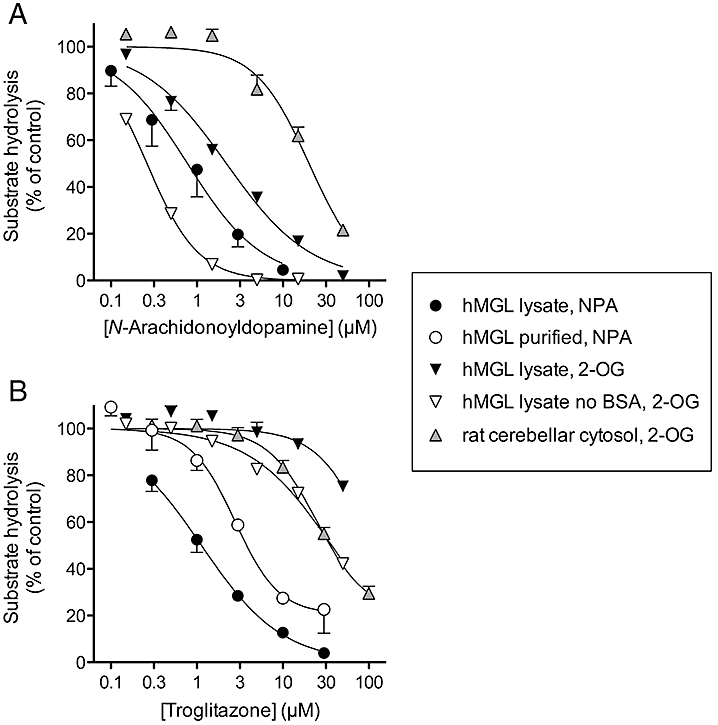
The potencies of N-arachidonoyl dopamine (A) and troglitazone (B) towards monoacylglycerol lipase (MGL) in different assays. The compounds were not pre-incubated with the enzyme preparations prior to addition of substrate. Shown are means and SEM (when not enclosed by the symbols), n = 3–6. The catalogue number of the lysates was #705194. Note that the curves for the human MGL lysate with NPA as substrate are the same as shown in Figure 1A for the two compounds.
For troglitazone (no pre-incubation), there was a large variation in the inter-assay potencies. With NPA as substrate, the compound inhibited the purified hMGL with a threefold lower potency than seen for the lysate, and a pI50 value of 4.61 ± 0.07 (IC50 value 25 µM, residual enzyme activity 21 ± 7%) was seen for rat cytosols with 2-OG as substrate (Figure 4B). Rosiglitazone was also less potent as an inhibitor of 2-OG hydrolysis by the cytosols than of NPA hydrolysis by hMGL lysates, producing 31 ± 1% inhibition at the highest concentration tested (100 µM) for the cytosols (data not shown) as compared to an IC50 value of 29 µM for the hMGL lysates (Table 2). Most surprisingly, troglitazone was a very poor inhibitor of the hMGL lysate when assayed radiochemically with 2-OG as substrate (Figure 4B). This was not due to the assay temperature used, as a single experiment with 2-OG carried out at room temperature gave an IC50 value of approximately 62 µM for the inhibition by troglitazone of the hMGL lysate-catalysed hydrolysis of 2-OG (data not shown).
Three possible reasons for the discrepancy in potencies seen for troglitazone in the different assays using the same enzyme source were investigated:
The effect of troglitazone upon NPA hydrolysis is an artefact of the assay. For all compounds, the initial reading at t = 0 min was made to ensure that the compounds did not produce a signal at the OD used, but it is possible that troglitazone can quench the signal produced by NPA hydrolysis in the samples. However, this would have been expected to affect equally the signal seen in assays with the lysates and the purified enzyme, and would not have been likely to produce the effects it did in the dilution experiments or to give a competitive type of inhibition. Nonetheless, the possibility was checked by adding troglitazone to assay mixtures containing the lysate and NPA after a 20 min incubation period and reading the plate immediately and 2 min later. Over the concentration range tested (0.1–30 µM), troglitazone was without any effect on the observed absorbance at 405 nM (data not shown), indicating that effect of troglitazone is not due to quenching of the 4-nitrophenol signal.
