

Abstract
It is notoriously difficult to grow membrane protein crystals and solve membrane protein structures. Improved detection and screening of membrane protein crystals are needed. We have shown here that second-order nonlinear optical imaging of chiral crystals based on second harmonic generation can provide sensitive and selective detection of two-dimensional protein crystalline arrays with sufficiently low background to enable crystal detection within the membranes of live cells. The method was validated using bacteriorhodopsin crystals generated in live Halobacterium halobium bacteria and confirmed by electron microscopy from the isolated crystals. Additional studies of alphavirus glycoproteins indicated the presence of localized crystalline domains associated with virus budding from mammalian cells. These results suggest that in vivo crystallization may provide a means for expediting membrane protein structure determination for proteins exhibiting propensities for two-dimensional crystal formation.
Introduction
Understanding of the atomic structures of membrane proteins is of crucial importance for an understanding of the molecular basis of numerous human diseases and the development of drugs to combat them or ameliorate their consequences. Although approximately one-fourth of the human genome encodes integral membrane proteins there are only ∼270 such structures of integral membrane proteins known (1). In comparison, there are >60,000 independent structures of soluble proteins in the Protein Data Bank (1).
A major reason for the relatively slow pace of discovery for membrane protein structures is simply one of dimension: membrane proteins naturally reside in a two-dimensional lipid environment when embedded within the cell membrane, but virtually all conventional crystallization protocols require extraction of the protein from the cell membrane (e.g., using detergents) into bulk three-dimensional aqueous solution followed by crystallization screening in vitro.
Electron diffraction from two-dimensional crystals provides an alternative route that maintains membrane proteins in a lipid bilayer environment. Since the pioneering work by Henderson and Unwin in 1975 (2) that elucidated the seven trans-membrane helices of bacteriorhodopsin from two-dimensional crystals of purple membrane, several membrane protein structures, including acetylcholine receptor (3,4), bacteriorhodopsin (5), plant light-harvesting complex (6), human red cell aquaporin-1 (7), eye lens aquaporin-0 (8), rat aquaporin-4 (9), glutathione transferase (10), acetylcholine receptor (11), and mercurial insensitive water channel (12) have been solved by electron diffraction to obtain atomic models. Structures solved from two-dimensional crystals formed from overexpressed proteins have also been reported (13–15).
Despite the promise of two-dimensional crystallization approaches, progress in the field has been slowed by major bottlenecks in detection of two-dimensional crystals by electron microscopy (EM) (16). These include:
-
1.
Tedious and time-consuming checking for the formation of two-dimensional crystals using EM. At least ∼30 min are necessary to prepare a negatively stained grid, load a grid into an EM column, search a sufficiently large number of areas of the EM grid to find two-dimensional crystals, and unload the sample grid from the EM.
-
2.
Destructive testing for crystals. An aliquot (2–4 μL) is typically periodically sacrificed to check for two-dimensional crystals using transmission electron microscopy (TEM).
-
3.
The need for relatively large amounts of protein, as each screened condition typically requires 20–100 μL volume for the periodic check by EM. Attempts at high-throughput methodologies have been proposed and implemented involving 96-well dialysis crystallization plates (17), robotic handling of EM grids (18–22), and automated EM imaging (19,23,24).
Although it is conceivable that all of these could be used in tandem to increase the throughput of two-dimensional crystallization screening with EM imaging, the time-consuming nature of EM detection method is challenging to scale-up dramatically and will limit two-dimensional membrane protein crystallization screening to far fewer conditions compared to methods targeting the screening of conditions for three-dimensional crystals.
The major challenges associated with two-dimensional crystal detection may potentially be addressed through the use of second-order nonlinear optical imaging of chiral crystals (SONICC), based on second harmonic generation (SHG) microscopy. SONICC has recently been demonstrated as a selective and sensitive technique for the detection of three-dimensional protein crystals (25,26). SONICC relies on the unique symmetry requirements of SHG, or the frequency doubling of light, for selective detection of protein crystal formation. The sensitivity of SONICC to protein crystal formation arises primarily from the nearly complete suppression of background from unordered media.
For molecules with no preferred orientation, the coherent summation over all molecular orientations produces perfect cancellation and a net response of zero for coherent SHG. However, for proteins oriented by incorporation into either a membrane or a crystalline lattice, the coherent summation leads to addition rather than cancellation, with the SHG intensity scaling with the square of the number of contributing ordered molecules.
Furthermore, the only major practical requirement on the sample cell is accessibility to visible and near-infrared light, such that essentially all crystallization platforms that allow optical imaging also are amenable to SONICC measurements without modification. In this study, SONICC is explored for the detection of two-dimensional crystal formation of integral membrane proteins without requiring labels or tags, and more significantly for the two-dimensional crystals formed while still within the membranes of living cells.
