

Abstract
BACKGROUND AND PURPOSE
Although the serum and glucocorticoid-inducible protein kinase 1 (SGK1) appears to be involved in controlling epithelial Na+ absorption, its role in this physiologically important ion transport process is undefined. As SGK1 activity is dependent upon target of rapamycin complex 2 (TORC2)-catalysed phosphorylation of SGK1-Ser422, we have explored the effects of inhibiting TORC2 and/or TORC1 upon the hormonal control of Na+ absorption.
EXPERIMENTAL APPROACH
Na+ absorption was quantified electrometrically in mouse cortical collecting duct cells (mpkCCD) grown to confluence on permeable membranes. Kinase activities were assessed by monitoring endogenous protein phosphorylation, with or without TORC1/2 inhibitors (TORIN1 and PP242) and the TORC1 inhibitor: rapamycin.
KEY RESULTS
Inhibition of TORC1/2 (TORIN1, PP242) suppressed basal SGK1 activity, prevented insulin- and dexamethasone-induced SGK1 activation, and caused modest (10–20%) inhibition of basal Na+ absorption and substantial (∼80%) inhibition of insulin/dexamethasone-induced Na+ transport. Inhibition of TORC1 did not impair SGK1 activation or insulin-induced Na+ transport, but did inhibit (∼80%) dexamethasone-induced Na+ absorption. Arginine vasopressin stimulated Na+ absorption via a TORC1/2-independent mechanism.
CONCLUSION AND IMPLICATIONS
Target of rapamycin complex 2, but not TORC1, is important to SGK1 activation. Signalling via phosphoinositide-3-kinase/TORC2/SGK1 can explain insulin-induced Na+ absorption. TORC2, but not TORC1, is also involved in glucocorticoid-induced SGK1 activation but its role is permissive. Glucocorticoid-induced Na+ transport displayed a requirement for TORC1 activity. Therefore, TORC1 and TORC2 contribute to the regulation of Na+ absorption. Pharmacological manipulation of TORC1/2 signalling may provide novel therapies for Na+-sensitive hypertension.
Keywords: kinase inhibitors, PP242, TORIN1, serum and glucocorticoid-inducibleprotein kinase 1, phosphoinositide-3-kinase, epithelial Na+ channel, cortical collecting duct
Introduction
Phosphoinositide-3-kinase (PI3K) controls many aspects of cellular physiology by catalysing the formation of phospholipid second messenger compounds, most notably phosphatidylinositol 3,4,5-trisphosphate (PIP3), which regulate downstream protein kinases such as protein kinase B (PKB, also known as Akt) and serum and glucocorticoid-inducible kinase 1 (SGK1) (see Cohen, 2006). Early evidence that PI3K is involved in the control of epithelial Na+ absorption came from the observation that PI3K inhibitors inhibit electrogenic Na+ transport by reducing the number of functional, epithelial Na+ channels (ENaC; nomenclature follows Alexander et al., 2009) in the apical membrane (Paunescu et al., 2000). This control over the apical ENaC expression appears to depend upon SGK1, a protein kinase that can prevent the internalization and degradation of ENaC subunits which normally seems to restrict apical Na+ conductance (Kobayashi and Cohen, 1999; Park et al., 1999; Debonneville et al., 2001; Wang et al., 2001; Snyder et al., 2004b). Indeed, SGK1-dependent control over apical ENaC expression seems to allow PI3K-coupled agonists, such as insulin and insulin-like growth factor 1(IGF-1), to stimulate Na+ transport (Blazer-Yost et al., 1998; 2003; Kobayashi and Cohen, 1999; Park et al., 1999; Debonneville et al., 2001; Wang et al., 2001; Snyder et al., 2004b; Huang et al., 2006; Gonzalez-Rodriguez et al., 2007; Mansley and Wilson, 2010), and a body of evidence also implicates SGK1 in the control of Na+ transport by mineralocorticoid and glucocorticoid hormones (Wang et al., 1991; Itani et al., 2002; Gonzalez-Rodriguez et al., 2007). Moreover, SGK1 may also contribute to the control of Na+ transport by cAMP/PKA (adenine-nucleotide-dependent protein kinase)-coupled hormones such as arginine vasopressin (AVP) (Gonzales-Robayana et al., 2000; Perrotti et al., 2001; Vasquez et al., 2008). However, not all data are consistent with the idea that SGK1 plays such a central role in the control of Na+ absorption as sgk1 gene deletion causes only mild dysfunction of renal Na+ handing (Wulff et al., 2002) without preventing the regulation of colon Na+ absorption (Rexhepaj et al., 2006).
Whilst the role of PI3K/SGK1 in the hormonal control of Na+ absorption is understood poorly (Lang et al., 2006; Loffing et al., 2006), it is clear that the PIP3-dependent activation of this protein kinase depends upon the phosphorylation of SGK1-Ser422 by the target of rapamycin signalling complex 2 (TORC2) (García-Martínez and Alessi, 2008). Pharmacological inhibition of TORC2 therefore provides a novel and effective way of inactivating SGK1 (García-Martínez and Alessi, 2008). In order to clarify the role of SGK1 in the control of Na+ absorption, we have, in the present study, explored the physiological consequences of exposing Na+ absorbing, mouse cortical collecting duct cells (mpkCCD) to novel inhibitors of TORC2, TORIN1 (Thoreen et al., 2009) and PP242 (Feldman et al., 2009).
Methods
All methods have been described elsewhere (Mansley and Wilson, 2010) and so only brief details are given here.
Experiments were undertaken using confluent mpkCCD cells (Bens et al., 1999) that had been deprived of hormones/growth factors for ∼48 h. Previous work (Bens et al., 1999) shows that these cells generate transepithelial current that is due, almost exclusively, to electrogenic Na+ absorption and this parameter (equivalent short circuit current, IEq) was therefore monitored under open circuit conditions. All experiments were terminated by adding 10 µM amiloride to the apical bath, and this always caused >90% inhibition of IEq and, unless otherwise stated, also increased transepithelial resistance (Rt) one to twofold.
Changes in endogenous SGK1 activity were monitored by quantifying (by Western blot analysis and densitometry) the phosphorylation of residues (Thr346/356/366) within the protein encoded by n-myc downstream regulated gene 1 (NDRG1) that are substrates for SGK1 but not for other closely related kinases (Murray et al., 2005; Inglis et al., 2009) whilst TORC1 and TORC2 activities were monitored by assaying the phosphorylation of the 70 kDa ribosomal S6 kinase (P70-S6K)-Thr389 (Proud, 2007) and protein kinase B (PKB)-Ser473 (Sarbassov et al., 2005) respectively.
Data analysis
Experiments were undertaken using strictly paired protocols in which control and experimental cells were age-matched and at identical passage. Pooled data are shown as mean ± SEM and values of n refer to the number of times a protocol was repeated using cells at different passages. Statistical significances were tested using Student's t-test or analysis of variance (anova), with Bonferroni post hoc test, as appropriate.
Materials
Amiloride, dexamethasone, AVP, insulin, culture reagents and general laboratory chemicals were from Sigma (Poole, Dorset, UK) whilst PP242 and TORIN1 were generous gifts from Prof D.R. Alessi (MRC Protein Phosphorylation Unit, University of Dundee, Dundee, UK) and Prof D.M. Sabatini (Whitehead Institute for Biomedical Research, Cambridge, MA, USA) respectively. Antibodies against Ser473-phosphorylated and total PKB, Ser133-phopsphorylated and total cAMP response element binding protein (CREB), and Thr389-phosphorylated and total P70-S6K were from Cell Signalling (Hertfordshire, UK), whilst antibodies against Thr346/356/366-phosphorylated and total forms of NDRG1 were generously provided by Prof D.R. Alessi.
Results
Insulin-induced phosphorylation of endogenous proteins
Hormone-deprived cells displayed basal phosphorylation of PKB-Ser473, and insulin stimulation (20 nM, 30 min) increased the abundance of this phosphoprotein without altering the overall PKB expression level (Figure 1A,B), indicating (Sarbassov et al., 2005) that this hormone normally activates TORC2. TORIN1 (0.03–1 µM, 30 min pre-incubation) caused a concentration-dependent decline in the abundance of Ser473-phosphorylated PKB both in hormone-deprived and insulin-stimulated cells, and concentrations ≥0.1 µM were fully effective (Figure 1A). As TORIN1 did not alter the overall PKB expression level (Figure 1B), this response must reflect reduced abundance of the Ser473-phosphorylated form of this protein. Although small responses to insulin persisted in the presence of 0.01 µM and 0.03 µM TORIN1, concentrations of TORIN1 ≥0.1 µM reduced the expression of Ser473-phosphorylated PKB to an undetectable level in hormone-deprived and insulin-stimulated cells indicating complete inhibition of TORC2 (Figure 1A,B). Hormone-deprived cells also displayed clearly detectible levels of Thr346/356/366-phosphorylated NDRG1, and insulin (20 nM, 30 min) also increased the abundance of this phosphoprotein without altering the overall expression level indicating activation of SGK1 (Figure 1C,D). TORIN1 reduced the abundance of Thr346/356/3566-phosphorylated NDRG1 in hormone-deprived and insulin-stimulated cells without altering overall expression (Figure 1C,D) indicating inactivation of SGK1. This effect was complete at ≥0.1 µM.