The manner in which the substrate is presented to the enzyme may be of importance, given the difference in lipophilicity between 2-OG and NPA. In order to investigate this possibility, the inhibitory potency of troglitazone was investigated when 2-OG was emulsified using Triton X-100 and compared to that in the standard BSA-containing assay. However, preliminary experiments indicated that the enzyme activity towards 2-OG in the presence of Triton X-100 was very much lower than seen in the standard BSA-containing assay (∼12% for both MGL lysates and for rat cytosolic preparations using a Triton X-100 concentration of 5 mM to emulsify the substrate, final assay concentration 0.625 mM, that is, 0.04% w v−1), and in the cytosolic preparations, there was no obvious difference in the sensitivity to troglitazone (data not shown).
Differences in fatty acid-free BSA concentration in the two assays affect the observed inhibitory potency of troglitazone. The presence of BSA is likely to reduce the free troglitazone concentration given its propensity to bind to albumin (>99.9% binding to human serum albumin, Shibukawa et al., 1995), and this may result in a reduced observed potency. In order to investigate this possibility, NPA assays were undertaken in the presence of different concentrations of fatty acid-free BSA (0–0.25% w v−1) and with N-arachidonoyl dopamine and troglitazone as test compounds. The experiments were complicated by the fact that serum albumins have esterase activity (see Salvi et al., 1997), and this can be clearly seen in the blank values obtained at each fatty acid-free BSA concentration (Figure 5A and B). At the high BSA concentrations, the blank values are so high relative to the MGL activity that accurate determination of inhibitory potencies is not possible. However, the difference between the blank and enzyme-containing controls is approximately the same at 0 and 0.125% w·v−1 BSA, and in these cases, the inhibition produced by 8 µM N-arachidonoyl dopamine and 10 µM troglitazone, appeared to be greater in the absence than in the presence of 0.125% w·v−1 fatty acid-free BSA. Experiments were also undertaken using the hMGL lysates and 2-OG in the radiochemical assay, but lacking fatty acid-free BSA. In these experiments, N-arachidonoyl dopamine was a very potent inhibitor of the 2-OG hydrolysis (pI50 value 6.58 ± 0.02, IC50 0.26 µM), that is, greater inhibition than seen in the presence of BSA (Figure 4A), although as a caveat it was noted that the vehicle used (ethanol) reduced the enzyme activity, compared to a DMSO vehicle. Troglitazone was also a better inhibitor of 2-OG hydrolysis by the lysates in the absence of BSA (pI50 4.43 ± 0.04, IC50 37 µM), although this value is still lower than seen with the lysates in the NPA assay (Figure 4B).
Figure 5.
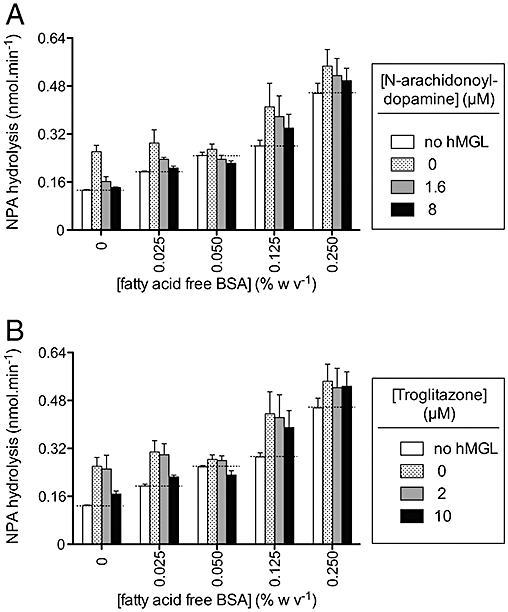
Effect of fatty acid-free bovine serum albumin (BSA) upon the rate of hydrolysis of 4-nitrophenyl acetate (NPA) and its inhibition by (A) N-arachidonoyl dopamine, (B) troglitazone. The compounds were not pre-incubated with the enzyme preparations prior to addition of substrate. Note that in the figure, the blank values (containing the same concentration of fatty acid-free bovine serum albumin, but no hMGL lysate) are not subtracted. Shown are means and SEM., n = 3. The catalogue number of the lysates was #705194.