Materials and Methods
Bacterial strains and cell culture conditions
Halobacterium halobium R1 was obtained (ATCC29341; American Type Culture Collection, Manassas, VA). Oxoid peptone medium, from Oesterhelt and Stoeckenlius's formula, contained the following components per liter: NaCl, 250 g; MgSO4, 9.8 g; KCl, 2.0 g; Tri-sodium citrate·2H2O, 3.0 g; Oxoid bacteriological peptone (Cat. No. LP0037, Oxoid, Basingstoke, Hampshire, UK), 10 g. Cell cultures were placed under 850 W flood light (∼50 μmol s−1 m−2), while shaken for 90–100 h at 37°C, 200 rpm, followed by static cultivation for two days. Cell growth was monitored by the ultraviolet/visible spectrometer (Nanodrop 2000c with special cuvette for cell density measurement, Thermo Scientific, Waltham, MA) at 660 nm.
Purple membrane purification
Cells from 1 L culture were harvested by centrifugation (15,000 g) for 15 min. Cell pastes were then resuspended in 25 mL basal salt (medium without peptone) and 50 μL DNase I (2000 U/mL, New England Biolabs, Ipswich, MA) and dialyzed against 2 L of deionized water overnight. The clean red dialysate was centrifuged at 50,000 g for 40 min and resuspended in 35 mL deionized water. After homogenization by a tissue grinder with a tight-fitting Teflon pestle, solution was centrifuged at 50,000 g. This step was repeated two times until the supernatant was nearly colorless.
The purple pellet was then resuspended in 1 mL deionized water. A linear 30–50% (w/w) with a 60% cushion sucrose density gradient centrifugation at 100,000 g, 22 h was used to separate the purple membrane and the red membrane at 15°C (see Fig. S1 in the Supporting Material). The isolated purple band was dialyzed against 1 L deionized water for 3 h with one water change. The dialysate was centrifuged at 14,000 g for 30 min, and finally resuspended in 250 μL deionized water and stored at −20°C.
Imaging of individual H. halobium and BHK-15 cells
SONICC images were acquired using a custom system performing beam scanning with a resonant vibrating mirror (7.8 kHz) to direct the beam along the fast scan axis, and a galvanometer-driven mirror for beam-scanning along the slow axis. The incident beam was generated from a Mai Tai laser, 100 fs, 80 Mhz, ∼50 mW average power at 1000 nm during imaging (Spectra-Physics, Mountain View, CA), focused onto the sample with a 100 × (1.45 N.A.) objective. The SHG was collected in both epi and transmission (XP2920PC; Burle Industries, Lancaster, PA), with dichroic mirrors used to reject the incident beam and narrow (20 nm) band-pass filters (HQ 500/20m-2p; Chroma Technology, Bellows Falls, VT) centered at ∼500 nm. Red fluorescence generated by red fluorescent protein (RFP) was detected with a bandpass filter (HQ 654/24x; Chroma Technology) centered at 654 nm.
H. halobium cells were rinsed and resuspended with basal salt solution to remove fluorescent materials present in the growth medium. A ∼10 μL drop of the bacteria was placed on a poly-L-lysine coated glass coverslip and sandwiched with a coverslip to prevent evaporation. The image shown in Fig. 1 is ∼140 pixels × 140 pixels, corresponding to a field of view of 130 μm × 140 μm, with an approximate acquisition time of 2 min. The BHK-15 cells were imaged in the wells that they were cultured in. The plane of focus was on the bottom of the well where the cells remained adhered to the plate.
Figure 1.

Representative 100× (a) SHG and (b) bright-field images of H. halobium cells, surface immobilized by poly-lysine coating (1000 nm incident with SHG detection at 500 nm).
SHG imaging of purified purple membrane proteins
An ∼2 μL aliquot of purified purple membrane fragments suspended in deionized water was imaged with 1000 nm incident through a 20× objective on a glass slide. A coverslip was placed on top of the drop to prevent drying of the sample. The fragments were concentrated such that there were multiple fragments being probed simultaneously in the focal volume. The transmission generated SHG was detected through a narrow (20 nm) band-pass filters (HQ 500/20m-2p; Chroma Technology) centered at ∼500 nm.
Growth curve of H. halobium
Cells were grown in the same condition as described above. A 1 mL aliquot of the culture was taken every 4–5 h, rinsed with a basal salt solution, and pelleted. The pelleted cells were sandwiched between a glass slide and coverslip coated in a thin layer of glycerol. The glycerol was added to prevent drying of the sample and salt formation during the acquisition time. A 1000-nm incident beam (100 mW average power) was focused with a 20× objective (0.75 N.A.) and scanned over a 400 μm × 500 μm area of the pelleted cells with a vibrating mirror (7.8 kHz) along the fast scan axis and a computerized stage (Optiscan II; Prior Scientific, Fulbourn, Cambridge, UK) for sample-scanning along the slow axis.
