

Abstract
BACKGROUND AND PURPOSE
Hydrogen sulphide (H2S), considered as a novel gas transmitter, is produced endogenously in mammalian tissue from L-cysteine by two enzymes, cystathionine β-synthase and cystathionine γ-lyase. Recently, it has been reported that H2S contributes to the local and systemic inflammation in several experimental animal models. We conducted this study to investigate on the signalling involved in H2S-induced inflammation.
EXPERIMENTAL APPROACH
L-cysteine or sodium hydrogen sulphide (NaHS) was injected into the mouse hind paw and oedema formation was evaluated for 60 min. In order to investigate H2S-induced oedema formation, we used 5-HT and histamine receptor antagonists, and inhibitors of KATP channels or arachidonic acid cascade. Prostaglandin levels were determined in hind paw exudates by radioimmunoassay. Paws injected with L-cysteine or NaHS were examined by histological methods.
KEY RESULTS
Both NaHS and L-cysteine caused oedema characterized by a fast onset which peaked at 30 min. This oedematogenic action was not associated with histamine or 5-HT release or KATP channel activation. However, oedema formation was significantly inhibited by the inhibition of cyclooxygenases and selective inhibition of phospholipase A2. Prostaglandin levels were significantly increased in exudates of hind paw injected with NaHS or L-cysteine. The histological examination clearly showed an inflammatory state with a loss of tissue organization following NaHS or L-cysteine injection.
CONCLUSIONS AND IMPLICATIONS
Phospholipase A2 and prostaglandin production are involved in pro-inflammatory effects of H2S in mouse hind paws. The present study contributes to the understanding of the role of L-cysteine/H2S pathway in inflammatory disease.
Keywords: hydrogen sulphide, L-cysteine, oedema, mice, prostaglandins, phospholipase A2
Introduction
Hydrogen sulphide (H2S) is known to be a highly toxic pollutant, but it has recently been recognized as the third gaseous physiological transmitter. H2S is synthesized endogenously from L-cysteine or L-methionine by two pyridoxal -5'-phosphate dependent enzymes, cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE). Several studies support a pro-inflammatory role for H2S in different experimental animal models (Hui et al., 2003; Bhatia et al. 2005a,b; Li et al., 2005; Mok and Moore, 2008; Tamizhselvi et al., 2008). In endotoxemic animals, the expression of both CBS and CSE was up-regulated with an augmented H2S biosynthesis in different organs, such as liver, kidney, lung and blood (Li et al., 2005; 2006;). Plasma levels of H2S were similarly increased in human septic shock (Li et al., 2005). Elevated levels of H2S in plasma in haemorrhagic shock was also associated with an increase in inflammatory parameters, along with neutrophil infiltration in liver and lung (Mok et al., 2004; Mok and Moore, 2008). The same authors have shown that the treatment with CSE inhibitors led to a reduction in cell migration as well as a rapid restoration in blood pressure and heart rate in rats (Mok et al., 2004; Mok and Moore, 2008). In a model of caereulin-induced pancreatitis and associated lung injury, blockade of CSE significantly reduced plasma level of amylase, neutrophil sequestration, pancreatic acinal cell and lung injury (Bhatia et al., 2005b). H2S also contributed to neurogenic inflammation, stimulating the sensory nerve to release substance P (Bhatia, 2010). In addition, rat hind paw H2S concentrations were elevated in formalin-induced nociceptive flinching and this effect was reversed by the inhibition of CSE (Lee et al., 2008). Moreover, the intraplantar injection of carrageenan in rats increased H2S levels, as well as myeloperoxidase activity, in the hind paw (Bhatia et al., 2005a).
All together, these findings suggest an important pro-inflammatory role of H2S but the signalling pathway(s) involved has not been extensively investigated. Therefore, in this study we have addressed this specific issue by using sodium hydrogen sulphide (NaHS) as an exogenous source of H2S to generate inflammation, as well as L-cysteine, the major endogenous substrate for CSE.
Methods
Animals
All animal care and experimental procedures in this study followed the Principles of Laboratory Animal Care (NIH publication no. 86-23, revised 1985), as well as the specific guidelines of the Italian (N. 116/1992) and European Council law (N. 86/609/CEE). Male mice (CD-1, Harlan, Udine, Italy, 38–40 g) were used for in vivo and ex vivo experiments. Animals were kept at temperatures of 23 ± 2°C, humidity range 40–70% and 12 h light/dark cycles. Food and water were provided ad libitum.