Discussion
In the present study, we used a high-capacity assay to investigate a series of compounds with respect to their MGL inhibitory properties, and characterized the two most potent in the screen with respect to their inter-assay variability. The screen identified several compounds which inhibited NPA hydrolysis by the hMGL lysates in the low micromolar range. With the exception of troglitazone (see below), the compounds were either arachidonoyl analogues (AM404, N-arachidonoyl dopamine) or compounds sharing the ability of 2-AG to interact with CB receptors (CP 55 940; JWH015; WIN 55,212-2). With respect to CP 55,940, JWH015 and WIN 55,212-2, the potencies towards the hMGL in the lysates are lower than the nanomolar concentrations required to interact with CB receptors (see McPartland et al., 2007), and so can best be described as ‘off-target’ actions of these compounds unlikely to contribute to their pharmacological effects. The same is probably true for N-arachidonoyl dopamine, which is a potent TRPV1 receptor agonist, although the IC50 value seen here for human MGL is similar to that seen for the interaction of the compound with CB1 receptors (Bisogno et al., 2000; Chu et al., 2003). The potency of AM404 towards hMGL in the lysates is similar to its potency as an inhibitor of endocannabinoid uptake (Beltramo et al., 1997), and so its effects upon MGL may contribute to the pharmacological actions of this compound in vivo. AM404 also inhibits the hydrolysis of 2-OG by rat cerebellar cytosolic fractions (IC50 value of ∼20 µM, Vandevoorde and Fowler, 2005).
Two other compounds, N-arachidonoyl serotonin and tetrahydrolipstatin, were found to be ‘hits’ in the original screen, but were not investigated further. Tetrahydrolipstatin was also evaluated by Muccioli et al. (2008) using the NPA assay and a different hMGL preparation. They found it to be quite a potent inhibitor of hMGL, but not to produce more than ∼50% inhibition of the enzyme, even at high (100 µM) concentrations. However, it had an IC50 value >10 µM in cytosolic fractions from monkey COS-7 cells when MGL was assayed with 2-AG as substrate (Ortar et al., 2008). N-arachidonoyl serotonin was reported not to inhibit the hydrolysis of 2-AG by rat cerebellar membranes (Saario et al., 2004), so it is possible that the effect seen here may be dependent upon the species and/or assay used. Muccioli et al. (2008) also tested a series of non-steroidal anti-inflammatory agents (including ibuprofen, naproxen, flubiprofen and diclofenac sodium), and found <50% (100 µM) and ≤33% (10 µM) inhibition in all cases. This lack of effect of this class of drugs upon hMGL is consistent with our data (see Table 1). Finally, the organic mercurial compound thimerosal produced 55% inhibition at 10 µM in our screen, a finding consistent with the ability of other organic mercurials to inhibit MGL activity (see Tornqvist and Belfrage, 1976; Chau and Tai, 1988; Tarzia et al., 2007).
The finding that the PPARγ agonist troglitazone inhibits NPA hydrolysis by the hMGL lysates extends previous work showing that PPARγ agonists can inhibit FAAH (Lenman et al., 2007), and that endocannabinoids and related biological lipids can interact with PPAR subtypes (see O'Sullivan, 2007). PPARγ is a ligand-activated nuclear receptor, and the PPARγ ligands rosiglitazone and pioglitazone are currently used for the treatment of type 2 diabetes. For the thiazolidinedione compounds tested here, the rank order of potencies for inhibition of MGL (troglitazone > ciglitazone > rosiglitazone) was different from that seen for either inhibition of FAAH (ciglitazone > rosiglitazone ≈ troglitazone, Lenman et al., 2007) or activation of PPARγ (rosiglitazone >> troglitazone > ciglitazone; Willson et al., 1996; 2000;). The initial inhibition of NPA hydrolysis in the hMGL lysates by troglitazone was competitive in nature (Ki 0.34 µM). Although the inhibition was not time dependent, dilution experiments indicated that the inhibition was not rapidly reversible.