Figure 1.
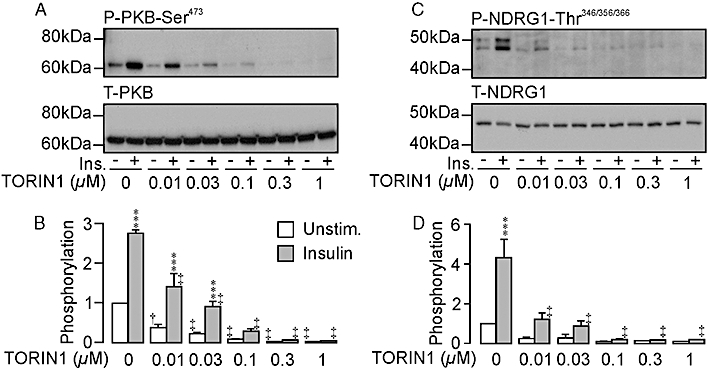
Effects of TORIN1 upon the phosphorylation of protein kinase B (PKB)-Ser473 and by n-myc downstream regulated gene 1-encoded protein 1 Thr346/356/366 (NDRG1-Thr346/356/366) in unstimulated (Unstim.) and insulin-stimulated (Ins.) cells. The blots in each figure illustrate the effects of TORIN 1 (0–1 µM, 30 min pre-incubation) upon the expression of (A) Ser473-phosphorylated (upper panel) and total (lower panel) PKB, and (C) Thr346/356/366-phosphorylated (upper panel) and total (lower panel) NDRG1, whilst the histograms include pooled data from six such experiments (mean ± SEM) which show the effects of insulin and/or TORIN1 upon the phosphorylation of PKB-Ser473 (B) and NDRG1-Thr 346/356/366 (D). ***P < 0.01; significant responses to insulin; Student's paired t-test. †P < 0.05; ‡P < 0.02; significant effects of TORIN1; anova /Bonferroni post hoc test).
Figure 2 shows the results of experiments that explored the effects of PP242 (1 µM, 30 min pre-incubation), another TORC2 inhibitor (Feldman et al., 2009). These data confirm that insulin normally increases the abundance of both Ser473-phosphorylated PKB and Thr346/356/366-phosphorylated NDRG1, and also show that PP242 completely abolished the expression of these phosphoproteins in hormone-deprived and insulin-stimulated cells with no effect upon the expression of the corresponding full length proteins.
Figure 2.
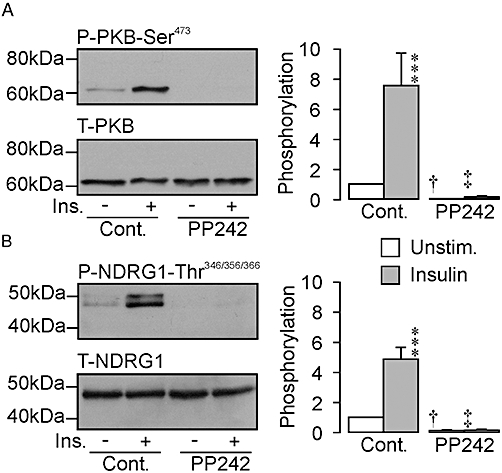
Effects of PP242 upon the phosphorylation of PKB-Ser473 and NDRG1-Thr346/356/366. The blots in each figure illustrate the effects of PP242 (1 µM, 30 min pre-incubation) upon the abundance of (A) Ser473-phosphorylated and total PKB and (B) Thr346/356/366-phosphorylated and total NDRG1 in hormone-deprived and insulin-stimulated (20 nM, 30 min) cells, whilst the histograms each show the pooled data derived from six such experiments. ***P < 0.01, significant effects of insulin upon the phosphorylation of these endogenous proteins. †P < 0.05, ‡P < 0.01, significant effects of PP242; anova /Bonferroni post hoc test). Cont., control; Ins., insulin-stimulated; NDRG1, protein encoded by n-myc downstream regulated gene 1, PKB, protein kinase B; Unstim., unstimulated.
Basal Na+ transport
Analysis of currents recorded from hormone-deprived cells showed that TORIN1 (0.1 µM) and PP242 (1 µM) caused slight (TORIN1: 10.8 ± 3.1%, n = 32; PP242: 22.3 ± 2.1%, n = 33) inhibition of electrogenic Na+ absorption. These effects became apparent after 2–3 min and developed over the following 10–15 min. Rapamycin (0.1 µM, n = 3), an inhibitor of TORC1, had no effect upon the currents recorded under such conditions. These compounds had no discernible effect upon Rt indicating that epithelial integrity is maintained.
Insulin-stimulated Na+ transport
Figure 3A shows currents recorded from hormone-deprived and insulin-stimulated (20 nM, basolateral) control cells whilst Figure 3B shows data from TORIN1-treated (0.1 µM, 30 min) cells. To ensure that the small effects on the basal current described above did not confound analysis of these data, these results are shown as deviations from the value of IEq measured over the 5 min period prior to hormone application. The magnitude of this basal current normally showed a slight tendency to decline with time but insulin consistently evoked an augmentation of IEq that became apparent after ∼5 min and reflected a hyperpolarization of the transepithelial voltage (Vt) that coincided with a small fall in Rt (not shown). The magnitude of this response was quantified by measuring the insulin-induced change in IEq and subtracting the spontaneously developing change measured in hormone-deprived cells. This analysis showed that insulin normally enhances the recorded current (Figure 3C) and, although a small response persisted in TORIN1-treated cells (Figure 5B, P < 0.05), the magnitude of this response was reduced by 73.3 ± 3.6%. Further experiments confirmed this effect of insulin (Figure 3D) and showed that PP242 (Figure 3E) caused 83.7 ± 6.7% inhibition of this response (Figure 3F). The magnitude of this effect did not differ from that quantified in TORIN1-treated cells.
Figure 3.
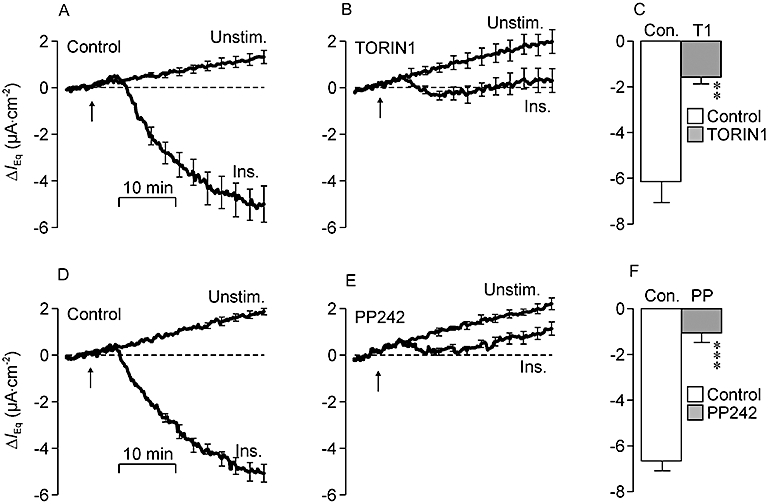
Effects of TORIN1 and PP242 upon the electrometric response to insulin. In this and in all subsequent such figures the values of equivalent short circuit current (IEq) measured during the initial 5 min of each recording period (i.e. the basal IEq) has been subtracted from all subsequently measured data points so that each presented trace shows the changes (mean ± SEM, n = 5) from this basal value (ΔIEq). (A) Values of IEq derived from unstimulated and insulin-stimulated (Ins.; 20 nM, basolateral, arrows) control cells. In this series of experiments the values of basal IEq quantified in the unstimulated (Unstim.) and insulin-stimulated cells were −14.4 ± 1.9 µA·cm−2 and −17.0 ± 2.0 µA·cm−2 respectively. (B) Data from age-matched cells pretreated (30 min) with 0.1 µM TORIN1 (basal IEq unstimulated cells: −13.6 ± 1.2 µA·cm−2; basal IEq insulin-stimulated cells: −14.6 ± 1.4 µA·cm−2). (C) Responses to insulin in control (Con.) and TORIN1-treated cells were quantified by measuring the change in IEq that developed during exposure to hormone and subtracting the spontaneous change measured in unstimulated cells. (D) Data showing the spontaneous and insulin-induced changes in IEq in control cells (basal IEq unstimulated cells: −17.5 ± 1.0 µA·cm−2; basal IEq insulin-stimulated cells: −20.1 ± 1.6 µA·cm−2). (E) Data from age-matched cells pretreated (30 min) with 1 µM PP242 (basal IEq unstimulated cells: −13.9 ± 1.1 µA·cm−2; basal IEq insulin-stimulated cells: −15.7 ± 1.3 µA·cm−2). (F) Responses to insulin measured in control and PP242-treated cells. **P < 0.01; ***P < 0.005, significant responses to insulin; Student's paired t-test.
Figure 5.