Measurement of mouse paw oedema
Mice received an intraplantar injection of NaHS (100–300 and 500 µg per paw) as exogenous source of H2S or vehicle [30 µL of potassium phosphate buffer (PPS), pH 7.4]. The same protocol was used for L-cysteine, as endogenous source of H2S, and the dose chosen on the basis of a preliminary study, was 500 µg per paw. Next, to be certain of a specific effect mediated by L-cysteine and in turn by H2S, via CBS and CSE, we injected d-cysteine (500 µg per paw) as negative control.
Pharmacological modulation was achieved with different inhibitors. In order to evaluate an involvement of histamine/5-HT in H2S-induced oedema, cyproheptadine (CPR; 5 mg·kg−1, i.p.), as a histamine/5-HT receptor antagonist was used. To assess the contribution of prostaglandins (PG), pharmacological inhibitors of the arachidonic acid cascade were tested such as indomethacin (INDO; a non-selective cyclooxygenase inhibitor, 10 mg·kg−1, per os), dexamethasone (DEX; a PLA2 inhibitor, 1 mg·kg−1, per os), 4-(4-octadecylphenyl)-4-oxobutenoic acid (OBAA; a selective PLA2 inhibitor, 0.1, 0.3 or 1 mg·kg−1, per os) or YM 26734 (secretory PLA2 inhibitor, 0.3, 1 or 3 mg·kg−1, per os). Glibenclamide (GLB; a blocker of KATP channels, 10 mg·kg−1, i.p.) was used in order to assess the effects of H2S mediated by KATP channel activation. The inhibitors were given orally as solutions in carboxymethyl-cellulose (0.5% w/v) and Tween 20 (10% v/v) aqueous solutions, at a final volume of 100 µL. All the inhibitors given per os were administered 90 min before the NaHS or L-cysteine injection (500 µg per paw) (Church and Miller, 1978; Marshall et al., 1989). CPR or GLB were administered 30 min before NaHS or L-cysteine (500 µg per paw) (Cirino et al., 1996; Zanardo et al., 2006). Paw oedema was measured by means of a plethysmometer (Ugo Basile, Comerio, Italy) at several time points (15, 30, 45 and 60 min) after the intraplantar injection of NaHS or L-cysteine. The results were expressed as increase in paw volume (µL).
Western blotting analysis
The animals were killed by cervical dislocation, and the left paws were removed and frozen at −80°C until assayed, as follows. The tissues were frozen in liquid nitrogen and then homogenized in modified RIPA buffer (Tris-HCl 50 mM, pH 7.4, Triton 1%, sodium deoxycholate 0.25%, NaCl 150 mM, EDTA 1 mM, phenylmethylsulphonyl fluoride 1 mM, aprotinin 10 µg mL−1, leupeptin 20 µM, NaF 1 mM, sodium orthovanadate 1 mM). After centrifugation of homogenates at 8000×g for 15 min, protein concentration was determined by Bradford assay using BSA as standard (Bio-Rad Laboratories, Milan, Italy). Denatured proteins (40 µg) were separated on 10% sodium dodecyl sulfate polyacrylamide gels and transferred to a polyvinylidene fluoride membrane. Membranes were blocked by incubation in phosphate-buffered saline (PBS) containing 0.1% v/v Tween 20 and 5% non-fat dried milk for 1 h at room temperature and then incubated with rabbit polyclonal antibody for CBS (1:1000; Santa Cruz Biotechnology, Inc., Heidelberg, Germany) and with mouse monoclonal antibody for CSE (1:1000; Santa Cruz Biotechnology, Inc.) overnight at 4°C. The membranes were washed extensively in PBS containing 0.1% v/v Tween-20 and then incubated for 2 h at 4°C with anti-rabbit or anti-mouse IgG-horseradish peroxidase conjugate (1:5000). The filters were then washed and the immunoreactive bands, visualized using the enhanced chemiluminescence substrate (Amersham Pharmacia Biotech, San Diego, CA, USA), were densitometrically analysed with a model GS-800 imaging densitometer (Biorad, Milan, Italy).
Assay of PGE2 levels in exudates of hind paws
Mice were killed with carbon dioxide at 30 min after NaHS (500 µg per paw) or vehicle (30 µL, PPS) administration. In order to obtain the exudates (supernatants) to measure PG levels, the paws were cut and were suspended from a hook in a tube and immediately centrifuged at 3000×g for 30 min. Exudates were collected with 100 µL of saline and used for PGE2 quantification (Posadas et al., 2004.). To determine PGE2 levels, proteins were removed from the exudates with ZnSO4 (30% for 15 min; Thomsen et al., 1990). PGE2 levels were determined in deproteinized exudates by radioimmunoassay according to manufacturers' instructions.