A degree of variation in potency is expected for a compound when tested in different assay systems, and in the case of JZL184, the primary variable influencing the observed potency is the pre-incubation time, as expected of an irreversible inhibitor. In the case of N-arachidonoyl dopamine, the variation in potency seems to be species-related, since the potencies were reasonably similar for the hMGL lysates when assayed with the two different substrates. For troglitazone, however, the pattern was at first sight more surprising, given that the compound inhibited the hydrolysis of NPA much more potently than the hydrolysis of 2-OG when the hMGL lysates were used. However, troglitazone has a very high degree of albumin binding (Shibukawa et al., 1995), and the difference in potency appears to be related in part to the concentration of fatty acid-free BSA in the assay medium, since its addition reduced the inhibition by both troglitazone and N-arachidonoyl dopamine of NPA hydrolysis by the hMGL lysates. The reverse assay (the use of 2-OG in the absence of fatty acid-free BSA) is more difficult to interpret, since the albumin concentration will affect the level of free substrate, and hence the observed inhibitory potency of a competitive inhibitor like troglitazone. We found that the sensitivity of hMGL to inhibition by N-arachidonoyl dopamine was indeed increased in the 2-OG assay when BSA was excluded. We have previously reported that the potency of VDM11 (an arachidonoyl-based uptake inhibitor with a structure close to AM404) and arachidonoyl serinol (serinol is 2-amino-1,3-propanediol) are roughly twice as potent as inhibitors of the hydrolysis of 2-OG by rat cerebellar membranes when assayed in the absence of fatty acid-free BSA compared to when assayed at 0.125% w·v−1 albumin (Vandevoorde and Fowler, 2005), which is entirely consistent with the present data with N-arachidonoyl dopamine and with the known ability of its analogues arachidonate and AEA to bind to albumin (Bojesen and Bojesen, 1994; Bojesen and Hansen, 2003; Oddi et al., 2009). Why the sensitivity of the 2-OG hydrolysis to inhibition by troglitazone is not similarly increased in the absence of BSA is unclear, but our current hypothesis is that 2-OG presents itself to the enzyme in a different manner to NPA and by doing so is less amenable to inhibition by troglitazone (but not by N-arachidonoyl dopamine or JZL184).
An obvious question raised by the present study is whether the ability of troglitazone to inhibit MGL is seen in man after normal dosing of the drug. Troglitazone was the first thiazolidinedione used clinically for the treatment of diabetes type 2. It was withdrawn from the US market at the request of the FDA in 2000 due to serious hepatotoxicity issues, the mechanism of which is controversial, but may involve the production of reactive metabolites and/or the ability of the compound to impair mitochondrial function (see Masubuchi, 2006). However, pharmacokinetic data were obtained prior to its withdrawal. In healthy subjects, 7 days of treatment with troglitazone at doses of 200, 400 and 600 mg (doses in the range that was used to reduce plasma glucose in patients with type 2 diabetes, Johnson et al., 1998) produced maximum serum concentrations of 0.90, 1.61 and 2.82 µg/mL, respectively, corresponding to concentrations of 2.0, 3.6 and 6.4 µM, respectively. The corresponding trough plasma concentrations were 0.3, 0.5 and 0.8 µM, respectively (Loi et al., 1999). Although these numbers do not consider the influence of the plasma protein binding of troglitazone (>99.8%, Shibukawa et al., 1995), they are at least in the same range as seen for the inhibition of NPA hydrolysis by the hMGL lysates, which in turn are similar to the potency of the compound for activation of human PPARγ in transactivation assays (EC50 value 0.55 µM, Willson et al., 2000). Given the variation in potency of troglitazone for the different MGL assays, a ‘best case’ speculation is that MGL may have been inhibited upon normal troglitazone dosing. On the other hand, it is highly unlikely that the enzyme is inhibited following normal rosiglitazone dosing to patients with type 2 diabetes, given that the compound is used in lower doses than troglitazone (the maximum plasma concentration obtained after an 8 mg tablet is ∼600 ng/mL, corresponding to 1.7 µM; Cox et al., 2000); is an order of magnitude more potent than troglitazone towards PPARγ; and is an order of magnitude less potent than troglitazone as an inhibitor of NPA hydrolysis by the hMGL lysates and of 2-OG hydrolysis by the rat cerebellar cytosol fractions.
In conclusion, the present study has investigated a series of compounds with primary biological actions in areas where the endocannabinoid system is known to operate, and identified the PPARγ ligand troglitazone as a reasonably potent inhibitor of NPA (but not 2-OG) hydrolysis by hMGL lysates. PPARγ ligands may be useful in the treatment of diseases such as arthritis and neuropathic pain (Fehrenbacher et al., 2009; Giaginis et al., 2009), diseases where modulators of the endocannabinoid system are also being explored as treatment strategies (see Yao et al., 2008). It may thus be possible to use troglitazone as a template for the design of novel compounds without its hepatotoxicity but which have potent effects upon both MGL and PPARγ, as potential novel analgesics and anti-arthritis drugs. The study also underlines the importance of investigating MGL inhibitory potency in several different assays, to ensure that the inhibitory properties of the compound are not assay dependent.