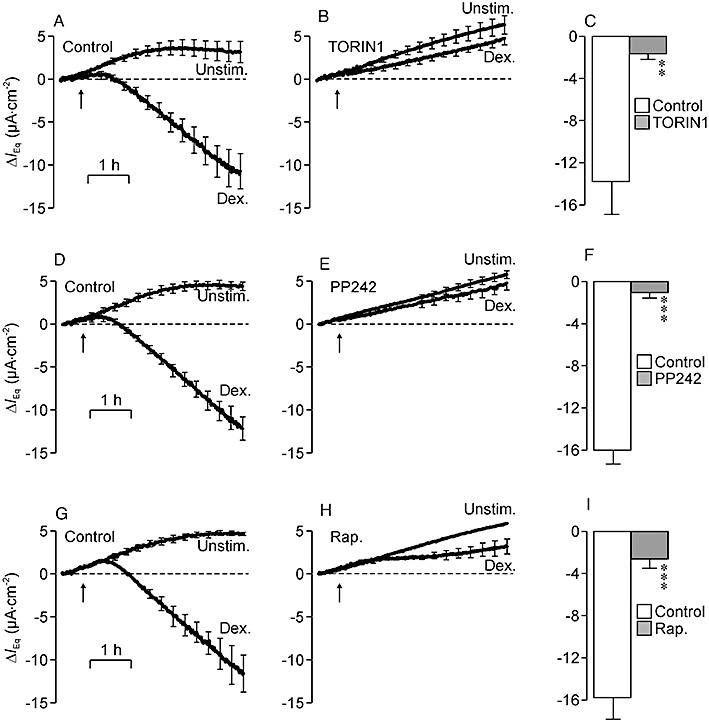
Effects of TORIN1, PP242 and rapamycin upon the dexamethasone-induced stimulation of Na+ transport. (A) Changes in equivalent short circuit current (IEq) (n = 6) measured in unstimulated (Unstim.) and dexamethasone-stimulated (Dex.; 0.2 µM, arrows) cells maintained under control conditions (basal IEq unstimulated cells: −20.7 ± 1.9 µA·cm−2; basal IEq dexamethasone-stimulated cells: −22.0 ± 2.8 µA·cm−2). (B) Data from age-matched cells at identical passage pretreated (30 min) with 0.1 µM TORIN1 (basal IEq unstimulated cells: −15.9 ± 1.8 µA·cm−2; basal IEq dexamethasone-stimulated cells: −18.9 ± 2.2 µA·cm−2). (C) Responses to dexamethasone in control and TORIN1-treated cells were quantified by measuring the change in current that developed during exposure to this hormone and subtracting the spontaneously developing change in IEq measured in unstimulated cells. (D) Data (n = 6) from separate experiments showing spontaneous and dexamethasone-induced changes in IEq in control cells (basal IEq unstimulated cells: −20.8 ± 1.0 µA·cm−2; basal IEq dexamethasone-stimulated cells: −20.8 ± 1.5 µA·cm−2). (E) Data from age-matched pretreated (30 min) with 1 µM PP242 (basal IEq unstimulated cells: −14.8 ± 1.0 µA·cm−2; basal IEq dexamethasone-stimulated cells: −14.7 ± 1.4 µA·cm−2). (F) Responses to dexamethasone quantified in control and PP242-treated cells. (G) Spontaneous and dexamethasone-induced changes in IEq measured under control conditions (n = 3) (basal IEq unstimulated cells: −18.7 ± 0.2 µA·cm−2; basal IEq dexamethasone-stimulated cells: −19.2 ± 1.7 µA·cm−2). (H) Data from age-matched cells pretreated (30 min) with 0.1 µM rapamycin (Rap.; basal IEq unstimulated cells: −14.9 ± 1.0 µA·cm−2; basal IEq dexamethasone-stimulated cells: −15.4 ± 1.9 µA·cm−2). (I) Responses to dexamethasone in control and rapamycin-treated cells. **P < 0.02, ***P < 0.01, significant effects of the inhibitors; Student's paired t-test.
Effects of aldosterone
Aldosterone could augment the amiloride-sensitive IEq by inducing a hyperpolarization of Vt that developed over 1–2 h and coincided with a small fall in Rt, and this hormone also increased the abundance of Thr346/356/366-phosphorylated NDRG1 without altering the expression of the full length protein indicating activation of SGK1 (not shown). However, such responses occurred only in cells exposed to aldosterone concentrations 15- to 500-fold greater (0.5–5 µM) than those encountered in vivo and the cells were insensitive to physiologically relevant concentrations (1–30 nM) of this hormone.
Dexamethasone-induced protein phosphorylation
The control data in Figure 4 show that dexamethasone (0.2 µM, 2.5 h) had no effect upon the abundance of Ser473-phosphorylated or total PKB (Figure 4A–C) indicating that this synthetic glucocorticoid did not activate TORC2. TORIN1 (0.1 µM, Figure 4A) and PP242 (1 µM, Figure 4B), on the other hand, reduced the expression of Ser473-phosphorylated PKB to an undetectable level in both hormone-deprived and dexamethasone-stimulated cells, without altering the overall expression level. Rapamycin had no such effect (Figure 4C). Further experiments showed that dexamethasone consistently increased the abundance of the Thr346/356/366-phosphorylated NDRG1 (Figure 4D–F) and, although this synthetic glucocorticoid had no effect upon the overall abundance of this protein, it did induce the appearance of a second, less mobile band in blots probed using the antibody against full length NDRG1 (Figure 4D–F). TORIN1 (Figure 4D) and PP242 (Figure 4E) virtually abolished the expression of Thr346/356/366-phosphorylated NDRG1 in hormone-deprived and dexamethasone-stimulated cells without altering the overall expression level. Whilst a small response to dexamethasone persisted in the presence of TORIN1, this did not reach statistical significance (Figure 4D). Rapamycin (0.1 µM) did not alter the phosphorylation status of NDRG1-Thr346/356/366 (Figure 4F). Further experiments (n = 4, not shown) showed that hormone-deprived cells normally displayed detectible levels of Thr389-phosphorylated P70-S6K indicating basal activity of TORC1. Dexamethasone (0.2 µM, 2.5 h) had no effect upon the phosphorylation of this residue establishing that this synthetic steroid did not activate TORC1. Rapamycin (0.1 µM), on the other hand, abolished the expression of Thr389-phosphorylated P70-S6K in hormone-deprived and dexamethasone-stimulated cells without altering the overall expression of this protein (not shown). Rapamycin therefore causes complete inactivation of TORC1 under the present conditions.
Figure 4.
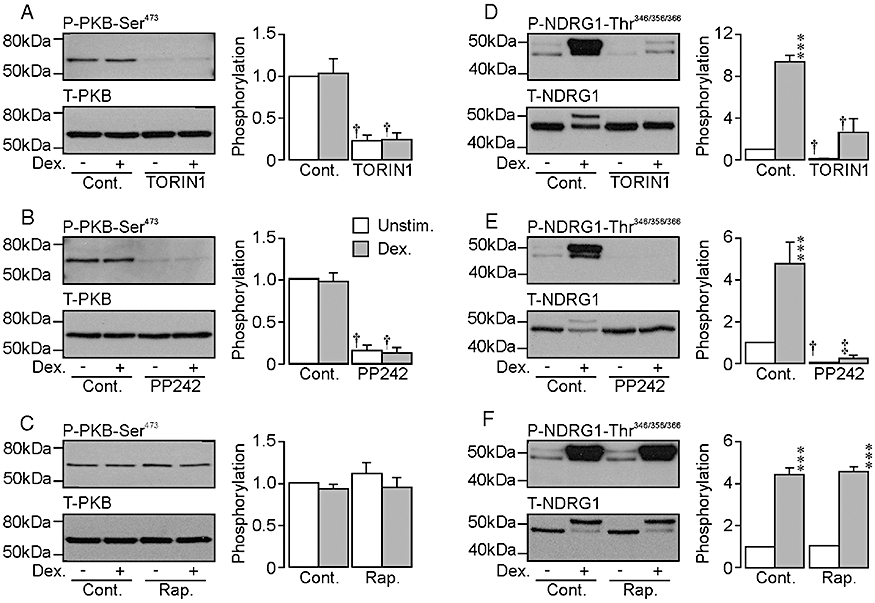
Effects of TORIN1 and PP242 upon the phosphorylation of PKB-Ser473 and NDRG1-Thr346/356/366 in unstimulated and dexamethasone-stimulated cells. The blots in each figure illustrate the effects of 0.1 µM TORIN1 (A, D) 1 µM PP242 (B, E) and 0.1 µM rapamycin (C, F) upon the abundance of Ser473-phosphorylated and total PKB (A, B, C) and Thr346/356/366-phosphoryated and total NDRG1 (D, E, F) measured in both hormone-deprived and dexamethasone-stimulated (0.2 µM, 2.5 h) cells. The pooled data (mean ± SEM) presented in the histograms are each derived from a total of six (TORIN1, PP242) or four (rapamycin) such experiments. ***P < 0.001, significant responses to dexamethasone; Student's paired t-test. †P < 0.05; ‡P < 0.001, significant effects of the TORC1/2 inhibitors; anova. Cont., control; Dex., dexamethasone-stimulated; NDRG1, protein encoded by n-myc downstream regulated gene 1, PKB, protein kinase B; Rap., rapamycin; Unstim., unstimulated.