Histological analysis
The animals were killed by cervical dislocation 30 min after the NaHS (500 µg per paw), L-cysteine (500 µg per paw) or vehicle intra-plantar injection. Mice paws were fixed in neutral buffered formalin before being embedded in paraffin. Sections (4 µm) were stained with haematoxylin-eosin and analysed under light microscopy, by an observer unaware of the treatment protocol.
Statistical analysis
The results were calculated as mean ± s.e., n = 12 mice for each treatment. Statistical analysis was performed using Student's t-test or two way anova followed by Bonferroni's test, as needed. P-values less than 0.05 were considered as significant.
Materials
Potassium phosphate buffer 0.1 M (pH 7.4) was prepared mixing appropriate volumes of K2HPO4 (0.1 M) and KH2PO4 (0.1 M). The reagents used were (sources in parentheses): mouse anti-CBS polyclonal antibody (Santa Cruz Biotechnology, DBA, Milan, Italy), mouse anti-CSE monoclonal antibody (Santa Cruz Biotechnology, DBA), sodium hydrogen sulphide (Sigma, Milan, Italy), CPR (Sigma), 4-(4-octadecylphenyl)-4-oxobutenoic acid (Tocris, Bristol, UK), DEX (Sigma), INDO (Sigma), GLB (Sigma), YM 26734 (Tocris), [3H]-PGE2 (PerkinElmer Life Sciences, Milan, Italy), PGE2 antibody (Sigma-Aldrich, Milan, Italy). The drug/molecular target nomenclature follows Alexander et al. (2009).
Results
CSE and CBS are expressed in mouse hind paw
Firstly we investigated the presence of both CBS and CSE in mouse hind paw, the enzymes responsible for the biosynthesis of H2S, in order to assess the contribution of the L-cysteine/H2S pathway in this tissue. Western blot analysis clearly showed the presence of both CBS and CSE under normal, non-inflamed conditions (Figure 1A).
Figure 1.
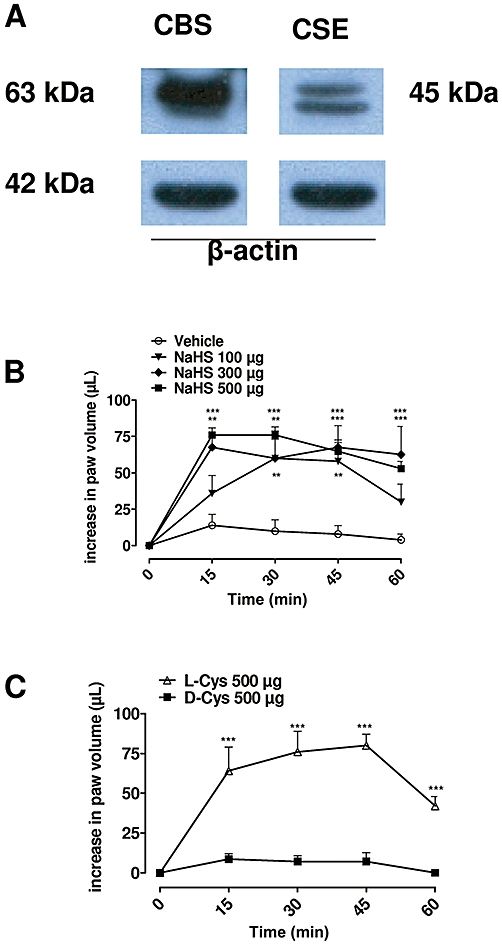
(A) Representative Western blot for cystathionine β-synthase (CBS) and CSE, cystathionine γ-lyase (CSE) in mouse hind paw. (B) Intra-plantar injection of sodium hydrogen sulphide (NaHS) (100, 300 and 500 µg per paw) in mouse hind paw caused a dose-dependent oedema, compared with vehicle (P < 0.0001). (C) Intra-plantar injection of L-cysteine (L-Cys; 500 µg per paw) in mouse hind paw but not D-cysteine (D-Cys; 500 µg per paw) caused significant oedema (P < 0.0001). The statistical analysis was performed by analysis of variance, and each time point was analysed by Bonferroni's post hoc test (**P < 0.01, ***P < 0.001).
Intra-plantar injection of NaHS or L-cysteine induced oedema formation
Intra-plantar injection of NaHS (100, 300 and 500 µg per paw) induced mouse paw oedema in a dose-dependent manner (Figure 1B, P < 0.0001). The peak of oedematogenic response, at the higher doses used, was evident as early as 15 min after injection, reaching a maximum at 30 min and declining by 60 min thereafter (Figure 1B). In order to evaluate the role of the biosynthesis of H2S, we injected intra-plantarly L-cysteine (500 µg per paw) the substrate of CSE/CBS, or d-cysteine (500 µg per paw) as a negative control. L-cysteine, but not d-cysteine, induced oedema (Figure 1C, P < 0.0001), suggesting that the paw tissue efficiently converted L-cysteine into H2S.