Acknowledgments
The authors would like to thank Britt Jacobsson and Eva Hallin for technical assistance with the radiochemical assays. This work was supported by grants from the Swedish Research Council (Grant no. 12158, medicine) and the Research Funds of the Medical Faculty, Umeå University.
Glossary
Abbreviations
- NPA
4-nitrophenyl acetate
- AEA
anandamide
- 2-AG
2-arachidonoylglycerol
- AM404
N-(4-hydroxyphenyl)-5Z,8Z,11Z,14Z-eicosatetraenamide
- BSA
bovine serum albumin
- CAY10415
(5-[[4-[2-(5-ethyl-2-pyridinyl)-2-oxoethoxy]phenyl]methyl]-2,4-thiazolidinedione
- CAY10514
8-hydroxy-8-[2-(pentyloxy)phenyl]-5-octynoic acid, methyl ester
- CB
cannabinoid
- CP55 940
(-)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol
- FAAH
fatty acid amide hydrolase
- JWH015
(2-methyl-1-propyl-1H-indol-3-yl)-1-naphthalenylmethanone
- JZL184
4-nitrophenyl-4-(dibenzo[d][1,3] dioxol-5-yl(hydroxy)methyl)piperidine-1-carboxylate
- MGL
monoacylglycerol lipase
- 2-OG
2-oleoylglycerol
- PPAR
peroxisome proliferator-activated receptor
- URB597
3′-(aminocarbonyl)[1,1′-biphenyl]-3-3-yl)-cyclohexylcarbamate
- WIN 55,212-2
(R)-(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo[1,2,3-de]-1,4-benzoxazin-6-yl]-1-naphthalenylmethanone mesylate
Conflicts of interest
None declared with respect to the present work.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC) Br J Pharmacol. (4th edn.) 2009;158(Suppl 1):S1–S25. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D. Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science. 1997;277:1094–1097. doi: 10.1126/science.277.5329.1094. [DOI] [PubMed] [Google Scholar]
- Bishay P, Schmidt H, Marian C, Häussler A, Wijnvoord N, Ziebell S, et al. R-flurbiprofen reduces neuropathic pain in rodents by restoring endogenous cannabinoids. PLoS ONE. 2010;5:e10628. doi: 10.1371/journal.pone.0010628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisogno T, Melck D, Bobrov M, Gretskaya N, Bezuglov V, De Petrocellis L, et al. N-acyl-dopamines: novel synthetic CB1 cannabinoid-receptor ligands and inhibitors of anandamide inactivation with cannabimimetic activity in vitro and in vivo. Biochem J. 2000;351:817–824. [PMC free article] [PubMed] [Google Scholar]
- Blankman JL, Simon GM, Cravatt BF. A comprehensive profile of brain enzymes that hydrolyze the endocannabinoid 2-arachidonoylglycerol. Chem Biol. 2007;14:1347–1356. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bojesen I, Bojesen E. Binding of arachidonate and oleate to bovine serum albumin. J Lipid Res. 1994;35:770–778. [PubMed] [Google Scholar]
- Bojesen I, Hansen H. Binding of anandamide to bovine serum albumin. J Lipid Res. 2003;44:1790–1794. doi: 10.1194/jlr.M300170-JLR200. [DOI] [PubMed] [Google Scholar]
- Boldrup L, Wilson SJ, Barbier AJ, Fowler CJ. A simple stopped assay for fatty acid amide hydrolase avoiding the use of a chloroform extraction phase. J Biochem Biophys Methods. 2004;60:171–177. doi: 10.1016/j.jbbm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Caijo F, Mosset P, Grée R, Audinot-Bouchez V, Boutin J, Renard P, et al. Synthesis of new carbo- and heterocyclic analogues of 8-HETE and evaluation of their activity towards the PPARs. Bioorg Med Chem Lett. 2005;15:4421–4426. doi: 10.1016/j.bmcl.2005.07.049. [DOI] [PubMed] [Google Scholar]
- Chau L-Y, Tai H-H. Monoglyceride and diglyceride lipases from human platelet microsomes. Biochim Biophys Acta. 1988;963:436–444. doi: 10.1016/0005-2760(88)90312-8. [DOI] [PubMed] [Google Scholar]