Dexamethasone-induced Na+ transport
The control data in Figure 5 show that dexamethasone (0.2 µM) increased IEq although this response developed more slowly than the response to insulin (see above), as it only became apparent after ∼40-min latency. Moreover, the present experiments were terminated after 2.5 h and the response had not reached a plateau value by this time (Figure 5A,D,G). Whilst there was some evidence of a dexamethasone-induced augmentation of IEq in TORIN1- (Figure 5B) and PP242- (Figure 5E) treated cells, these responses were smaller than control (Figure 5C,F) and analysis showed that these substances caused 87.4 ± 3.8% and 92.1 ± 3.9% inhibition respectively. Although rapamycin did not affect the dexamethasone-induced activation of SGK1 (Figure 4F), this substance suppressed the dexamethasone-induced Na+ transport (Figure 5H) as effectively (84.2 ± 3.4% inhibition) as TORIN1 (Figure 5B) or PP242 (Figure 5E).
Responses to AVP
Figure 6A shows continuous records of Vt derived from confluent cells. Standard pulses of transepithelial current (−10 µA·cm−2, 20 s) were repeatedly injected throughout all such experiments so that Rt could be calculated, and analysis of these records showed that Vt was normally ∼−50 mV (Figure 6C) and, as Rt was ∼2 kΩcm2 (Figure 6D), IEq was normally ∼−25 µA·cm−2 (Figure 6F). Apical amiloride (10 µM) essentially abolished Vt and increased Rt, and only negligible current (−1.5 ± 0.1 µA·cm−2) persisted in the presence of this ENaC blocker. Figure 6B shows that AVP induced a depolarization of Vt that became apparent immediately and developed over ∼30 min (Figure 6C). However, this depolarization was accompanied by a substantial fall in Rt (Figure 6B,D), and analysis using Ohm's Law showed that AVP actually caused a substantial augmentation of IEq (Figure 6E). Subsequent application of amiloride essentially abolished Vt (Figure 6B) and reduced IEq to a negligible level (−2.2 ± 1.0 µA·cm−2). Amiloride was added after 30 min exposure to AVP and the fact that this drug essentially abolishes the recorded current shows that the response to AVP measured at this time point must reflect a stimulation of electrogenic Na+ transport. Amiloride did not, however, increase Rt in AVP-stimulated cells (Figure 6B) and the fall in resistance evoked by this peptide hormone cannot therefore be attributed to ENaC activation. The mechanism of the AVP-induced fall in Rt was not investigated further. AVP (10 nM, 30 min) also evoked a significant increase in the abundance of Ser133-phosphorylated CREB that occurred with no change to overall expression of this protein (Figure 6F,G). As CREB-Ser133 is a substrate for PKA, this result shows that AVP activates the cAMP/PKA-dependent signalling pathway.
Figure 6.
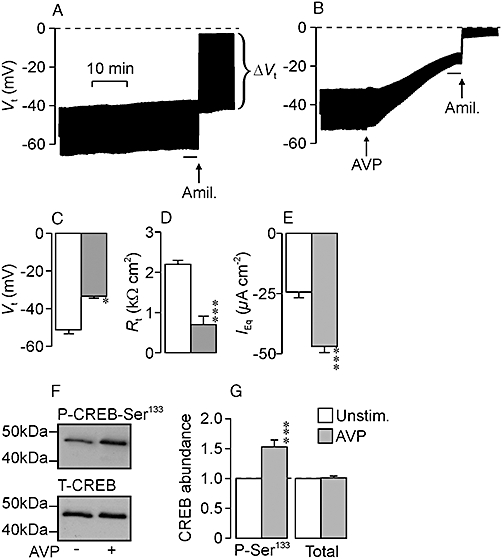
The response to arginine vasopressin (AVP). The upper panels show records of transepithelial voltage (Vt) derived from unstimulated (A) and AVP-treated (B, 10 nM, arrows) cells that were age-matched and at identical passage. Experiments were terminated by adding apical amiloride (Amil.; 10 µM, arrows). Throughout all such experiments pulses of transepithelial current (−10 µA·cm−2, 20 s) were injected every minute so that the transepithelial resistance (Rt) could be determined (Rt=ΔVt/ΔI) which allowed the equivalent short circuit current (IEq, i.e. the current required to hold Vt at 0 mV) to be calculated (IEq=Vt/Rt). The middle panels show pooled values (mean ± SEM) of Vt (C), Rt (D) and IEq (E) derived from a series (n = 9) of such experiments. These data are derived from analysis of data recorded immediately prior to the addition of amiloride. (F) Typical Western blots from experiments that explored the effects of AVP (10 nM, 30 min) upon the abundance of Ser133-phosphorylated (upper panel) and total (lower panel) cAMP response element binding protein (CREB). (G) Pooled data showing the effects of AVP upon the abundance of Ser133-phosphorylated (P-Ser133) and total CREB (n = 14). *P < 0.05, ***P < 0.001, significant effects of AVP; Student's paired t-test.
Effects of AVP upon the phosphorylation of NDRG1-Thr346/356/366
The control data in Figure 7 show AVP (10 nM, 30 min) slightly reduced (20–30%) the abundance of Thr346/356/366-phosphorylated NDRG1 without altering overall expression level. The AVP-induced stimulation of Na+ transport (Figure 6) thus appears to be associated with slight loss of SGK1 activity (Figure 7). Analysis of protein extracted from TORIN1- (Figure 7A,B) or PP242-treated (Figure 7C,D) cells confirmed that these substances abolished NDRG1-Thr346/356/366 phosphorylation, and showed that AVP had no effect upon the phosphorylation status of these residues in TORIN1/PP242-treated cells (Figure 7A,B).
Figure 7.
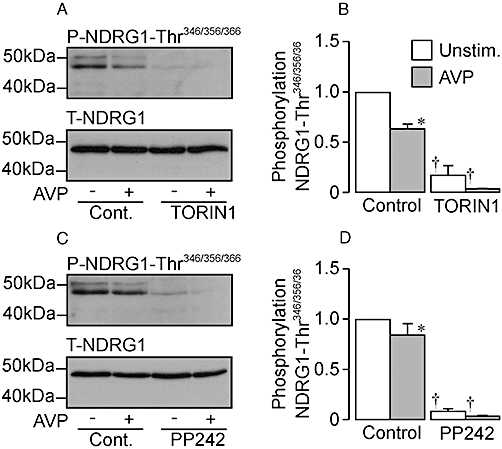
Effects of arginine vasopressin (AVP) upon the phosphorylation of Thr346/356/366 in protein encoded by n-myc downstream regulated gene 1 (NDRG1) in control, TORIN1-treated and PP242-treated cells. (A) Typical Western blots showing the effects of AVP (10 nM, 30 min exposure) upon the abundance of the Thr346/356/366-phosphorylated (upper panels) and total NDRG1 (lower panels) in control (Cont.) and TORIN1-pretreated (0.1 µM, 30 min) cells. (B) Pooled data showing the effects of AVP upon the abundance of Thr346/356/366-phosphorylated NDRG1 in control and TORIN1-treated cells (n = 6). (C) Western blots showing the effects of AVP (10 nM, 30 min exposure) upon the abundance of the Thr346/356/366-phosphorylated (upper panels) and total NDRG1 (lower panels) in control and PP242-pretreated (1 µM, 30 min) cells. (D) Pooled data showing the effects of AVP upon NDRG1-Thr346/356/366 phosphorylation in control and PP242-treated cells (n = 6). *P < 0.05, significant effects of AVP; Student's paired t-test. †P < 0.05, significant effects of TORIN1 or PP242; one-way anova, Bonferroni post hoc test). Unstim., unstimulated.
AVP-induced Na+ transport
Figure 8 shows that the AVP induced augmentation of IEq developed more rapidly than the equivalent response to insulin and was preceded by a transient suppression of IEq. TORIN1 (Figure 8B) and PP242 (Figure 8F) had no effect upon this response (Figure 8C,G).
Figure 8.