The SHG was collected in epi and transmission at 500 nm, summed, and averaged over the scanned area to generate the values in the growth curve plot (Fig. 2). The optical density of the cells was measured by UV-VIS at 660 nm (Nanodrop 2000c; Thermo Scientific). The expression levels of purple membrane were also monitored by depleting retinals (27) from 9 mL aliquots of the cell culture taken every ∼8 h and measured with UV-VIS absorption difference at 568 nm. At each time point, three measurements of 3 mL aliquots were taken and the average was calculated.
Figure 2.

SHG image of purified purple membrane fragments (a) and Cryo-EM image of the purified purple membrane fragments (b), and computed Fourier transform (c) of the fragment (pointed to by an arrow in panel b) shows a diffraction pattern, confirming the crystalline order.
Spectra of H. halobium and purified protein
Emission spectra were acquired by focusing the incident laser beam (100 mW) with a 20× objective (0.75 N.A.) on a sample of pelleted cells or concentrated purified protein. The beam was scanned in the horizontal direction with a vibrating mirror to prevent sample heating and to acquire data from a distribution of cells/protein molecules (∼500 cells were sampled per spectra). The epi-generated emission was separated from the incident beam with a longpass dichroic mirror (Chroma) and focused onto the slit of a monochromator (Spectra Pro 150; Acton, Roper Industries, Sarasota, FL). The emission spectra were generated by scanning a grating (1200 groves/mm, 500 nm blaze angle) and detecting emission with a photomultiplier tube (XP2920PC; Burle). Emission spectra were acquired as a function of incident wavelength by tuning the center wavelength of the Ti:sapphire laser.
Cryo-EM and data processing
Relatively large (∼1 μm2) two-dimensional crystals of purple membranes that had been annealed and fused through Henderson's method (28) were used for imaging by TEM. A quantity of 5 μL of sample suspension and 5 μL 1% trehalose solution were mixed on an aged 400 mesh/inch Cu grid with continuous carbon film which was supported by holey carbon prepared in house. The grid was blotted manually and plunged into liquid ethane and then transferred to liquid nitrogen. Images were recorded on a 4 K × 4 K charge-coupled device camera (Gatan, Pleasanton, CA) with the CM200 TEM (Philips, Andover, MA) operating at 200 kV at liquid nitrogen temperature. The effective magnification was 88,860× (equivalent to 1.69 Å/pixel on the specimen). A single image was used for processing through fast Fourier transform and unbending by the 2DX software suite (29). The two-dimensional crystal lattice of the purple membrane was 65 Å × 65 Å with P3 space group.
Transfection and growth of BHK-15-15 cells
Baby hamster kidney (BHK-15-15) cells were grown in Eagle's minimal essential medium (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum at 37°C in the presence of 5% CO2. All the E2 glycoprotein mutations were constructed from pToto64, a full-length cDNA clone of SINV (30). Two SINV cDNA mutants were constructed in pToto64 with double amino-acid substitutions (Y400A, L402A and C416A, C417A) in the cdE2 region. The mutant cDNA clones and pToto64 were then tagged with RFP on the N-terminus of E2 (J. Jose and R. J. Kuhn, unpublished results).
The wild-type and mutant cDNA clones were linearized with SacI and in vitro transcribed with SP6 RNA polymerase (Amersham Biosciences, Piscataway, NJ). For electroporation, BHK-15-15 cells grown in T-75 culture flasks were harvested by trypsinization and washed twice with phosphate-buffered saline. The cells were then resuspended in 400 μL of phosphate-buffered saline and were mixed with the in vitro transcribed RNA, placed in a 2-mm gap cuvette (BioRad,Hercules, CA), and electroporated (two pulses at settings of 1.5 kV, 25 μF and 200 Ω) using a GenePulser II electroporation apparatus (BioRad). The cells were then suspended in Eagle's minimal essential medium supplemented with 10% fetal bovine serum and plated onto 30-mm wells of six well plates.
Results and Discussion
Second-order nonlinear optical imaging of chiral crystals (SONICC) was used to image H. halobium cells grown under conditions known to promote high expression levels of bacteriorhodopsin (bR), leading to purple membrane patches (i.e., two-dimensional crystals of bR) in the cell membrane (31) (Fig. 1). Live cells were fixed to a poly-L-lysine coated microscope slide and imaged with a 100× microscope objective. Bright SHG was observed from several cells (Fig. 1), with substantial cell-to-cell variation. Throughout the measurements, the bacteria remained adhered to the poly-L-lysine coated glass substrate with no visible changes in morphology.
To validate the specific detection of two-dimensional crystalline bacteriorhodopsin in H. halobium cells by SHG, both SHG microscopy and EM measurements were performed on purified purple membrane fragments (Fig. 2). Strong SHG with an approximate signal/background ratio of 50 was detected for the purified purple membrane fragments (Fig. 2 a). Fourier transformation of the EM image of isolated purple membrane fragments yielded a hexagonal diffraction pattern with a lattice spacing of 65 Å (Fig. 2 c), consistent with the expected spacing for crystalline bR (32).