- Chu CJ, Huang SM, De Petrocellis L, Bisogno T, Ewing SA, Miller JD, et al. N-oleoyldopamine, a novel endogenous capsaicin-like lipid that produces hyperalgesia. J Biol Chem. 2003;278:13633–13639. doi: 10.1074/jbc.M211231200. [DOI] [PubMed] [Google Scholar]
- Cox PJ, Ryan DA, Hollis FJ, Harris AM, Miller AK, Vousden M, et al. Absorption, disposition, and metabolism of rosiglitazone, a potent thiazolidinedione insulin sensitizer, in humans. Drug Metab Dispos. 2000;28:772–780. [PubMed] [Google Scholar]
- Di Marzo V, Bisogno T, De Petrocellis L, Melck D, Orlando P, Wagner J, et al. Biosynthesis and inactivation of the endocannabinoid 2-arachidonoylglycerol in circulating and tumoral macrophages. Eur J Biochem. 1999;264:258–267. doi: 10.1046/j.1432-1327.1999.00631.x. [DOI] [PubMed] [Google Scholar]
- Dinh T, Carpenter D, Leslie F, Freund T, Katona I, Sensi S, et al. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fehrenbacher J, LoVerme J, Clarke W, Hargreaves K, Piomelli D, Taylor B. Rapid pain modulation with nuclear receptor ligands. Brain Res Rev. 2009;60:114–124. doi: 10.1016/j.brainresrev.2008.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler C, Tiger G, Stenström A. Ibuprofen inhibits rat brain deamidation of anandamide at pharmacologically relevant concentrations. Mode of inhibition and structure–activity relationship. J Pharmacol Exp Ther. 1997;283:729–734. [PubMed] [Google Scholar]
- Ghafouri N, Tiger G, Razdan RK, Mahadevan A, Pertwee RG, Martin BR, et al. Inhibition of monoacylglycerol lipase and fatty acid amide hydrolase by analogues of 2-arachidonoylglycerol. Br J Pharmacol. 2004;143:774–784. doi: 10.1038/sj.bjp.0705948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaginis C, Giagini A, Theocharis S. Peroxisome proliferator-activated receptor-γ (PPAR-γ) ligands as potential therapeutic agents to treat arthritis. Pharmacol Res. 2009;60:160–169. doi: 10.1016/j.phrs.2009.02.005. [DOI] [PubMed] [Google Scholar]
- Gobbi G, Bambico F, Mangieri R, Bortolato M, Campolongo P, Solinas M, et al. Antidepressant-like activity and modulation of brain monoaminergic transmission by blockade of anandamide hydrolysis. Proc Natl Acad Sci USA. 2005;102:18620–18625. doi: 10.1073/pnas.0509591102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goparaju S, Ueda N, Taniguchi K, Yamamoto S. Enzymes of porcine brain hydrolyzing 2-arachidonoylglycerol, an endogenous ligand of cannabinoid receptors. Biochem Pharmacol. 1999;57:417–423. doi: 10.1016/s0006-2952(98)00314-1. [DOI] [PubMed] [Google Scholar]
- Goparaju S, Ueda N, Yamaguchi H, Yamamoto S. Anandamide amidohydrolase reacting with 2-arachidonoylglycerol, another cannabinoid receptor ligand. FEBS Lett. 1998;422:69–73. doi: 10.1016/s0014-5793(97)01603-7. [DOI] [PubMed] [Google Scholar]
- Gühring H, Hamza M, Sergejeva M, Ates M, Kotalla C, Ledent C, et al. A role for endocannabinoids in indomethacin-induced spinal antinociception. Eur J Pharmacol. 2002;454:153–163. doi: 10.1016/s0014-2999(02)02485-8. [DOI] [PubMed] [Google Scholar]
- Guindon J, De Léan A, Beaulieu P. Local interactions between anandamide, an endocannabinoid, and ibuprofen, a nonsteroidal anti-inflammatory drug, in acute and inflammatory pain. Pain. 2006;121:85–93. doi: 10.1016/j.pain.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Holt S, Comelli F, Costa B, Fowler CJ. Inhibitors of fatty acid amide hydrolase reduce carrageenan-induced hind paw inflammation in pentobarbital-treated mice: comparison with indomethacin and possible involvement of cannabinoid receptors. Br J Pharmacol. 2005;146:467–476. doi: 10.1038/sj.bjp.0706348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huffman J, Liddle J, Yu S, Aung M, Abood M, Wiley J, et al. 3-(1′,1′.Dimethylbutyl)-1-deoxy-Δ8-THC and related compounds: synthesis of selective ligands for the CB2 receptor. Bioorg Med Chem. 1999;7:2905–2914. doi: 10.1016/s0968-0896(99)00219-9. [DOI] [PubMed] [Google Scholar]