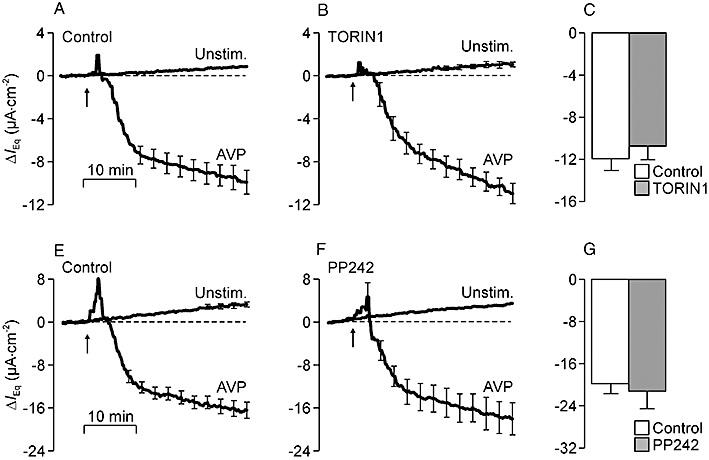
Effects of TORIN1 and PP242 upon the electrometric response to arginine vasopressin (AVP). (A) Changes in equivalent short circuit current (IEq) (n = 4) measured in studies of unstimulated (Unstim.) and AVP-stimulated (10 nM, basolateral, arrows) control cells (basal IEq unstimulated cells: −15.8 ± 0.6 µA·cm−2; basal IEq AVP-stimulated cells: −13.1 ± 1.0 µA·cm−2). (B) Data from age-matched cells that were pretreated (30 min) with 100 nM TORIN1 (basal IEq unstimulated cells: −12.8 ± 1.5 µA·cm−2; basal IEq AVP-stimulated cells: −13.3 ± 2.0 µA·cm−2). (C) Responses to AVP in control and TORIN1-treated cells were quantified by measuring the change in IEq that developed during exposure to AVP and subtracting the spontaneous change measured in unstimulated cells. (E) Data (n = 5) from separate control experiments showing the spontaneous and AVP-induced changes in IEq (basal IEq unstimulated cells: −27.5 ± 2.8 µA·cm−2; basal IEq AVP-stimulated cells: −30.3 ± 1.4 µA·cm−2). (F) Data from age-matched cells at identical passage that had been pretreated (30 min) with 1 µM PP242 (basal IEq unstimulated cells: −20.8 ± 1.8 µA·cm−2; basal IEq AVP-stimulated cells: −22.4 ± 1.6 µA·cm−2). (G) Responses to AVP quantified in control and PP242-treated cells.
Discussion
Effectiveness of TORC2 inhibitors
The aim of initial experiments was to ensure that the TORC2 inhibitors were effective under the conditions used in the present study and, as TORC2 catalyses the PI3K-dependent phosphorylation of PKB-Ser473 (Sarbassov et al., 2005), we therefore explored the effects of TORIN1 upon the phosphorylation of this residue. These studies showed that TORIN1 caused a concentration-dependent decline in the abundance of Ser473-phosphorylated PKB in hormone-deprived and insulin-stimulated cells suggesting strongly that TORIN1 does suppress TORC2 activity in mpkCCD cells. However, there is substantial homology between the catalytic domains of TORC1, TORC2 and PI3K, which makes it difficult to design compounds that block these kinases selectively (Brunn et al., 1993; Bain et al., 2007). Moreover, inhibition of PI3K or TORC2 would both prevent the phosphorylation of PKB-Ser473 (Bayascas and Alessi, 2005; Sarbassov et al., 2005), which raises the possibility that the present effect of TORIN1 may reflect direct inhibition of PI3K. Thoreen et al. (2009) addressed this issue by monitoring the phosphorylation of an alternative residue within PKB (Thr308) that is catalysed in a PIP3-dependent manner via a mechanism that does not involve TORC2 (Biondi et al., 2001; Bayascas and Alessi, 2005). However, as the TORC2-catalysed phosphorylation of PKB-Ser473 can allosterically enhance the phosphorylation of PKB-Thr308 (Biondi et al., 2001; Sarbassov et al., 2005), these studies were undertaken using cells lacking a critical component of the TORC2 complex to ensure that the phosphorylation status of PKB-Thr308 would provide a readout of PI3K activity (Thoreen et al., 2009). Whilst these studies showed that TORIN1 could inhibit PI3K, this was seen only in cells exposed to >1 µM TORIN1 whilst the inhibition of TORC2 was complete at <0.1 µM (Thoreen et al., 2009). The fact that the present effect on the phosphorylation status of PKB-Ser473 was complete at 0.1 µM TORIN1 therefore shows that this cannot be due to inhibition of PI3K. Moreover, although TORIN1 inhibits TORC1 and TORC2 (Thoreen et al., 2009), it is clear that rapamycin, an inhibitor of TORC1 (Bain et al., 2007), does not alter the phosphorylation status of PKB-Ser473 (present study, Sarbassov et al., 2005; Mansley and Wilson, 2010). Moreover, experiments that monitored the phosphorylation status of P70-S6K-Thr389 clearly showed that rapamycin did cause complete inhibition of TORC1 under the present conditions (Mansley and Wilson, 2010), whilst detailed in vitro studies have shown that, even when used at 1 µM, rapamycin does not suppress the activity of any member of a panel of 70 different protein kinases. It is therefore clear that rapamycin is an extremely selective inhibitor of the TORC1 signalling complex (Bain et al., 2007). The present data, in common with earlier findings (Thoreen et al., 2009) therefore show that TORC2 is fully inactivated by 0.1 µM TORIN1, and the fact that TORIN1 also blocked the phosphorylation of an endogenous SGK1 substrate (NDRG1-Thr346/356/366) supports the view that TORC2 is critical to the PI3K-dependent activation of SGK1 (García-Martínez and Alessi, 2008). Further evidence of this comes from the fact that PP242, a structurally unrelated compound that also inhibits TORC1/2 (Feldman et al., 2009), reproduced these effects of TORIN1. Exposing cells to TORIN1/PP242 therefore causes essentially complete loss of SGK1 activity, and subsequent experiments therefore explored the effects of these compounds upon the hormonal control of electrogenic Na+ absorption.
Hormone-deprived cells
Hormone-deprived mpkCCD cells generate IEq via an apparently spontaneous mechanism dependent upon ENaC (Bens et al., 1999; Mansley and Wilson, 2010) and, whilst the TORC2 inhibitors used in the present study caused full inactivation of SGK1 (see above), these substances caused only 10–20% inhibition of basal Na+ transport. As rapamycin was ineffective, these small effects must reflect inhibition of TORC2 and, because highly selective inhibitors of PI3K and SGK1 also have little effect upon basal Na+ absorption (Mansley and Wilson, 2010), it now appears that signalling via PI3K – TORC2 – SGK1 makes only a very small contribution to this basal Na+ transport process in this cell type. This accords well with the observation that deletion of the sgk1 gene does not alter renal Na+ handling in Na+ replete animals (Wulff et al., 2002).
The response to insulin
Insulin induced a clear stimulation of electrogenic Na+ transport that was accompanied by unambiguous activation of TORC2 and SGK1 (see also Mansley and Wilson, 2010) and these findings accord well with the view that insulin stimulates Na+ absorption within the distal nephron in addition to its well characterized role in carbohydrate metabolism (see e.g. Atchley et al., 1936; Miller and Bogdonoff, 1954; Defronzo et al., 1975; Tiwari et al., 2007; Loffing and Korbmacher, 2009). TORIN1 and PP242 completely abolished the insulin-induced activation of TORC2 and SGK1 and also reduced (∼80% inhibition) the stimulation of Na+ transport. As these responses to insulin were unaffected by rapamycin (Mansley and Wilson, 2010), these data show that this hormone stimulates Na+ absorption by activating the PI3K/TORC2/SGK1 pathway. This accords well with earlier data which show that insulin stimulates the trafficking of ENaC to the apical membrane via a mechanism dependent upon PI3K/SGK1 (see e.g. Blazer-Yost et al., 1998; 2003; Record et al., 1998; Wang et al., 2001; Arteaga and Canessa, 2006), and with the observation that sgk1 gene deletion blocks insulin-induced Na+ retention in vivo (Huang et al., 2006). However, whilst TORIN1 and PP242 effectively abolished SGK1 activity, small responses to insulin did persist in TORIN1/PP242-treated cells. Whilst the physiological basis of these residual responses was not investigated, TORC1/2 inhibition would not prevent PI3K-catalysed formation of PIP3 (Feldman et al., 2009; Thoreen et al., 2009) and such phospholipid second messengers can directly control ENaC activity (see e.g. Yue et al., 2002; Markadieu et al., 2004). It is therefore possible that PIP3 may mediate the residual responses to insulin in TORIN1/PP242-treated cells.
Effects of aldosterone
In our hands, mpkCCD cells were insensitive to physiologically relevant concentrations of aldosterone and this contrasts with the situation documented in the original description of this cell line which documents responses to 0.1–50 nM aldosterone (Bens et al., 1999). However, even in this study, high aldosterone concentrations (0.5 µM) were needed to achieve a maximal response and these authors concluded that the effects of this hormone were largely mediated via glucocorticoid receptors rather than via mineralocorticoid receptors (Bens et al., 1999) and such high concentrations of aldosterone have been used in subsequent studies of these cells (see e.g. Lu et al., 2010). The fact that we observe clear responses to 0.2 µM dexamethasone, a selective glucocorticoid receptor agonist, confirms that functional glucocorticoid receptors are present, and the reason why we did not see responses to physiological concentrations of aldosterone is unknown. However, aldosterone regulates Na+ transport in the distal nephron by binding to mineralocorticoid receptors, and the simplest explanation of our data is that the cellular expression of such receptors may not be maintained in vitro. Although this suggestion raises the possibility that such permanent cell lines may display modified responses to the other Na+ retaining hormones, it is clear that functional glucocorticoid receptors are present in the aldosterone-sensitive epithelia of the distal nephron (Loffing and Korbmacher, 2009). Indeed, glucocorticoid-receptor mediated Na+ retention may contribute to the development of hypertension in pathophysiological conditions, such as obesity and chronic stress, characterized by over activity of the hypothalamic – pituitary – adrenal axis (Bailey et al., 2009). Our subsequent experiments therefore characterize the effects of TORIN1/PP242 upon the response to this dexamethasone in order to define the role of TORC2 in such glucocorticoid receptor mediated Na+ transport.