To further assess the specificity of SHG signal from two-dimensional crystals of bacteriorhodopsin in H. halobium cells, the growth curves of cell density (OD660nm), the amount of extracted purple membrane, and the SHG signal of cells were measured independently (Fig. 3). Consistent with previous findings (31), the purple membrane growth curve was delayed compared to the cell growth curve. Once the cell culture reaches the stationary phase of growth there is a sharp increase of bacteriorhodopsin expression and thus formation of crystalline purple membrane. The SHG curve closely follows the purple membrane curve, suggesting that SHG signals of the whole cells originate predominantly from the crystalline purple membrane throughout the entire cell growth time course.
Figure 3.
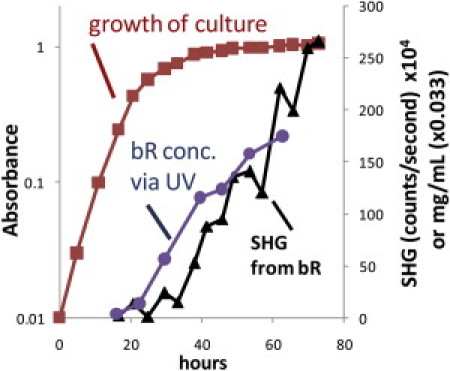
Growth curve of H. halobium cell population and expression levels of crystalline bR monitored by UV-VIS absorbance and SHG. The expression levels of bR exponentially increases after 25 h of growth time as tracked by SHG in real time and UV measurements on bleached aliquots.
Although oriented but noncrystalline proteins contained within a lipid bilayer are also noncentrosymmetric and can potentially produce background SHG, the quadratic dependence on number density combined with the tight dependence on orientational order suggest that the strongest signals can be expected from highly ordered and tightly packed domains such as arising in two-dimensional crystals. Consistent with this prediction, negligible SHG is observed from H. halobium during the early stages of growth (Fig. 3) even when there are high average concentrations of red membrane protein present. The SHG only increases when the purple protein begins to be expressed within the cell, supporting selectivity for crystalline protein versus high concentrations of unordered membrane proteins.
The ability to selectively detect SHG from whole cells hinges on the reduction of autofluorescence at the wavelength of detection. As evident in the emission spectra for live H. halobium cells (Fig. 4 a), there was strong nonspecific fluorescent emission from the cells when excited at 800 nm (equivalent to 400-nm two-photon excited fluorescence). However, when the wavelength of the laser was tuned to lower energies, the autofluorescence was significantly reduced such that there was negligible interference from the bacteria.
Figure 4.
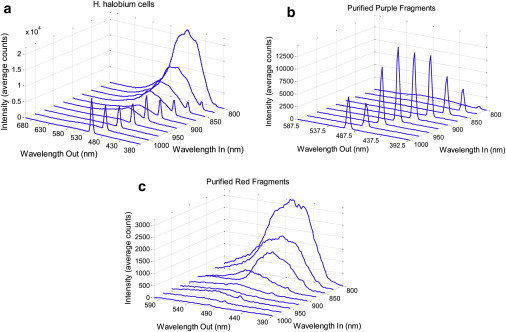
Emission spectra as functions of the excitation wavelength, tuned from 1025 nm to 800 nm for (a) H. halobium prepared with high bR expression levels, (b) isolated purple membrane fragments, and (c) isolated red fragments. The sharp spikes correspond to SHG, while the broad emission arises from autofluorescence. The intensity scale on panel c is approximately fivefold lower than panels a and b.
The bacteria were also rinsed and resuspended in an isotonic salt buffer before measurements to reduce the amount of fluorescence from the nutrients and metals contained in the growth medium. Emission spectra were also acquired for the purified purple and red membrane fragments (Fig. 4, b and c). Strong SHG is detected for the whole cells as well as the purified purple membrane fragments. However, the fragments from the red fraction containing noncrystalline proteins (bacterioruberin) produced no detectable SHG. The small SHG peaks visible from the purified red protein sample in Fig. 4 c are attributed to an incomplete separation of the purple and red protein during purification, as evident from use of sodium-dodecyl-sulfate polyacrylamide-gel electrophoresis (Fig. S2).
Calculations and measurements were performed to assess possible deleterious influences of ultrafast laser exposure on the live cells investigated, building on extensive prior studies (33–35). From studies by Sacconi et al. (33) using changes in cell transmembrane potential as a sensitive indicator for cell perturbation and correcting for the lower numerical aperture and exposure times used in our studies, it was conservatively estimated that the exposures and powers used correspond to changes of ∼1/10,000 in transmembrane potential per minute of continuous imaging. This number represents an upper bound of expected cellular damage for system in the studies examined in this article, and is still three orders-of-magnitude less than the measured voltage changes expected for the initial indicators of cell damage.
Consistent with this estimation, no lift-off of bacteria from the cell surface or changes in cell morphology were observed, both of which would be expected if the laser intensity was obviously damaging. These initial indications suggest that the bacteria remain viable after imaging. The acquisition time is also amenable to routine screening of cells. A 100-μm2 image, as shown in Fig. 1, required 3 min to acquire using an electronics package not optimized for speed, which is already substantially faster than the amount of time it would take to image the cells with electron microscopy. Video-rate nonlinear microscopy is readily achievable (36–38), such that analysis time is likely not to be a significant limiting factor.