Inhibition of 5-HT and histamine receptors and of KATP channels
Pretreatment with CPR (5 mg·kg−1) did not affect the oedematogenic response to NaHS or L-cysteine, excluding the contribution of preformed histamine and 5-HT release to the oedema (Figure 2A,B). Similarly, GLB (10 mg·kg−1) did not modify the NaHS or L-cysteine-induced oedema (Figure 2A,B), suggesting that KATP channel activation was not involved as well.
Figure 2.
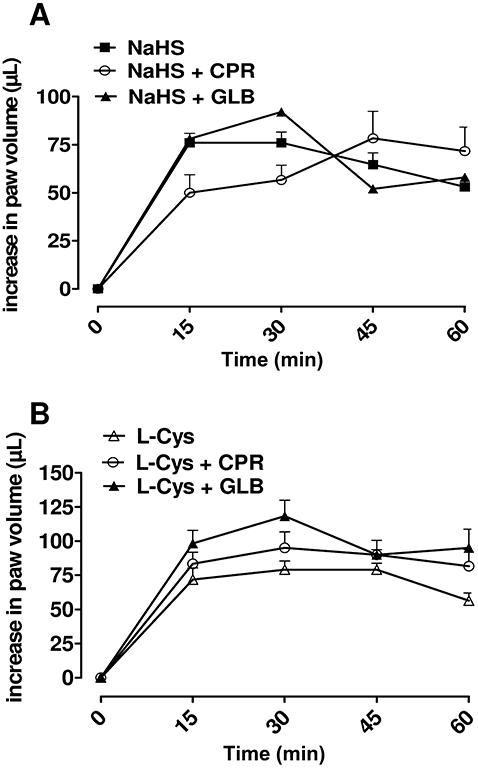
Cyproheptadine (CPR, 5 mg·kg−1) or glibenclamide (GLB, 10 mg·kg−1) did not affect NaHS (A) or L-cysteine (B)-induced oedema at dose of 500 µg per paw. Data were analysed by two way analysis of variance, and each time point was analyzed by Bonferroni's post hoc test.
Modulation of the arachidonic acid cascade
In order to evaluate the involvement of PG, which are the major arachidonic acid metabolites contributing to the formation of oedema, we inhibited different steps in the arachidonic acid cascade by blocking either COX or PLA2 enzymes.
INDO (10 mg·kg−1), a non-selective COX inhibitor, as well as DEX (1 mg·kg−1) significantly (Figure 3A, P < 0.0001), reduced NaHS-induced oedema. Pretreatment with OBAA (0.1, 0.3 and 1 mg·kg−1), a non-selective PLA2 inhibitor, reduced in a dose-dependent manner the oedematogenic response to NaHS (Figure 3B, P < 0.0001). Specific inhibition of the secretory PLA2 isoform (sPLA2) by YM 26734 (0.3, 1 and 3 mg·kg−1) resulted in a dose-dependent reduction of NaHS-induced oedema (P < 0.0001, Figure 3C). Similarly, L-cysteine-induced oedema was inhibited by DEX pretreatment (Figure 3D, P < 0.0001) as well as by OBAA or YM 26734 (data not shown).
Figure 3.