The response to dexamethasone
Dexamethasone caused robust activation of SGK1 and this response was anticipated because SGK1 was originally identified as a glucocorticoid-inducible protein kinase (Lang et al., 2006; Loffing et al., 2006). This response was essentially abolished by TORIN1/PP242 but unaffected by rapamycin, and this activation of SGK1, in common with the response to insulin, therefore depends upon TORC2 and not TORC1. However, whilst insulin consistently caused a clear increase in TORC2 activity, the glucocorticoid-induced activation of SGK1 did not coincide with any discernible change to the activity of this kinase. Dexamethasone and insulin therefore activate SGK1 via different molecular mechanisms and it is therefore interesting that dexamethasone does seem to increase the expression of SGK1 protein far more effectively than insulin (Wang et al., 2001; Gonzalez-Rodriguez et al., 2007). This would be expected as the promoter region of SGK1 gene contains a glucocorticoid response element that would allow dexamethasone to regulate transcription (Itani et al., 2002). However, even if the response to dexamethasone was entirely dependent upon the de novo synthesis SGK1, TORC2 would still be needed because it is this kinase which confers catalytic activity upon the newly synthesized protein (Kobayashi and Cohen, 1999; Park et al., 1999; García-Martínez and Alessi, 2008). Whilst TORC2 is essential to the glucocorticoid-induced activation of SGK1, our data therefore suggest that the role of this signalling complex is permissive and it is therefore interesting that work recently published by Lu et al. (2010) shows that PP242 can inhibit the current generated by mpkCCD cells that have been exposed to a combination of aldosterone and insulin. As rapamycin did not alter this current, these authors also concluded that TORC2 is critical to the hormonal control of ENaC function in these cells. Moreover, these authors (Lu et al., 2010) showed that this hormone-stimulated current is also inhibited by suppressing the expression of a critical component of the TORC2 signalling complex, and present evidence confirms (García-Martínez and Alessi, 2008) that it is TORC2, and not TORC1, that catalyse the phosphorylation of SGK1-Ser422. All data in this report are derived from cells exposed to aldosterone at a concentration (1 µM) much higher than that encountered in vivo and the responses described in this study will therefore be mediated, at least in part, via glucocorticoid receptors (see e.g. Bens et al., 1999).
Although it did not alter the amount of NDRG1 in the cells, dexamethasone induced the appearance of a second, less mobile band on blots probed with the antibody against the full length protein. Two such bands were also identified in protein extracted from HeLa cells (Murray et al., 2005), and it appears that the phosphorylation of NDRG1-Thr346/356/366 converts this protein into a substrate for glycogen synthase kinase 3 (GSK3) which can then phosphorylate NDRG1 at Ser342/352/362. The second, less mobile band seems to correspond to the GSK3-phosphorylation form of NDRG1 (Murray et al., 2005), and the fact that dexamethasone, but not insulin, induced the formation of this band raises the possibility that glucocorticoids might activate GSK3 as well as SGK1.
Dexamethasone caused a clear and consistent stimulation of Na+ absorption and, although this response developed more slowly than the response to insulin, the responses to both hormones were antagonized by TORIN1/PP242. However, whilst the response to insulin was unaffected by rapamycin (Mansley and Wilson, 2010), this TORC1 inhibitor did suppress the dexamethasone-induced stimulation of Na+ transport, despite the fact that it did not prevent the accompanying activation of SGK1. This surprising result therefore identifies a further difference between the mechanisms that allow insulin and dexamethasone to regulate Na+ transport and, despite the evidence implicating SGK1 in the control of Na+ transport, the present data show that the glucocorticoid-induced activation of SGK1 does not provide a stimulus sufficient to drive increased Na+ absorption. Although the hormone-sensitive currents described by Lu et al. (2010) were insensitive to rapamycin, the cells used in this study were all exposed to a high concentration aldosterone in combination with insulin, and this experimental design implies that a specific effect of rapamycin upon the responses to steroid hormones may not have been detected.
The mechanisms that allow TORC1 to contribute to the glucocorticoid-dependent control of Na+ transport is unknown although it may be relevant that steroid hormones activate other signalling pathways involved in the regulation of ENaC activity. Such hormones thus evoke expression of glucocorticoid-inducible leucine zipper proteins (GILZ1-3) that appear to stimulate Na+ transport by suppressing the activity of extracellular-signal regulated kinases (ERK1/2) (Soundararajan et al., 2005). Moreover, it has recently been suggested that GILZ1 and SGK1 act synergistically to increase the abundance of ENaC in the plasma membrane (Soundararajan et al., 2009). Since our data suggest that inhibition of TORC1 suppresses glucocorticoid-induced Na+ transport without preventing the activation of SGK1, it is possible that TORC1 may play a role in the induction of GILZ1. Otulakowski et al. (2007), on the other hand, report that α-ENaC protein expression is dependent upon a signalling mechanism downstream to TORC1, and their studies of foetal distal lung epithelial cells, in common with the present data, show that electrogenic Na+ transport can be inhibited by rapamycin under certain conditions. Further evidence of a role for TORC1 comes from studies with mice with a mutation in the adenomatous polyposis (APC) gene. This mutation induces a complex phenotype that includes hyper-absorption of Na+ in the colon (Koehl et al., 2010) and it is therefore relevant that APC gene mutation causes activation of TORC1 and that the apparent hyper-activation of ENaC is corrected by rapamycin (Koehl et al., 2010). Although the mechanism is obscure, at least two other lines of evidence implicate TORC1 in the control of ENaC function (Otulakowski et al., 2007; Koehl et al., 2010).
The response to AVP
Arginine vasopressin evoked changes in Vt and Rt that were very different to those induced by insulin (Mansley and Wilson, 2010) or dexamethasone as this hormone caused a depolarization of Vt that was associated with a marked fall in Rt. However, further analysis clearly showed that AVP did increase amiloride-sensitive IEq and this accords with the body of work indicating that this peptide hormone exerts a Na+ retaining action in addition to its better-characterized anti-diuretic effect. Perhaps the best evidence of this comes from recent studies of micro-dissected mouse cortical collecting ducts which revealed acute (2–3 min) activation of ENaC in response to AVP and showed that this response was dependent upon the cAMP/PKA-dependent signalling pathway (Bugaj et al., 2009). Studies of several renal epithelial cell lines have similarly shown that the Na+ retaining effect of AVP is mediated via cAMP/PKA (see Loffing and Korbmacher, 2009) and our observation that AVP evokes CREB-Ser133 phosphorylation provides further evidence of this. However, despite these clear findings, studies of A6 cells suggest that PI3K/SGK1 may play a critically important role in this response (Edinger et al., 1999), and cAMP-coupled hormones do seem to stimulate SGK1 expression/phosphorylation in ovarian granulosa cells (Gonzales-Robayana et al., 2000) whilst PKA appears able to activate recombinant SGK1 (Perrotti et al., 2001). It has therefore been suggested that cAMP/PKA-coupled agonists might control Na+ transport by activating SGK1 (Perrotti et al., 2001; Thomas et al., 2004; Vasquez et al., 2008).
The present data, however, show that AVP-induced Na+ transport is not associated with increased phosphorylation of NDRG1-Thr346/356/366. Indeed, AVP actually reduced the abundance of this phosphoprotein suggesting slight inhibition of SGK1. Moreover, although TORIN1 and PP242 clearly inactivated SGK1, these substances did not alter AVP-induced Na+ transport and our data therefore indicate that cAMP-coupled agonists stimulate Na+ absorption via a mechanisms completely independent of SGK1 (Inglis et al., 2009). It is therefore interesting that Snyder et al. (2004a) have shown that LY294002, an inhibitor of PI3K, does not prevent the cAMP-induced activation of ENaC expressed in Fisher rat thyroid cells. Indeed, these authors suggested that PKA stimulates Na+ transport by phosphorylating and inactivating neural precursor cell expressed, developmentally down-regulated protein 4-2 (Nedd-4/2), a ubiquitin ligase that can target the ENaC channel complex for internalization/degradation. Since Nedd-4/2 is also phosphorylated by SGK1 (Debonneville et al., 2001; Snyder et al., 2002), this protein may represent an important point of convergence between the PI3K and the PKA-dependent pathways (Snyder et al., 2004a).