SONICC measurements were performed on mammalian cells containing alphaviruses to assess the capabilities of SONICC for detection of two-dimensional protein crystals in eukaryotes. Baby hamster kidney (BHK-15) cells were infected or transfected with in vitro transcribed RNA from either wild-type or nonbudding mutant alphaviruses, respectively, and analyzed for two-dimensional crystal formation using SHG (Fig. 5, b and c). Highly localized areas of SHG were detected both in the forward and backward direction for the cells transfected by alphavirus RNA. These SHG signals are attributed to viral glycoprotein two-dimensional crystals as shown in previous EM images of virus infected cells exhibiting propensities for two-dimensional crystal formation (39).
Figure 5.
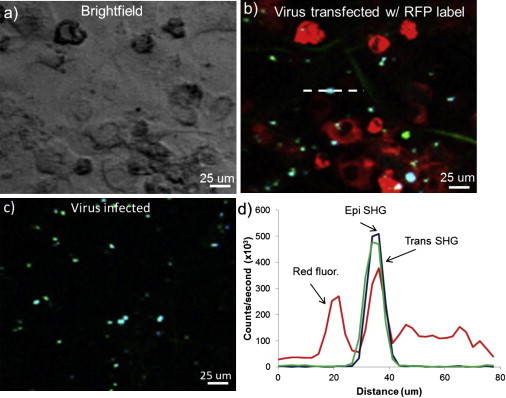
Bright-field image (a) and corresponding SHG and two-photon excitation-fluorescence overlaid images (b) of BHK-15 cells transfected by nonbudding mutant alphavirus with RFP-fused envelope protein E2. SHG image of unlabeled alphavirus-infected BHK-15 cells shown in panel c. Red channel corresponds to two-photon excitation fluorescence from RFP-E2, blue is epi SHG, and green is trans SHG. (d) Line trace of the region (dashed line) in panel b for all three channels.
In addition, much weaker and more diffused SHG signals primarily in transmission were observed in these cells. In contrast, the naïve cells only generated these weaker and diffused SHG signals (Fig. S3) but not the brighter localized SHG signals. We attribute these weaker diffused SHG signals from cellular microfilaments with one-dimensional repeating structures. The one-dimensional structures have much fewer number of repeating units in unit volume compared to two-dimensional crystals, which is consistent with the observed weaker SHG signals from the one-dimensional structures. This is also consistent with the above observations of a lack of similar weak and diffuse SHG signals in bacterial cells because the microfilament structures are unique to eukaryotic cells.
To further correlate the localized bright SHG signals in cells transfected with viral RNA, the cells were transfected with viral RNA of nonbudding mutant alphavirus with RFP fused to the glycoprotein. Alphavirus budding from the plasma membrane of infected cells is driven by the specific interaction of the cytoplasmic domain of the envelope spike glycoprotein E2 (cdE2) and nucleocapsids from the cytoplasm (40–42). During wild-type virus infection, the viral membrane proteins are captured efficiently by budding nucleocapsids and hence it is expected that no large pool of free glycoprotein spikes exists at the plasma membrane (43).
Several mutations in the cdE2 can efficiently prevent the alphavirus budding by selectively interfering with the spike glycoprotein and nucleocapsid interactions (30,40,44). Although during wild-type virus infection, the viral membrane proteins are captured efficiently by budding nucleocapsids, there is still a steady state of glycoprotein accumulation that would produce SHG. The localized SHG signals were found to colocalize with the red fluorescence signals from the RFP-tagged glycoprotein, indicating that the SHG signals originate from the ordered alphavirus glycoproteins (Fig. 5 d). It is interesting to note that there is no obvious difference in the distribution of glycoproteins for the wild and mutated cells in Fig. 5, b and c, suggesting that the mutation and RFP labeling does not interfere with the glycoprotein assembly. In addition, only a small fraction of the RFP-glycoprotein regions generates SHG signals. We attribute the SHG-silent fractions of RFP-glycoprotein regions to noncrystalline monomers or aggregates of alphavirus RFP-glycoprotein.
Additional evidence supporting the assignment of the localized SHG to glycoprotein two-dimensional crystal formation can be obtained from the thickness-dependence of SHG. SHG-active features are less than the backward coherence length of the propagating light field (e.g., 150 nm based on the refractive index of 1.3 for similar proteins (45)), generating SHG equally strong in the forward and backward directions. As the features increase in thickness approaching the wavelength of the doubled light, the fraction of the total light generated in the forward direction generally increases (46).