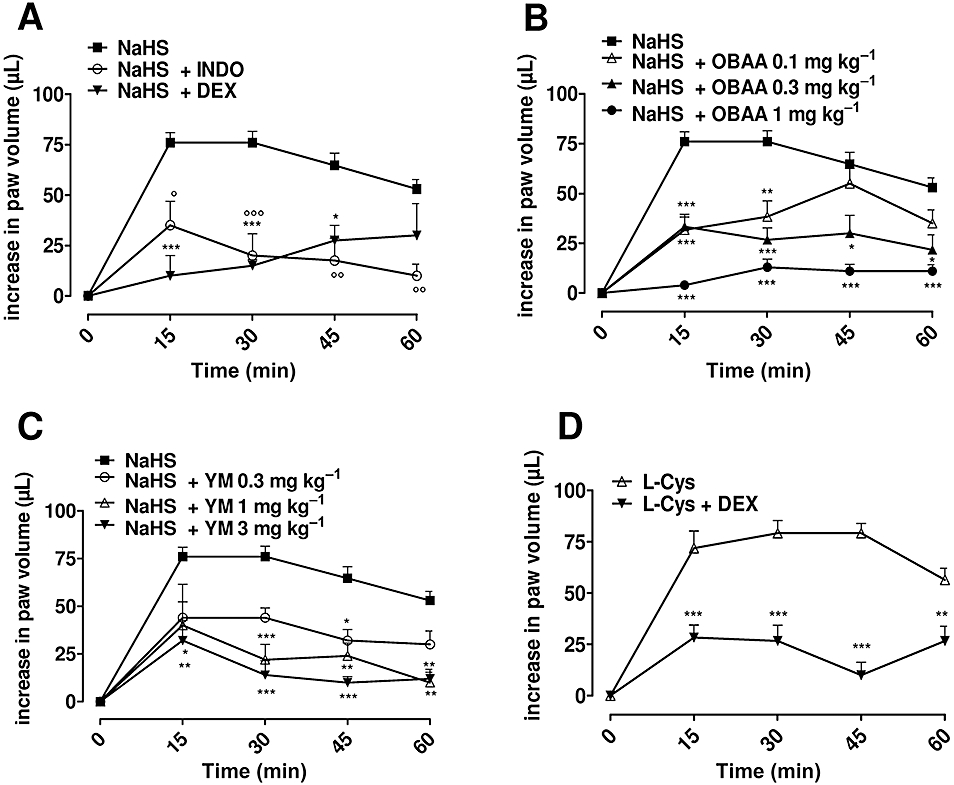
(A) Indomethacin (INDO, 10 mg·kg−1) or dexamethasone (DEX, 1 mg·kg−1) inhibited significantly sodium hydrogen sulphide (NaHS)-induced oedema at dose of 500 µg per paw (P < 0.0001). (B) 4-(4-octadecylphenyl)-4-oxobutenoic acid (OBAA; 0.1, 0.3 and 1 mg·kg−1), PLA2 inhibitor, or (C) YM 26734 (YM; 0.3, 1 and 3 mg·kg−1), sPLA2 inhibitor, reduced significantly in a dose-dependent manner, the NaHS-induced oedema (500 µg per paw; P < 0.0001). (D) DEX (1 mg·kg−1) inhibited significantly the L-cysteine-induced oedema (500 µg per paw; P < 0.0001). The statistical analysis was performed by analysis of variance, and each time point was analysed by Bonferroni's post hoc test (*P < 0.05; **P < 0.01; ***P < 0.001).
Intraplantar injection of NaHS increased PGE2 levels in paw exudates
To further assess the involvement of PG in NaHS-induced paw oedema, we measured PGE2 levels in vehicle- and NaHS-treated mice. Intraplantar injection of NaHS (500 µg per paw) induced a significant increase in PGE2 exudate levels (P < 0.05, Figure 4).
Figure 4.
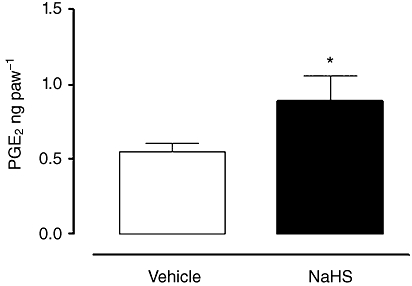
Prostaglandin (PGE2) levels increased in exudates of hind paws collected 30 min after NaHS injection (500 µg per paw), compared with vehicle (*P < 0.05). Results were analysed by Student t-test for unpaired data.
Histological study
Paws injected with L-cysteine, but not those injected with vehicle, clearly showed the loss of tissue organization characteristic of oedema and inflammation (Figure 5A,B). NaHS injection induced a similar pattern of inflammation (Figure 5C,D).
Figure 5.