Significance of present findings
The regulated re-absorption of Na+ within the distal nephron determines the amount of Na+ lost in urine and is therefore critical to whole body Na+ and water balance and to the control of blood pressure. Inappropriate stimulation of this Na+ transport process underlies several forms of hypertension (Loffing and Korbmacher, 2009) and may complicate the clinical management of patients with type 2 diabetes who receive insulin-sensitizing drugs (Buckingham and Hanna, 2007). Our data confirm that TORC2 is of central importance to SGK1 activity (García-Martínez and Alessi, 2008) and indicate that insulin stimulates Na+ transport by activating the PI3K – TORC2 – SGK1 pathway. Whilst this pathway is also essential to the glucocorticoid-induced activation of SGK1, its role in this response appears to be entirely permissive, whilst AVP-induced Na+ transport seems to involve a separate mechanism. Moreover, whilst TORC1 activity is not needed for the insulin-dependent control of Na+ transport, TORC1 does seem to be essential for glucocorticoid-induced Na+ retention. Future studies must therefore clarify the way in which TORC1 contributes to the control of Na+ transport by glucocorticoids and use alternative experimental systems that are sensitive to physiologically relevant concentrations of aldosterone, to define the role/s of TORC1/2 in the response to this critically important hormone. The present data do, however, raise the possibility of treating Na+-sensitive hypertension by pharmacological manipulation of TORC1/2.
Acknowledgments
This work was supported by an MRC Project grant (SMW) and by a postgraduate studentship (MKM) from the MRC doctoral training scheme. The authors are grateful to Professors Dario Alessi, D. Sabatini and Phillip Cohen for the provision of many reagents and for valuable advice concerning their use.
Glossary
Abbreviations
- APC
adenomatous polyposis
- AVP
arginine vasopressin
- CREB
cAMP response element binding protein
- ENaC
epithelial sodium channel
- GSK3
glycogen synthase kinase 3
- IEq
equivalent short circuit current
- IGF-1
insulin-like growth factor 1
- mpkCCD
mouse cortical collecting duct cells
- NDRG1
protein encoded by n-myc downstream regulated gene 1
- Nedd-4/2
neural precursor cell expressed, developmentally down-regulated protein 4-2
- P70-S6K
70 kDa ribosomal S6 kinase
- PDK1
phosphoinositide-dependent protein kinase 1
- PI3K
phosphoinositide-3-kinase
- PIP3
phosphatidylinositol 3,4,5-trisphosphate
- PKA
adenine-nucleotide-dependent protein kinase
- PKB
protein kinase B (also known as Akt)
- Rt
transepithelial resistance
- SGK1
serum and glucocorticoid-inducible protein kinase 1
- TORC1
and 2, target of rapamycin complex 1 and 2
- Vt
transepithelial voltage
Conflict of interest
The authors report no conflicts of interest.
Supporting Information
Teaching Materials; Figs 1–8 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arteaga MF, Canessa C. Functional specificity of Sgk1 and Akt1 on ENaC activity. Am J Physiol Renal Physiol. 2006;289:90–96. doi: 10.1152/ajprenal.00390.2004. [DOI] [PubMed] [Google Scholar]
- Atchley D, Loeb RF, Richards DW, Benedict EM, Driscoll ME. On diabetic acicosis. A detailed study of electrolyte balances following the withdrawal and reestablishment of insulin therapy. J Clin Invest. 1936;12:297–326. doi: 10.1172/JCI100504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey MA, Mullins JJ, Kenyon CJ. Mineralocorticoid and glucocorticoid receptors stimulate epithelial sodium channel activity in a mouse model of cushing syndrome. Hypertension. 2009;54:890–896. doi: 10.1161/HYPERTENSIONAHA.109.134973. [DOI] [PubMed] [Google Scholar]
- Bain J, Plater L, Elliott M, Shpiro N, Hastie J, McClauchlan H, et al. The selectivity of protein kinase inhibitors: a further update. Biochem J. 2007;408:297–315. doi: 10.1042/BJ20070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayascas JR, Alessi DR. Regulation of Akt/PKB Ser473 phosphorylation. Mol Cell. 2005;18:143–145. doi: 10.1016/j.molcel.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Bens M, Vallet V, Cluzeaud F, Padcula-Letallec L, Kahn A, Rafestin-Oblin ME, et al. Corticosteroid-dependent sodium transport in a novel immortalized mouse collecting duct principal cell line. J Am Soc Nephrol. 1999;10:923–934. doi: 10.1681/ASN.V105923. [DOI] [PubMed] [Google Scholar]
- Biondi RM, Kieloch A, Currie RA, Deak M, Alessi DR. The PIF-binding pocket in PDK1 is essential for activation of S6K and SGK, but not PKB. EMBO J. 2001;20:4380–4390. doi: 10.1093/emboj/20.16.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blazer-Yost BL, Liu XH, Helman SI. Hormonal regulation of ENaCs: insulin and aldosterone. Am J Physiol Cell Physiol. 1998;274:1373–1379. doi: 10.1152/ajpcell.1998.274.5.C1373. [DOI] [PubMed] [Google Scholar]
- Blazer-Yost BL, Esterman MA, Vlahos CJ. Insulin-stimulated trafficking of ENaC in renal cells requires PI3-kinase activity. Am J Physiol Cell Physiol. 2003;284:C1645–C1653. doi: 10.1152/ajpcell.00372.2002. [DOI] [PubMed] [Google Scholar]
- Brunn GJ, Williams J, Sabers C, Wiederrecht G, Lawrence JC, Abraham RT. Direct inhibition of the signaling functions of the mammalian target of rapamycin by the phosphoinositide 3-kinase inhibitors, wortmannin and LY294002. EMBO J. 1993;15:5256–5267. [PMC free article] [PubMed] [Google Scholar]
- Buckingham RE, Hanna A. Thiazolidinedione insulin sensitizers and the heart: a tale of two organs. Diabetes Obes Metab. 2007;10:312–328. doi: 10.1111/j.1463-1326.2006.00700.x. [DOI] [PubMed] [Google Scholar]
- Bugaj V, Pochynyuk O, Stockand JD. Activation of the epithelial Na+ channel in the collecting duct by vasopressin contributes to water reabsorption. Am J Physiol Renal Physiol. 2009;297:F1411–F1418. doi: 10.1152/ajprenal.00371.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P. Timeline – The twentieth century struggle to decipher insulin signalling. Nature Rev Mol Cell Biol. 2006;7:867–873. doi: 10.1038/nrm2043. [DOI] [PubMed] [Google Scholar]
- Debonneville C, Flores SY, Kamynina E, Plant PJ, Tauxe C, Thomas MA, et al. Phosphorylation of Nedd4-2 by Sgk1 regulates epithelial channel surface expression. EMBO J. 2001;20:7052–7059. doi: 10.1093/emboj/20.24.7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Defronzo RA, Cooke CR, Andres R, Faloona GR, Davis PJ. Effect of insulin on renal handling of sodium, potassium, calcium, and phosphate in man. J Clin Invest. 1975;55:845–855. doi: 10.1172/JCI107996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edinger RS, Rokaw MD, Johnson JP. Vasopressin stimulates sodium transport in A6 cells via a phosphatidylinositide 3-kinase-dependent pathway. Am J Physiol Renal Physiol. 1999;277:F575–F579. doi: 10.1152/ajprenal.1999.277.4.F575. [DOI] [PubMed] [Google Scholar]
- Feldman ME, Apsel B, Uotila A, Loewith R, Knight ZA, Ruggero D, et al. Active-site inhibitors of mTOR target rapamycin-resistant outputs of mTORC1 and mTORC2. Plos Biol. 2009;7:371–383. doi: 10.1371/journal.pbio.1000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Martínez JM, Alessi DR. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-inducible protein kinase 1 (SGK1) Biochem J. 2008;416:375–385. doi: 10.1042/BJ20081668. [DOI] [PubMed] [Google Scholar]
- Gonzales-Robayana IJ, Falender AE, Ochsner S, Firestone GL, Richards JS. Follicle-stimulating hormone (FSH) stimulates phopsphorylation / activation of protein kinase B (PKB/Akt) and serum and glucocorticoid-induced kinase (Sgk): evidence for A kinase-independent signalling by FSH in granulosa cells. Mol Endocrinol. 2000;14:1283–1300. doi: 10.1210/mend.14.8.0500. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Rodriguez E, Gaeggeler HP, Rossier BC. IGF-1 vs insulin: respective roles in modulating sodium transport via the PI-3 kinase/Sgk1 pathway in a cortical collecting duct cell line. Kidney Int. 2007;71:116–125. doi: 10.1038/sj.ki.5002018. [DOI] [PubMed] [Google Scholar]
- Huang DY, Boini KM, Freidrich B, Metzger M, Just L, Osswald H, et al. Blunted hypertensive effect of combined fructose and high salt intake in gene targeted mice lacking serum and gluccocorticoid-inducible kinase SGK1. Am J Physiol Regul Integr Comp Physiol. 2006;290:R935–R944. doi: 10.1152/ajpregu.00382.2005. [DOI] [PubMed] [Google Scholar]