Thus, features generating SHG equally in the forward and backward direction can be reasonably assumed to be much less than the wavelength of light in thickness. The much weaker diffused SHG signals in the BHK-15 cells are predominantly forward-propagating SHG, which is consistent with thicker structures (collectively >∼150 nm in thickness) exhibiting fairly low orientational order. The localized areas of signal from virus-infected cells and the bacteriorhodopsin in H. halobium generate light propagating in both the epi and transmission directions are indicative of feature thickness of <150 nm. This observation is consistent with the assignment of SHG arising from ultrathin two-dimensional crystalline arrays.
Our data suggest that the SONICC method can sensitively and specifically detect two-dimensional crystals even in complex surrounding milieu of noncrystalline intracellular and extracellular proteins in live cells. SONICC is highly selective for proteins ordered over distances approaching optical wavelengths (>100 nm), exhibiting no detectable background signals from disordered proteins or protein aggregates. It is known that many membrane proteins from a wide range of organisms form two-dimensional crystals in vivo (2,47,48), suggesting possible merits of performing larger-scale SONICC screenings of membrane protein two-dimensional crystals at the very earliest stages with the membrane proteins still within the live cells. Many factors, such as the intrinsic propensity of the protein, induced overexpression, and controlled cell culture conditions can potentially lead to formation of such membrane two-dimensional crystals in vivo in live cells.
A rough estimate of the lower limit for the smallest two-dimensional unlabeled membrane protein crystal that can be reliably detected by SONICC can be assessed by inspection of the images shown in Fig. 1 and Fig. 5 b. In the case of bR, the single-pixel signals from crystalline patches corresponded to ∼60 counts. Because of the quadratic dependence of SONICC on the incident intensity, only the proteins present within the confocal volume probed in each pixel contribute to the detected signal. In this case, single pixel patches of bR and E2 membrane protein crystals no larger than 1 μm × 1 μm × 1 molecular monolayer in dimension produced SONICC signal to background ratios of 32 and 420, respectively. These dimensions correspond to a maximum of ∼25,000 protein copies within the crystalline array. Significant improvements in these values can be reasonably expected in more pristine crystallization matrices, but this is already approaching the lower practical limit for patch sizes amenable to high-resolution electron diffraction characterization.
Facile label-free detection of two-dimensional crystals has the potential to significantly influence several pipelines for membrane protein structure determination. Optical detection of two-dimensional crystal formation addresses a key bottleneck in screening and optimization of conditions for electron diffraction analysis, in combination with advances in EM instrumentation and sample preparation methods. Even in efforts targeting the generation of three-dimensional crystals for x-ray diffraction analysis, generation of Type I three-dimensional crystals of membrane proteins hinge on the formation of ordered two-dimensional assemblies serving as the repeat units within the crystals.
Under favorable conditions such as those realized for bR, in vivo detection may serve as an early indicator for promise in subsequent purification, stabilization, and crystallization efforts targeting the formation of Type I crystals.
Finally, two-dimensional crystallization may even enable initial structural studies by EM directly on the isolated crystalline fragments without extraction and recrystallization of the monomeric protein, provided the patches can be isolated intact from a lysate as in the case of bR. However, the generality of this last approach remains largely undetermined. Additional experiments aided by the availability of facile two-dimensional crystal detection demonstrated herein may provide insights into its potential viability.
Acknowledgments
The authors are grateful to William A. Cramer for helpful discussions and suggestions. The EM images of purple membrane were taken in the Purdue Biological Electron Microscopy Facility.
W.J. and G.J.S. acknowledge financial support from the National Institutes of Health (P50 award No. 1P50GM088499-01). R.J.K. acknowledges support from National Institute of General Medical Sciences award No. GM56279.
Supporting Material
References
- 1.Raman P., Cherezov V., Caffrey M. The membrane Protein Data Bank. Cell. Mol. Life Sci. 2006;63:36–51. doi: 10.1007/s00018-005-5350-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Henderson R., Unwin P.N.T. Three-dimensional model of purple membrane obtained by electron microscopy. Nature. 1975;257:28–32. doi: 10.1038/257028a0. [DOI] [PubMed] [Google Scholar]