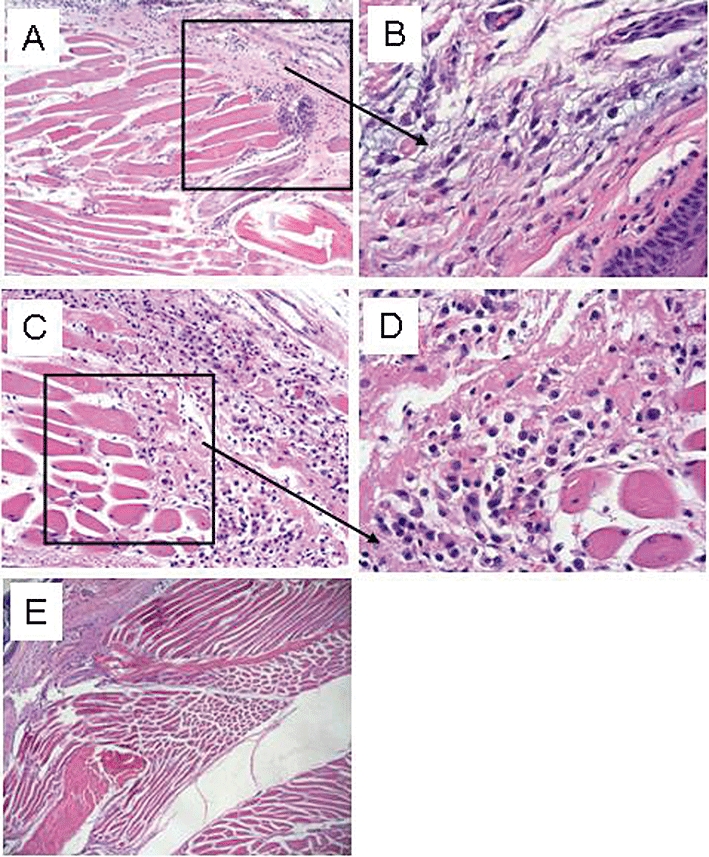
Histological study with haematoxylin-eosin. (A) L-cysteine (500 µg per paw) induced an inflammatory state with a loss of tissue organization, characteristic of oedema (magnification 10×). (B) Higher magnification of boxed area in A (magnification 40×). (C) Sodium hydrogen sulphide (500 µg per paw) induced an inflammatory state with a loss of tissue organization characteristic of oedema (magnification 10×). (D) Higher magnification of boxed area in C (magnification 40×). (E) Absence of inflammatory state following vehicle administration (magnification 10×).
Discussion
The pro-inflammatory role of H2S has been well documented in different animal models (Hui et al., 2003; Bhatia et al., 2005b; Li et al., 2005; Mok and Moore, 2008; Tamizhselvi et al., 2008). Bhatia and co-authors reported a marked increase in H2S production in rat hind paw injected with carrageenan that was significantly reduced by the treatment with propargyl glycine (Bhatia et al., 2005a). However, the intracellular signalling underlying the H2S pro-inflammatory action is still not clear. Here we have assessed the intracellular signalling involved in the H2S pro-inflammatory effect in mice following a local administration of either an exogenous source of H2S (NaHS) or the substrate (L-cysteine).
The Western blot analysis showed that CBS and CSE, the enzymes involved in H2S biosynthesis, were both expressed in mouse hind paw under normal, physiological conditions, implying a role for the L-cysteine/H2S pathway in this tissue. Intra-plantar injection of either NaHS or L-cysteine induced oedema with a rapid onset, implying the involvement of the L-cysteine/H2S pathway in the early phases of the inflammatory reaction (0–60 min).
Intra-plantar injection of L-cysteine, but not d-cysteine, caused oedema, demonstrating that this pro-inflammatory effect was associated with an efficient conversion of L-cysteine to H2S, catalyzed by CBS and/or CSE. Interestingly, both L-cysteine- and NaHS-induced oedema showed a similar profile with a rapid onset which peaked at 30 min. Histological analysis of the paw tissue injected with NaHS or L-cysteine showed a loss of tissue organization and inflammation, characteristic of oedema. In order to gain insights into the intracellular signalling of the pro-inflammatory actions of H2S, we used pharmacological tools. The rapid onset of H2S-induced paw oedema (maximum peak 30 min) suggested an involvement of preformed mediators such as histamine and 5-HT. Pretreatment of the mice with CPR, a 5-HT and histamine receptor antagonist, did not affect either NaHS or L-cysteine-induced oedema. This result excludes the involvement of preformed histamine and 5-HT in the pro-inflammatory effects of H2S.
It is well established that the action of H2S involves activation of KATP channels (Zhao and Wang, 2002). Thus, to assess if this vasodilating property was involved in the pro-inflammatory actions of H2S, we treated mice with GLB, a KATP channel inhibitor. GLB did not affect oedematogenic responses to H2S, suggesting a direct and specific role of H2S in inflammation over and above its vasoactive property. Next, we have evaluated the involvement of the arachidonic acid cascade in the pro-inflammatory actions of H2S. INDO and DEX, inhibitors of the COXs and PLA2 activity, respectively, significantly inhibited H2S induced-oedema. Interestingly, injection of PLA2 in the mouse hind paw resulted in a significant dose-dependent rise in paw oedema, with a rapid onset, within 15–30 min, and a progressive reduction by 1 h after injection (Marshall et al., 1989). The finding that the generation of PLA2-induced oedema had a similar profile to that of H2S-induced oedema suggested that PLA2 activation could be a crucial event in the pro-inflammatory actions of H2S. OBAA, a non-selective PLA2 inhibitor, reduced in a dose-dependent manner H2S-induced oedema in mice, confirming a role for PLA2 in this phenomenon. As it is well known that among the PLA2 isoforms, the sPLA2 plays a major role in the inflammatory process (Landucci et al., 2000; Thimmegowda et al., 2007), mice were treated with YM26734, a selective inhibitor of sPLA2. In our model, YM26734 reduced H2S-induced oedema, dose dependently. These results clearly imply that sPLA2 activation is essential for the pro-inflammatory effects of H2S in the mouse hind paw. This hypothesis was further supported by the increased PGE2 levels measured in exudates from H2S-injected paws. This latter result fits well with experimental evidence showing that PGE2 injection into mouse paw produced a dose- and time-dependent oedema (Claudino et al., 2006), with a similar profile of onset and duration to that observed with H2S.