- Inglis SK, Gallacher M, Brown SG, McTavish N, Getty J, Husband EM, et al. SGK1 activity in Na+ asorbing human airway epithelial cells monitored by assaying NDRG1-Thr346/356/366 phosphorylation. Pflugers Arch. 2009;457:1287–1301. doi: 10.1007/s00424-008-0587-1. [DOI] [PubMed] [Google Scholar]
- Itani OA, Liu KZ, Cornish KL, Campbell JR, Thomas CP. Glucocorticoids stimulate human sgk1 gene expression by activation of a GRE in its 5'-flanking region. Am J Physiol Endocinol Metab. 2002;283:E971–E979. doi: 10.1152/ajpendo.00021.2002. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinases by agonists that activate phosphatidylinositol 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase 1 (PDK1) and PDK2. Biochem J. 1999;339:319–328. [PMC free article] [PubMed] [Google Scholar]
- Koehl GE, Spitzner M, Ousingsawat J, Schreiber R, Geissler EK, Kunzelmann K. Rapamycin inhibits oncogenic intestinal ion channels and neoplasia in APCMin/+ mice. Oncogene. 2010;29:1553–1560. doi: 10.1038/onc.2009.435. [DOI] [PubMed] [Google Scholar]
- Lang F, Böhmer C, Palmada M, Seebohm G, Strutz-Seebohm N, Vallon V. Patho)physiological significance of the serum and glucocorticoid-inducible kinase isoforms. Physiol Rev. 2006;86:1151–1178. doi: 10.1152/physrev.00050.2005. [DOI] [PubMed] [Google Scholar]
- Loffing J, Korbmacher C. Regulated sodium transport in the renal collecting tubule (CNT) via the epithelial sodium channel (ENaC) Pflugers Arch. 2009;458:111–135. doi: 10.1007/s00424-009-0656-0. [DOI] [PubMed] [Google Scholar]
- Loffing J, Flores SY, Staub O. SGK kinases and their role in epithelial transport. Annu Rev Physiol. 2006;68:461–430. doi: 10.1146/annurev.physiol.68.040104.131654. [DOI] [PubMed] [Google Scholar]
- Lu M, Wang J, Jones KT, Ives HE, Feldman ME, Yao L-J, et al. mTOR complex-2 activates ENaC by phosphorylating SGK1. J Am Soc Nephrol. 2010;21:811–818. doi: 10.1681/ASN.2009111168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansley MK, Wilson SM. Effects of nominally selective inhibitors of the kinases PI3K, SGK1 and PKB upon the insulin-dependent control of epithelial Na+ absorption. Br J Pharmacol. 2010;161:571–588. doi: 10.1111/j.1476-5381.2010.00898.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markadieu N, Blero D, Boom A, Erneux C, Beauwens R. Phosphatidylinositol 3,4,5-trisphosphate: an early mediator of insulin-stimulated sodium transport in A6 cells. Am J Physiol Renal Physiol. 2004;287:F319–F328. doi: 10.1152/ajprenal.00314.2003. [DOI] [PubMed] [Google Scholar]
- Miller JH, Bogdonoff MD. Antidiuresis associated with administration of insulin. J Appl Physiol. 1954;6:509–512. doi: 10.1152/jappl.1954.6.8.509. [DOI] [PubMed] [Google Scholar]
- Murray JT, Cambell DG, Morrice N, Auld G, Shpiro N, Marquez R, et al. Exploitation of KESTREL to identify NDRG family members as physiological substrates of SGK1 and GSK3. Biochem J. 2005;385:1–12. doi: 10.1042/BJ20041057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otulakowski G, Duan W, Gandhi S, O'Brodovich H. Steroid and oxygen effects of eIF4F Complex, mTOR and ENaC translation in fetal lung epithelium. Am J Respir Cell Mol Biol. 2007;37:457–466. doi: 10.1165/rcmb.2007-0055OC. [DOI] [PubMed] [Google Scholar]
- Park J, Leong MLL, Buse P, Maiyar AC, Firestone GL, Hemmings BA. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 1999;18:3024–3033. doi: 10.1093/emboj/18.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paunescu TG, Blazer-Yost BL, Vlahos CJ, Helman SI. LY-294002-inhibitable PI 3-Kinase and regulation of baseline rates of Na+ transport in A6 epithelia. Am J Physiol Cell Physiol. 2000;279:C236–C247. doi: 10.1152/ajpcell.2000.279.1.C236. [DOI] [PubMed] [Google Scholar]
- Perrotti N, He RA, Phillips SA, Haft CR, Taylor SI. Activation of serum- and glucocorticoid-induced protein kinase (Sgk) by cyclic AMP and insulin. J Biol Chem. 2001;276:6406–9412. doi: 10.1074/jbc.M007052200. [DOI] [PubMed] [Google Scholar]
- Proud CG. Signalling to translation: how signal transduction pathways control the protein synthetic machinery. Biochem J. 2007;403:217–234. doi: 10.1042/BJ20070024. [DOI] [PubMed] [Google Scholar]
- Record RD, Froelich LL, Vlahos CJ, Blazer-Yost BL. Phoshatidylinositol 3-kinase activation is required for insulin-stimulated sodium transport in A6 cells. Am J Physiol Endocinol Metab. 1998;274:E611–E617. doi: 10.1152/ajpendo.1998.274.4.E611. [DOI] [PubMed] [Google Scholar]
- Rexhepaj R, Artunc F, Grahammer F, Nasir O, Sandu C, Freidrich B, et al. SGK1 is not required for regulation of colonic ENaC activity. Pflugers Arch. 2006;453:97–105. doi: 10.1007/s00424-006-0111-4. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–1101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- Snyder PM, Olsen DR, Thomas BC. Serum and glucocorticoid-regulated kinase modulates Nedd4-2-mediated inhibition of the epithelial Na+ channel. J Biol Chem. 2002;277:5–8. doi: 10.1074/jbc.C100623200. [DOI] [PubMed] [Google Scholar]
- Snyder PM, Olson DR, Kabra R, Zhou R, Steines JC. cAMP and serum and glucocorticoid-inducible kinase (SGK) regulate the epithelial Na+ channel through convergent phosphorylation of Nedd4-2. J Biol Chem. 2004a;279:45753–45758. doi: 10.1074/jbc.M407858200. [DOI] [PubMed] [Google Scholar]
- Snyder PM, Steines JC, Olson DR. Relative contribution of Nedd4 and Nedd4-2 to ENaC regulation in epithelia determined by RNA interference. J Biol Chem. 2004b;279:5042–5046. doi: 10.1074/jbc.M312477200. [DOI] [PubMed] [Google Scholar]
- Soundararajan R, Zhang TT, Wanerwall A, Pearce D. A novel role fof Glucocorticoidininduced leucine zipper protein in epithelial sodium channel-mediated sodium transport. J Biol Chem. 2005;280:39970–39981. doi: 10.1074/jbc.M508658200. [DOI] [PubMed] [Google Scholar]
- Soundararajan R, Melters D, Shih I-C, Wang J, Pearce D. Epithelial sodium channel regulated by differential composition of a signaling complex. Proc Natl Acad Sci USA. 2009;106:7804–7809. doi: 10.1073/pnas.0809892106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CP, Campbell JR, Wright PJ, Husted RF. cAMP-stimulated Na+ transport in H441 distal lung epithelial cells: role of PKA, phosphatidylinositol 3-kinase, and sgk1. Am J Physiol Lung Cell Mol Physiol. 2004;287:L843–L851. doi: 10.1152/ajplung.00340.2003. [DOI] [PubMed] [Google Scholar]
- Thoreen CC, Kang SA, Chang JW, Liu QS, Zhang JM, Gao Y, et al. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284:8023–8032. doi: 10.1074/jbc.M900301200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari S, Riazi S, Ecelbarger CA. Insulin's impact on renal sodium transport and blood pressure in health, obesity and diabetes. Am J Physiol Renal Physiol. 2007;293:F974–F984. doi: 10.1152/ajprenal.00149.2007. [DOI] [PubMed] [Google Scholar]
- Vasquez MM, Castro R, Seidner SR, Henson BM, Ashton DJ, Mustafa SB. Induction of Serum- and Glucocorticoid-Induced Kinase-1 (SGK1) by cAMP regulates increases in alpha-ENaC. J Cell Physiol. 2008;217:632–642. doi: 10.1002/jcp.21534. [DOI] [PubMed] [Google Scholar]
- Wang D, Huang N, Gonzalez FA, Heppel LA. Multiple signal transduction pathways lead to ATP-stimulated mitogenesis in mammalian cells: 1. Involvement of protein kinase C-dependent and independent pathways. J Cell Physiol. 1991;146:473–482. doi: 10.1002/jcp.1041460319. [DOI] [PubMed] [Google Scholar]
- Wang J, Barbary P, Maiyar A, Rozansky DJ, Bhargava A, Leong M, et al. SGK integrates insulin and mineralocorticoid regulation of epithelial sodium transport. Am J Physiol Renal Physiol. 2001;280:F303–F313. doi: 10.1152/ajprenal.2001.280.2.F303. [DOI] [PubMed] [Google Scholar]
- Wulff P, Vallon V, Huang DY, Volkl H, Yu F, Richter K, et al. Impaired renal Na+ retention in the sgk1-knockout mouse. J Clin Invest. 2002;110:1263–1268. doi: 10.1172/JCI15696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue G, Malik B, Yue G, Eaton DC. Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. J Biol Chem. 2002;277:11965–11969. doi: 10.1074/jbc.M108951200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.