- Jayamanne A, Greenwood R, Mitchell VA, Aslan S, Piomelli D, Vaughan CW. Actions of the FAAH inhibitor URB597 in neuropathic and inflammatory chronic pain models. Br J Pharmacol. 2006;147:281–288. doi: 10.1038/sj.bjp.0706510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MD, Campbell LK, Campbell RK. Troglitazone: review and assessment of its role in the treatment of patients with impaired glucose tolerance and diabetes mellitus. Ann Pharmacother. 1998;32:337–348. doi: 10.1345/aph.17046. [DOI] [PubMed] [Google Scholar]
- Kathuria S, Gaetani S, Fegley D, Valiño F, Duranti A, Tontini A, et al. Modulation of anxiety through blockade of anandamide hydrolysis. Nat Med. 2003;9:76–78. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- Kinsey SG, Long JZ, O'Neal ST, Abdullah RA, Poklis JL, Boger DL, et al. Blockade of endocannabinoid-degrading enzymes attenuates neuropathic pain. J Pharmacol Exp Ther. 2009;330:902–910. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kliewer S, Lenhard J, Willson T, Patel I, Morris D, Lehmann J. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor γ and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- Lenman A, Fowler CJ. Interaction of ligands for the peroxisome proliferator-activated receptor γ with the endocannabinoid system. Br J Pharmacol. 2007;151:1343–1351. doi: 10.1038/sj.bjp.0707352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loi CM, Alvey CW, Vassos AB, Randinitis EJ, Sedman AJ, Koup JR. Steady-state pharmacokinetics and dose proportionality of troglitazone and its metabolites. J Clin Pharmacol. 1999;39:920–926. doi: 10.1177/00912709922008533. [DOI] [PubMed] [Google Scholar]
- Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, et al. Selective blockade of 2-arachidonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol. 2009a;5:37–44. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, et al. Dual blockade of FAAH and MAGL identifies behavioral processes regulated by endocannabinoid crosstalk in vivo. Proc Natl Acad Sci USA. 2009b;106:20270–20275. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masubuchi Y. Metabolic and non-metabolic factors determining troglitazone hepatotoxicity: a review. Drug Metab Pharmacokinet. 2006;21:347–356. doi: 10.2133/dmpk.21.347. [DOI] [PubMed] [Google Scholar]
- McPartland J, Glass M, Pertwee R. Meta-analysis of cannabinoid ligand binding affinity and receptor distribution: interspecies differences. Br J Pharmacol. 2007;152:583–593. doi: 10.1038/sj.bjp.0707399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muccioli G, Labar G, Lambert D. CAY10499, a novel monoglyceride lipase inhibitor evidenced by an expeditious MGL assay. Chembiochem. 2008;9:2704–2710. doi: 10.1002/cbic.200800428. [DOI] [PubMed] [Google Scholar]
- Naidu PS, Booker L, Cravatt BF, Lichtman AH. Synergy between enzyme inhibitors of fatty acid amide hydrolase and cyclooxygenase in visceral nociception. J Pharmacol Exp Ther. 2009;329:48–56. doi: 10.1124/jpet.108.143487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Sullivan SE. Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol. 2007;152:576–582. doi: 10.1038/sj.bjp.0707423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddi S, Fezza F, Pasquariello N, D'Agostino A, Catanzaro G, De Simone C, et al. Molecular identification of albumin and Hsp70 as cytosolic anandamide-binding proteins. Chem Biol. 2009;16:624–632. doi: 10.1016/j.chembiol.2009.05.004. [DOI] [PubMed] [Google Scholar]