- 3.Miyazawa A., Fujiyoshi Y., Unwin N. Nicotinic acetylcholine receptor at 4.6 Å resolution: transverse tunnels in the channel wall. J. Mol. Biol. 1999;288:765–786. doi: 10.1006/jmbi.1999.2721. [DOI] [PubMed] [Google Scholar]
- 4.Ross M.J., Klymkowsky M.W., Stroud R.M. Structural studies of a membrane-bound acetylcholine receptor from Torpedo californica. J. Mol. Biol. 1977;116:635–659. doi: 10.1016/0022-2836(77)90264-9. [DOI] [PubMed] [Google Scholar]
- 5.Henderson R., Baldwin J.M., Downing K.H. Model for the structure of bacteriorhodopsin based on high-resolution electron cryo-microscopy. J. Mol. Biol. 1990;213:899–929. doi: 10.1016/S0022-2836(05)80271-2. [DOI] [PubMed] [Google Scholar]
- 6.Kühlbrandt W., Wang D.N., Fujiyoshi Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature. 1994;367:614–621. doi: 10.1038/367614a0. [DOI] [PubMed] [Google Scholar]
- 7.Murata K., Mitsuoka K., Fujiyoshi Y. Structural determinants of water permeation through aquaporin-1. Nature. 2000;407:599–605. doi: 10.1038/35036519. [DOI] [PubMed] [Google Scholar]
- 8.Gonen T., Cheng Y.F., Walz T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tani K., Mitsuma T., Fujiyoshi Y. Mechanism of aquaporin-4's fast and highly selective water conduction and proton exclusion. J. Mol. Biol. 2009;389:694–706. doi: 10.1016/j.jmb.2009.04.049. [DOI] [PubMed] [Google Scholar]
- 10.Holm P.J., Bhakat P., Hebert H. Structural basis for detoxification and oxidative stress protection in membranes. J. Mol. Biol. 2006;360:934–945. doi: 10.1016/j.jmb.2006.05.056. [DOI] [PubMed] [Google Scholar]
- 11.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4Å resolution. J. Mol. Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]
- 12.Yang B.X., Brown D., Verkman A.S. The mercurial insensitive water channel (AQP-4) forms orthogonal arrays in stably transfected Chinese hamster ovary cells. J. Biol. Chem. 1996;271:4577–4580. [PubMed] [Google Scholar]
- 13.Appel M., Hizlan D., Kühlbrandt W. Conformations of NhaA, the Na/H exchanger from Escherichia coli, in the pH-activated and ion-translocating states. J. Mol. Biol. 2009;386:351–365. doi: 10.1016/j.jmb.2008.12.042. [DOI] [PubMed] [Google Scholar]
- 14.Bodey A.J., Kikkawa M., Moores C.A. 9-Ångström structure of a microtubule-bound mitotic motor. J. Mol. Biol. 2009;388:218–224. doi: 10.1016/j.jmb.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 15.Jegerschöld C., Pawelzik S.C., Hebert H. Structural basis for induced formation of the inflammatory mediator prostaglandin E2. Proc. Natl. Acad. Sci. USA. 2008;105:11110–11115. doi: 10.1073/pnas.0802894105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Renault L., Chou H.T., Stahlberg H. Milestones in electron crystallography. J. Comput. Aided Mol. Des. 2006;20:519–527. doi: 10.1007/s10822-006-9075-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vink M., Derr K., Ubarretxena-Belandia I. A high-throughput strategy to screen 2D crystallization trials of membrane proteins. J. Struct. Biol. 2007;160:295–304. doi: 10.1016/j.jsb.2007.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Potter C.S., Pulokas J., Carragher B. Robotic grid loading system for a transmission electron microscope. J. Struct. Biol. 2004;146:431–440. doi: 10.1016/j.jsb.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 19.Stagg S.M., Lander G.C., Potter C.S. Automated cryoEM data acquisition and analysis of 284742 particles of GroEL. J. Struct. Biol. 2006;155:470–481. doi: 10.1016/j.jsb.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 20.Cheng A., Leung A., Potter C.S. Towards automated screening of two-dimensional crystals. J. Struct. Biol. 2007;160:324–331. doi: 10.1016/j.jsb.2007.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lefman J., Morrison R., Subramaniam S. Automated 100-position specimen loader and image acquisition system for transmission electron microscopy. J. Struct. Biol. 2007;158:318–326. doi: 10.1016/j.jsb.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu M.H., Vink M., Stokes D. Automated electron microscopy for evaluating two-dimensional crystallization of membrane proteins. J. Struct. Biol. 2010;171:102–110. doi: 10.1016/j.jsb.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Potter C.S., Chu H., Carragher B. LEGINON: a system for fully automated acquisition of 1000 electron micrographs a day. Ultramicroscopy. 1999;77:153–161. doi: 10.1016/s0304-3991(99)00043-1. [DOI] [PubMed] [Google Scholar]
- 24.Suloway C., Pulokas J., Carragher B. Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 2005;151:41–60. doi: 10.1016/j.jsb.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 25.Wampler R.D., Kissick D.J., Simpson G.J. Selective detection of protein crystals by second harmonic microscopy. J. Am. Chem. Soc. 2008;130:14076–14077. doi: 10.1021/ja805983b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kissick D.J., Gualtieri E.J., Cherezov V. Nonlinear optical imaging of integral membrane protein crystals in lipidic mesophases. Anal. Chem. 2010;82:491–497. doi: 10.1021/ac902139w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shand R.F., Betlach M.C. Expression of the bop gene cluster of Halobacterium halobium is induced by low oxygen tension and by light. J. Bacteriol. 1991;173:4692–4699. doi: 10.1128/jb.173.15.4692-4699.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baldwin J., Henderson R. Measurement and evaluation of electron-diffraction patterns from 2-dimensional crystals. Ultramicroscopy. 1984;14:319–335. [Google Scholar]