The activation of sPLA2 proceeds through the orientation of a water molecule by hydrogen bonding to the active site histidine. Adjacent to this histidine, there is a conserved aspartate residue, which, together with a Ca2+-binding loop, acts as a ligand cage for Ca2+ (Murakami and Kudo, 2002). Thus, Ca2+ and water are two key elements implicated in PLA2 activation. Interestingly, H2S could provide both these elements necessary to PLA2 activation. H2S has a three-dimensional structure close to that of H2O, but weaker intermolecular forces, and H2S also induced entry of extracellular Ca2+ (Zhao and Wang, 2002). Thus, it is feasible that H2S could activate PLA2, either through Ca2+entry and/or substituting for a molecule of H2O. Additionally, oxidative modification of phospholipids can alter the physiological state of the membrane, which in turn affects the susceptibility of oxygenated and non-oxygenated fatty acid residues towards sPLA2 attack in a multifaceted way (Murakami and Kudo, 2002). Therefore, an alternative or additional explanation for PLA2 activation by H2S could be that H2S, being a reducing agent, may alter the cellular redox status, triggering prostaglandin production. Moreover, because the isoform of PLA2 involved in the inflammatory process depends on the type of inflammation considered, H2S could interact with both secretory or cytosolic PLA2, without distinguishing between the isoforms.
H2S has been shown to have both pro and anti-inflammatory effects in different experimental in vivo settings (Li et al., 2006). The role of H2S varies depending upon the route of administration, the experimental model used (pathological versus physiological), and if endogenous H2S is modulated (use of inhibitors) or an exogenous source is used (substrates such as L-cysteine or a donor such as NaHS).
In conclusion, we have demonstrated that the pro-inflammatory action of H2S in mouse hind paw involves PLA2 activation with a consequent increase in PGE2 levels. This effect appears to be independent of preformed mediators such as histamine and 5-HT. Furthermore, the present study contributes to the clarification of the controversy on the pro-and anti-inflammatory roles of H2S.
Acknowledgments
We are grateful to medical veterinary Dr Antonio Baiano, Giovanni Esposito and Angelo Russo for animal care assistance.
We thank Prof Teodora Cicala for the English revision of the manuscript.
Glossary
Abbreviations
- CBS
cystathionine β-synthase
- COX
cyclooxygenase
- CSE
cystathionine γ-lyase
- OBAA
4-(4-octadecylphenyl)-4-oxobutenoic acid
- PG
prostaglandin
- PLA2
phospholipase A2
Conflicts of interest
The authors declare no conflicts of interest.
Supporting Information
Teaching Materials; Figs 1–5 as PowerPoint slide.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 4th edn. Br J Pharmacol. 2009;158(Suppl 1):S1–S254. doi: 10.1111/j.1476-5381.2009.00499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M. Hydrogen sulfide and substance P in inflammation. Antioxid Redox Signal. 2010;12:1191–1202. doi: 10.1089/ars.2009.2927. [DOI] [PubMed] [Google Scholar]
- Bhatia M, Sidhapuriwala J, Moochhala SM, Moore PK. Hydrogen sulphide is a mediator of carrageenan-induced hindpaw oedema in the rat. Br J Pharmacol. 2005a;145:141–144. doi: 10.1038/sj.bjp.0706186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M, Wong FL, Fu D, Lau HY, Moochhala SM, Moore PK. Role of hydrogen sulfide in acute pancreatitis and associated lung injury. FASEB J. 2005b;19:623–625. doi: 10.1096/fj.04-3023fje. [DOI] [PubMed] [Google Scholar]
- Church MK, Miller P. Time courses of the anti-anaphylactic and anti-inflammatory effects of dexamethasone in the rat and mouse. Br J Pharmacol. 1978;62:481–486. doi: 10.1111/j.1476-5381.1978.tb07751.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirino G, Cicala C, Bucci MR, Sorrentino L, Maraganore JM, Stone SR. Thrombin functions as an inflammatory mediator through activation of its receptor. J Exp Med. 1996;183:821–827. doi: 10.1084/jem.183.3.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claudino RF, Kassuya CA, Ferreira J, Calixto JB. Pharmacological and molecular characterization of the mechanisms involved in prostaglandin E2-induced mouse paw oedema. Pharmacol Exp Ther. 2006;318:611–618. doi: 10.1124/jpet.106.102806. [DOI] [PubMed] [Google Scholar]