- Ortar G, Bisogno T, Ligresti A, Morera E, Nalli M, Di Marzo V. Tetrahydrolipstatin analogues as modulators of endocannabinoid 2-arachidonoylglycerol metabolism. J Med Chem. 2008;51:6970–6979. doi: 10.1021/jm800978m. [DOI] [PubMed] [Google Scholar]
- Pacher P, Bátkai S, Kunos G. The endocannabinoid system as an emerging target of pharmacotherapy. Pharmacol Rev. 2006;58:389–462. doi: 10.1124/pr.58.3.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Wohlfeil E, Rademacher D, Carrier E, Perry L, Kundu A, et al. The general anesthetic propofol increases brain N-arachidonoylethanolamine (anandamide) content and inhibits fatty acid amide hydrolase. Br J Pharmacol. 2003;139:1005–1013. doi: 10.1038/sj.bjp.0705334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saario S, Savinainen J, Laitinen J, Järvinen T, Niemi R. Monoglyceride lipase-like enzymatic activity is responsible for hydrolysis of 2-arachidonoylglycerol in rat cerebellar membranes. Biochem Pharmacol. 2004;67:1381–1387. doi: 10.1016/j.bcp.2003.12.003. [DOI] [PubMed] [Google Scholar]
- Salvi A, Carrupt PA, Mayer JM, Testa B. Esterase-like activity of human serum albumin toward prodrug esters of nicotinic acid. Drug Metab Dispos. 1997;25:395–398. [PubMed] [Google Scholar]
- Shibukawa A, Sawada T, Nakao C, Izumi T, Nakagawa T. High-performance frontal analysis for the study of protein binding of troglitazone (CS-045) in albumin solution and in human plasma. J Chromatogr A. 1995;697:337–343. doi: 10.1016/0021-9673(94)00929-4. [DOI] [PubMed] [Google Scholar]
- Tanis SP, Parker TT, Colca JR, Fisher RM, Kletzein RF. Synthesis and biological activity of metabolites of the antidiabetic, antihyperglycemic agent pioglitazone. J Med Chem. 1996;39:5053–5063. doi: 10.1021/jm9605694. [DOI] [PubMed] [Google Scholar]
- Tarzia G, Antonietti F, Duranti A, Tontini A, Rivara S, Traldi P, et al. Identification of a bioactive impurity in a commercial sample of 6-methyl-2-p-tolylaminobenzo[d][1,3]oxazin-4-one (URB754) Ann Chim. 2007;97:887–894. doi: 10.1002/adic.200790073. [DOI] [PubMed] [Google Scholar]
- Tornqvist H, Belfrage P. Purification and some properties of a monoacylglycerol-hydrolysing enzyme of rat adipose tissue. J Biol Chem. 1976;251:813–819. [PubMed] [Google Scholar]
- Vandevoorde S, Fowler CJ. Inhibition of fatty acid amide hydrolase and monoacylglycerol lipase by the anandamide uptake inhibitor VDM11: evidence that VDM11 acts as an FAAH substrate. Br J Pharmacol. 2005;145:885–893. doi: 10.1038/sj.bjp.0706253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willson T, Cobb J, Cowan D, Wiethe R, Correa I, Prakash S, et al. The structure–activity relationship between peroxisome proliferator-activated receptor γ agonism and the antihyperglycemic activity of thiazolidinediones. J Med Chem. 1996;39:665–668. doi: 10.1021/jm950395a. [DOI] [PubMed] [Google Scholar]
- Willson T, Brown P, Sternbach D, Henke B. The PPARs: from orphan receptors to drug discovery. J Med Chem. 2000;43:527–550. doi: 10.1021/jm990554g. [DOI] [PubMed] [Google Scholar]
- Yao B, Hsieh G, Frost J, Fan Y, Garrison T, Daza A, et al. In vitro and in vivo characterization of A-796260: a selective cannabinoid CB2 receptor agonist exhibiting analgesic activity in rodent pain models. Br J Pharmacol. 2008;153:390–401. doi: 10.1038/sj.bjp.0707568. [DOI] [PMC free article] [PubMed] [Google Scholar]