- 29.Gipson B., Zeng X., Stahlberg H. 2DX—user-friendly image processing for 2D crystals. J. Struct. Biol. 2007;157:64–72. doi: 10.1016/j.jsb.2006.07.020. [DOI] [PubMed] [Google Scholar]
- 30.Owen K.E., Kuhn R.J. Alphavirus budding is dependent on the interaction between the nucleocapsid and hydrophobic amino acids on the cytoplasmic domain of the E2 envelope glycoprotein. Virology. 1997;230:187–196. doi: 10.1006/viro.1997.8480. [DOI] [PubMed] [Google Scholar]
- 31.Oesterhelt D., Stoeckenius W. Isolation of the cell membrane of Halobacterium halobium and its fractionation into red and purple membrane. Methods Enzymol. 1974;31(Pt A):667–678. doi: 10.1016/0076-6879(74)31072-5. [DOI] [PubMed] [Google Scholar]
- 32.Henderson R. The purple membrane from Halobacterium halobium. Annu. Rev. Biophys. Bioeng. 1977;6:87–109. doi: 10.1146/annurev.bb.06.060177.000511. [DOI] [PubMed] [Google Scholar]
- 33.Sacconi L., Dombeck D.A., Webb W.W. Overcoming photodamage in second-harmonic generation microscopy: real-time optical recording of neuronal action potentials. Proc. Natl. Acad. Sci. USA. 2006;103:3124–3129. doi: 10.1073/pnas.0511338103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang H.F., Fu Y., Cheng J.X. Experimental observation and theoretical analysis of Raman resonance-enhanced photodamage in coherent anti-Stokes Raman scattering microscopy. J. Opt. Soc. Am. B. 2007;24:544–552. [Google Scholar]
- 35.Fu Y., Wang H.F., Cheng J.X. Characterization of photodamage in coherent anti-Stokes Raman scattering microscopy. Opt. Express. 2006;14:3942–3951. doi: 10.1364/oe.14.003942. [DOI] [PubMed] [Google Scholar]
- 36.Roorda R.D., Hohl T.M., Miesenböck G. Video-rate nonlinear microscopy of neuronal membrane dynamics with genetically encoded probes. J. Neurophysiol. 2004;92:609–621. doi: 10.1152/jn.00087.2004. [DOI] [PubMed] [Google Scholar]
- 37.Evans C.L., Potma E.O., Xie X.S. Chemical imaging of tissue in vivo with video-rate coherent anti-Stokes Raman scattering microscopy. Proc. Natl. Acad. Sci. USA. 2005;102:16807–16812. doi: 10.1073/pnas.0508282102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Veilleux I., Spencer J.A., Lin C.P. In vivo cell tracking with video rate multimodality laser scanning microscopy. IEEE J. Sel. Top. Quantum Electron. 2008;14:10–18. [Google Scholar]
- 39.von Bonsdorff C.H., Harrison S.C. Hexagonal glycoprotein arrays from Sindbis virus membranes. J. Virol. 1978;28:578–583. doi: 10.1128/jvi.28.2.578-583.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao H.X., Lindqvist B., Liljeström P. A tyrosine-based motif in the cytoplasmic domain of the alphavirus envelope protein is essential for budding. EMBO J. 1994;13:4204–4211. doi: 10.1002/j.1460-2075.1994.tb06740.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee S., Owen K.E., Kuhn R.J. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure. 1996;4:531–541. doi: 10.1016/s0969-2126(96)00059-7. [DOI] [PubMed] [Google Scholar]
- 42.Mukhopadhyay S., Zhang W., Rossmann M.G. Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses. Structure. 2006;14:63–73. doi: 10.1016/j.str.2005.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Strauss J.H., Strauss E.G. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 1994;58:491–562. doi: 10.1128/mr.58.3.491-562.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ivanova L., Schlesinger M.J. Site-directed mutations in the Sindbis virus E2 glycoprotein identify palmitoylation sites and affect virus budding. J. Virol. 1993;67:2546–2551. doi: 10.1128/jvi.67.5.2546-2551.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cole T., Kathman A., McPherson A. Determination of local refractive index for protein and virus crystals in solution by Mach-Zehnder interferometry. Anal. Biochem. 1995;231:92–98. doi: 10.1006/abio.1995.1507. [DOI] [PubMed] [Google Scholar]
- 46.Lacomb R., Nadiarnykh O., Campagnola P.J. Phase matching considerations in second harmonic generation from tissues: effects on emission directionality, conversion efficiency and observed morphology. Opt. Commun. 2008;281:1823–1832. doi: 10.1016/j.optcom.2007.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fotiadis D., Jenö P., Engel A. Structural characterization of two aquaporins isolated from native spinach leaf plasma membranes. J. Biol. Chem. 2001;276:1707–1714. doi: 10.1074/jbc.M009383200. [DOI] [PubMed] [Google Scholar]
- 48.Wille H., Michelitsch M.D., Prusiner S.B. Structural studies of the scrapie prion protein by electron crystallography. Proc. Natl. Acad. Sci. USA. 2002;99:3563–3568. doi: 10.1073/pnas.052703499. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.