- Hui Y, Du J, Tang C, Bin G, Jiang H. Changes in arterial hydrogen sulfide (H(2)S) content during septic shock and endotoxin shock in rats. J Infect. 2003;47:155–160. doi: 10.1016/s0163-4453(03)00043-4. [DOI] [PubMed] [Google Scholar]
- Landucci EC, Toyama M, Marangoni S, Oliveira B, Cirino G, Antunes E, et al. Effect of crotapotin and heparin on the rat paw oedema induced by different secretory phospholipases A2. Toxicon. 2000;38:199–208. doi: 10.1016/s0041-0101(99)00143-9. [DOI] [PubMed] [Google Scholar]
- Lee AT, Shah JJ, Li L, Cheng Y, Moore PK, Khanna S. A nociceptive-intensity-dependent role for hydrogen sulphide in the formalin model of persistent inflammatory pain. Neuroscience. 2008;152:89–96. doi: 10.1016/j.neuroscience.2007.11.052. [DOI] [PubMed] [Google Scholar]
- Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath RD, Wang ZJ, et al. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005;19:1196–1198. doi: 10.1096/fj.04-3583fje. [DOI] [PubMed] [Google Scholar]
- Li L, Bhatia M, Moore PK. Hydrogen sulphide a novel mediator of inflammation? Curr Opin Pharmacol. 2006;6:125–129. doi: 10.1016/j.coph.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Marshall LA, Chang JY, Calhoun W, Yu J, Carlson RP. Preliminary studies on phospholipase A2-induced mouse paw oedema as a model to evaluate antiinflammatory agents. J Cell Biochem. 1989;40:147–155. doi: 10.1002/jcb.240400203. [DOI] [PubMed] [Google Scholar]
- Mok YY, Moore PK. Hydrogen sulphide is pro-inflammatory in haemorrhagic shock. Inflamm Res. 2008;57:512–518. doi: 10.1007/s00011-008-7231-6. [DOI] [PubMed] [Google Scholar]
- Mok YY, Atan MS, Yoke Ping C, Zhong Jing W, Bhatia M, Moochhala S, et al. Role of hydrogen sulphide in haemorrhagic shock in the rat: protective effect of inhibitors of hydrogen sulphide biosynthesis. Br J Pharmacol. 2004;143:881–889. doi: 10.1038/sj.bjp.0706014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami M, Kudo I. Phospholipase A2. Biochemistry. 2002;31:285–292. doi: 10.1093/oxfordjournals.jbchem.a003101. [DOI] [PubMed] [Google Scholar]
- Posadas I, Bucci M, Roviezzo F, Rossi A, Parente L, Sautebin L, et al. Carrageenan-induced mouse paw oedema is biphasic, age-weight dependent and displays differential nitric oxide cyclooxygenase-2 expression. Br J Pharmacol. 2004;142:331–338. doi: 10.1038/sj.bjp.0705650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamizhselvi R, Moore PK, Bhatia M. Inhibition of hydrogen sulfide synthesis attenuates chemokine production and protects mice against acute pancreatitis and associated lung injury. Pancreas. 2008;36:24–31. doi: 10.1097/MPA.0b013e31816857bb. [DOI] [PubMed] [Google Scholar]
- Thimmegowda NR, Dharmappa KK, Kumar CS, Sadashiva MP, Sathish AD, Nanda BL, et al. Synthesis and evaluation of tricyclic dipyrido diazepinone derivatives as inhibitors of secretory phospholipase A2 with anti-inflammatory activity. Curr Top Med Chem. 2007;7:811–820. doi: 10.2174/156802607780487650. [DOI] [PubMed] [Google Scholar]
- Thomsen LL, Ching LM, Baguley BC. Evidence for the production of nitric oxide by activated macrophages treated with the antitumor agents flavone-8-acetic acid and xanthenone-4-acetic acid. Cancer Res. 1990;50:6966–6970. [PubMed] [Google Scholar]
- Zanardo RC, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006;20:2118–2120. doi: 10.1096/fj.06-6270fje. [DOI] [PubMed] [Google Scholar]
- Zhao W, Wang R. H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol. 2002;283:H474–H480. doi: 10.1152/ajpheart.00013.2002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.